

Abstract
Protein synthesis inhibitor antibiotics are widely used to produce amnesia, and have been recognized to inhibit general or global mRNA translation in the basic translational machinery. For instance, anisomycin interferes with protein synthesis by inhibiting peptidyl transferase or the 80S ribosomal function. Therefore, de novo general or global protein synthesis has been thought to be necessary for long-term memory formation. However, it is unclear which mode of translation—gene-specific translation or general/global translation—is actually crucial for the memory consolidation process in mammalian brains. Here, we generated a conditional transgenic mouse strain in which double-strand RNA-dependent protein kinase (PKR)-mediated phosphorylation of eIF2α, a key translation initiation protein, was specifically increased in hippocampal CA1 pyramidal cells by the chemical inducer AP20187. Administration of AP20187 significantly increased activating transcription factor 4 (ATF4) translation and concomitantly suppressed CREB-dependent pathways in CA1 cells; this led to impaired hippocampal late-phase LTP and memory consolidation, with no obvious reduction in general translation. Conversely, inhibition of general translation by low-dose anisomycin failed to block hippocampal-dependent memory consolidation. Together, these results indicated that CA1-restricted genetic manipulation of particular mRNA translations is sufficient to impair the consolidation and that consolidation of memories in CA1 pyramidal cells through eIF2α dephosphorylation depends more on transcription/translation of particular genes than on overall levels of general translation. The present study sheds light on the critical importance of gene-specific translations for hippocampal memory consolidation.
Introduction
Proper neuronal functions are highly dependent on precise control of mRNA translation. Broadly defined, translational control falls into two categories: general or global translation, which affects most mRNAs through the regulation of basic translational machinery, and gene-specific translation by way of a specific cis-acting element in the mRNAs. Notably, in neurons, external signals induce translational derepression by regulating both general translational machinery and mRNA-specific repressors (Besse and Ephrussi, 2008). For instance, glutamate receptor activation triggers eEF2 phosphorylation, by which general protein synthesis is suppressed through phospho-eEF2-mediated peptide elongation arrest. Concomitantly, translation of a subset of mRNAs, including αCaMKII and Arc/Arg3.1, is selectively increased to trigger plastic change and memory consolidation (Scheetz et al., 2000; Park et al., 2008). Other studies suggests that general translation initiation occurs uniformly in response to multiple factors and neural activities, including BDNF, NMDA, and L-LTP induction (Kelleher et al., 2004). Therefore, the relative contributions of general and gene-specific translations to long-term plasticity and memory have yet to be delineated.
Reversible phosphorylation of the α subunit of eIF2 on Ser51 is one of the best characterized rate-limiting steps at the translational initiation level, and it controls the rates of both gene-specific translation and general translation (Hinnebusch, 2000; Dever, 2002; Sonenberg and Dever, 2003). When bound to GTP and the initiator Met-tRNAiMet in the cytoplasm, eIF2 forms a functional eIF2·GTP·Met-tRNAiMet ternary complex that delivers Met-tRNAiMet to the 40S ribosomal subunit. Ser51 phosphorylation of eIF2α precludes ternary complex formation and thus blocks general translation initiation. Paradoxically, the decrease in ternary complexes stimulates translation of specific mRNAs that contain upstream open reading frames (uORFs), including activating transcription factor 4 (ATF4) (Harding et al., 2000; Vattem and Wek, 2004) and β-site APP (amyloid precursor protein)-cleaving enzyme 1 (BACE1) (O'Connor et al., 2008). Notably, ATF4, also known as cAMP-responsive element binding protein-2 (CREB-2), is a negative regulator (Karpinski et al., 1992) of the CRE-dependent transcription critical for memory consolidation (Silva et al., 1998; Pittenger et al., 2002). Interestingly, behavioral training triggers Ser51 dephosphorylation of eIF2α, suggesting that eIF2α phosphorylation acts as a brake for memory consolidation (Costa-Mattioli et al., 2007). Indeed, genetic reduction of eIF2α phosphorylation, either in GCN2 knock-out mice (Costa-Mattioli et al., 2005) (one major eIF2α kinase) or in eIF2α+/S51A knock-in mice (Costa-Mattioli et al., 2007), reduces the induction threshold of both L-LTP and learning in behavioral tasks. Conversely, preventing training-induced eIF2α dephosphorylation with Sal003, an eIF2α phosphatase inhibitor, results in a consolidation impairment (Costa-Mattioli et al., 2007). These reports suggest that translational control through eIF2α phosphorylation is critical for establishing long-lasting synaptic changes and memory formation (Costa-Mattioli et al., 2009). However, upon training-induced eIF2α dephosphorylation, it remains unclear which translation mechanisms, general translation or gene-specific translation, efficiently gates the transition from short-term memory to long-term memory formation. To address this issue, we generated and analyzed a conditional double-stranded RNA-dependent protein kinase (PKR) transgenic mouse strain, in which translation of a particular protein—ATF4—through eIF2α phosphorylation in the hippocampal CA1 neurons was enhanced by a chemical inducer.
Materials and Methods
All experimental procedures were performed in accordance with the guidelines published in the National Research Council Guide for the Care and Use of Laboratory Animals, and were approved by the National Institute of Mental Health Animal Care and Use Committee. All efforts were made to minimize the number of animals used and their suffering.
Generation of transgenic mice
To manipulate translational machinery in the mouse brain, we generated loxP-flanked (“floxed”)-PKR transgenic mouse lines. We used a FK506 binding protein 1A (FKBP-12)-based chemically induced dimerization system (ARIAD Pharmaceuticals) to control the dimerization of the PKR kinase domain. The chemical dimerization inducer AP20187 binds with high affinity (subnanomolar concentration) to the mutant FKBP-12 with a single amino acid substitution (F36V), while binding affinity to the wild-type protein is 1000-fold lower (Clackson et al., 1998). A kinase domain cDNA of human PKR was excised from pC939 (a gift from Thomas E. Dever, National Institute of Child Health and Human Development, Bethesda, MD) with BamHI and HindIII, then fused in frame to a F36V mutant cDNA encoding FK506-binding protein 1A (FKBP-12 or FKBP), which was isolated from human MGC verified full-length cDNA clones (Open Biosystems) and was then mutated by PCR-based site-directed mutagenesis. The FKBP-PKR cDNA was inserted into pNN265 plasmid that carries a 5′ intron and a 3′ intron plus poly-A signal from SV40 (provided originally by Nobuki Nakanishi, Burnham Institute for Medical Research, La Jolla, CA) through the EcoRV site. A 3.6 kb fragment of n(nuclear localization signal)LacZ-stop-pA, which was isolated from 3.1-actin-nLacZ plasmid (from Eric H. Mercer, California Institute of Technology, Pasadena, CA) with SalI and BglII, was flanked by two loxP sequences, and placed in front of FKBP-PKR cDNA in pNN265. Finally, the entire DNA fragment with 5′ intron-loxP-nLacZ-stop-loxP-FKBP-PKR-3′ intron/pA was cut out from pNN265 vector using NotI and ligated under CaMKIIα promoter, which was derived from pJTCRE (a gift from Joe Z. Tsien, Medical College of Georgia, Atlanta, GA), to make pJPKR5 plasmid. The 15.2 kb linearized DNA fragment with SalI digestion of pJPKR5, which carried CaMKIIα promoter, loxP-nlsLacZ-stop-loxP sequence, and FKBP-PKR-pA, was injected into fertilized C57BL/6N mouse oocytes. Germline transmission of the transgenic construct was confirmed with PCR and Southern blots using tail DNA. Primers for genotyping of floxed-PKR transgenic lines were as follows: 5′-TTCTCCGTTTGCACTCAG-3′ (PKR primer 1) and 5′-CACTTTGCGTTTCTTCTTGGG-3′ (PKR primer 2). One of the regionally expressed floxed-PKR lines, fPKR#6, was crossed with a forebrain-specific Cre recombinase line, T29-1(Tsien et al., 1996). Double transgenic offspring (fPKR+/−; Cre+/−) were genotyped by PCR with the above PKR primers, and with a Cre primer set (5′-CCGGGCTGCCACGACCAA-3′ and 5′-GGCGCGGCAACACCATTTTT-3′) to detect the Cre transgene (Scheel et al., 2003). Both fPKR control mice and fPKR/Cre double hemizygous mutant mice were viable and fertile without obvious behavioral abnormalities.
In vivo drug administration
Intracerebroventricular administration of AP20187 (a gift from ARIAD Pharmaceuticals Inc.) into the cannulated mice was performed as previously described (Karpova et al., 2005). AP20187 was dissolved in ethanol with the concentration of 62.5 mg/ml and further diluted to the final concentration of 1 nmol/μl with saline before use. One nanomole of AP20187 was injected over a period of 1 min using a microinjection unit (Stoelting). Anisomycin (Sigma) was dissolved in normal saline with HCl and adjusted to pH 7.4 with NaOH for a final concentration of 17.5 mg/ml, and then subcutaneously injected into mice at a dose of 5 mg/kg, 10 mg/kg, or 100 mg/kg body weight 10 min before fear conditioning or 1 h before [35S]-methionine intraperitoneal injection.
RT-PCR
Hippocampi or hippocampal CA1 from fPKR control and mutant mice were dissected out and the total RNA was extracted using Trizol reagent (Invitrogen) and purified with RNeasy clean up kit (QIAGEN) after treatment with RQ1 DNase (Promega). RNA concentration and quality were examined by Nano drop (Thermo Fisher Scientific). Messenger RNA was reverse transcribed into cDNA with Superscript II reverse transcriptase (Invitrogen) using oligo(dT)15 as primers in a 20 μl reaction containing 1 μg of total RNA. The reaction mixtures were diluted 100-fold and then subjected to PCR amplification. A 25 μl PCR contained 100 pmol primers and 1 μCi 32P-dCTP (PerkinElmer). The PCR products were separated by electrophoresis on 8% polyacrylamide gels and were visualized and quantified by PhosphorImager analysis (GE Healthcare). The primers were as follows: BDNF promoter IV-specific primers, 5′-TGCGAGTATTACCTCCGCCAT-3′ and 5′-AGGATGGTCATCACTCTTCTC-3′; ATF4 primers 5′-TTCCTGAACAGCGAAGTGTTGG-3′ and 5′-CTAAAGGAATGCTCTGGAGTGG-3′; c-fos primers 5′-TGGTGAAGACCGTGTCAGGA-3′ and 5′-GCAGCCATCTTATTCCGTTCC-3′; zif268 primers 5′-CCTTCCAGTGTCGAATCTGCA-3′ and 5′-CTGGCAAACTTCCTCCCACA-3′; C/EBPβ primers 5′-GGGACTTGATGCAATCCGG-3′ and 5′-AACCCCGCAGGAACATCTTT-3′; GluR1 (GRIA1) primers 5′-CAAATATGCCTACCTCCTGG-3′ and 5′-ACCCCTTGGGTGTTGCAATG-3′; NR2A (GRIN2A) primers 5′-AAGCCCCCTTCGTCATCGTA-3′ and 5′-CACATTCATCCCTTCGTTGG-3′; PSD95 primers 5′-AGCCCCAGGATATGAGTTGC-3′ and 5′-GGTGATAAAGATGGATGGGTCG-3′. PCR amplification was performed for 27 cycles for the primers above. GAPDH primers 5′-TTGTCAGCAATGCATCCTGCAC-3′and 5′-GTTGCTGTTGAAGTCACAGGAGAC-3′ and β-actin primers 5′-TGCGTGACATCAAAGAGAAG-3′ and 5′-GATGCCACAGGATTCCATA-3′ were used as internal controls which were amplified for 23 cycles.
Digoxygenin in situ hybridization
RNA probes specific for human PKR mRNA were complementary to nucleotides 1399–1515 and nucleotides 1885–1985 in the human PKR mRNA (GenBank accession #NM_002759). In situ hybridization was performed as previously described (Schaeren-Wiemers and Gerfin-Moser, 1993; Matsunami and Buck, 1997). Briefly, digoxigenin (DIG)-labeled antisense and sense RNA probes were hybridized with frozen brain sections from 12-week-old mutants. After washing, DIG probes were reacted with alkaline phosphatase-conjugated anti-DIG, and signals were detected using NBT-BCIP.
Western blotting
Western blots were performed as previously described (Jiang et al., 2003). Antibodies including rabbit polyclonal anti-eIF2α (sc-11386), anti-BDNF (sc-546), and anti-CaMKIIα (sc-32288) were obtained from Santa Cruz Biotechnology, anti-phospho-eIF2α (#9721) was obtained from Cell Signaling Technology, anti-α-tubulin (T6199) was obtained from Sigma, anti-human PKR (#1511-1) was obtained from Epitomics, anti-ATF4 (#10834-1-AP) was obtained from ProteinTech Group, and anti-BACE1 (#18711) was obtained from IBL. Western blots results were quantified with KODAK Molecular Imaging Software (Carestream Health Molecular Imaging).
X-gal staining
Floxed-PKR mice and mutants were perfused with 30 ml of cold 5 mm EGTA/0.9% saline, then 100 ml of cold 4% paraformaldehyde in PBS. After being postfixed for 15 min, the fixed brains were sectioned by Vibratome (Leica VT1000S) in cold PBS with 50 μm thickness. The sections were washed in cold rinse buffer (1 × PBS, pH 7.6, 2 mm MgCl2, 0.01% SDS, and 0.02% NP-40) and then subjected to X-Gal staining overnight for up to 24 h at 37°C. The X-Gal staining solution contained 0.5 mg/ml X-Gal (4-chloro-5-bromo-3-indolyl-β-galactosidase), 5 mm K4Fe(CN)6·3H2O, 5 mm K3Fe(CN)6, and 2 mm MgCl2 in PBS. The X-Gal-stained slides were washed with PBS and postfixed with 10% formalin for 2 h at room temperature, then mounted to precoated Probe-On glass-slides (Fisher Scientific). After air drying completely, the sections were counterstained with Safranin O (Nissl staining).
Fluoro-Jade B staining
Fluoro-Jade B staining was performed according to the manufacturer's instructions (Histo-Chem). Briefly, sections were initially mounted, rehydrated with 80% ethanol and 70% ethanol, washed with distilled water, and incubated in 0.06% potassium permanganate solution for 10 min. The sections were then incubated in 0.0004% Fluoro-Jade B solution containing 0.1% glacial acetic acid for 15 min at room temperature, washed, and mounted with distyrene plasticizer xylene (DPX; Electron Microscopy Sciences).
Immunohistochemistry
Mutant and control fPKR male mice were perfused with cold PBS followed by 4% paraformaldehyde. After being postfixed with same solution overnight, the brains were sectioned by Vibratome at a 30 μm thickness. Single staining immunohistochemistry for bright-field microscope was performed as follows: brain sections were washed with PBS three times and blocked with 10% normal goat serum (NGS)/2% albumin in PBS, followed by overnight incubation at 4°C with gentle shaking in primary antibodies. After three washes in PBS, sections were further incubated with a biotinylated anti-rabbit IgG (1:200; Vector Laboratories) at room temperature for 2 h, and then with streptavidin-biotin/horseradish peroxidase complex (Vector Laboratories) for 30 min. Immunoreaction was visualized with 3,3′-diaminobenzidine (DAB) (Sigma). For double-labeling immunohistochemistry, slices were incubated in the dark in Cy3-conjugated goat anti-rabbit-IgG (1:200; Invitrogen) or Alexa 488-conjugated anti-mouse-IgG (1:200; Invitrogen) for 2 h following primary antibody incubation. Sections were mounted in Vectashield (Vector Laboratories). Primary antibodies used were rabbit anti-c-Fos (Ab-5) (1:20k; Calbiochem), rabbit anti-phospho-eIF2α (clone 119A11) (1:100; Cell Signaling Technology), rabbit anti-ATF4 (sc-200, 1:1000; Santa Cruz Biotechnology), rabbit anti-β-galactosidase (1:2000; MP Biomedicals), mouse anti-GAD67 (clone 1G10.2) (1:5000; Millipore Bioscience Research Reagents), mouse anti-CaMKIIα (clone 6G9) (1:5000; Millipore Bioscience Research Reagents), and mouse anti-MAP2a,b (clone AP20) (1: 200; Lab Vision). To test the specificity of ATF4 signals from double staining, an ATF4 antibody was incubated with ATF4 blocking peptide (sc-200p, Santa Cruz Biotechnology) for 1 h, and then subjected to immunohistochemistry. Confocal images were obtained on a Zeiss LSM 510 located at the National Institute of Neurological Disorders and Stroke Light-Imaging Facility.
Evaluation of de novo protein synthesis
Cannulated male mutant mice and their control fPKR and T29-1 Cre littermates received intracerebroventricular infusion with AP20187 (1 nmol). Two or eight hours after AP20187 administration or 1 h after anisomycin injection (Flood et al., 1973), 500 μCi/kg [35S]methionine (PerkinElmer) in saline (200 μl) was intraperitoneally injected. Thirty minutes later, brains were rapidly removed and serial fresh-frozen sections were cut and fixed as previously described (Qin et al., 2005) with some modifications. Images were obtained with Scion Image 1.62C following film exposure and analyzed with NIH ImageJ (http://rsbweb.nih.gov/ij/). The mean gray values of hippocampal area CA1 and CA3 were quantified from 3 sections of each animal, 4 animals from each group.
Electrophysiology
Transverse hippocampal slices were prepared from age-matched littermates (8–12 weeks old) and field EPSP (fEPSP) recordings were conducted as described previously (Lu et al., 2007). Extracellular fEPSPs were evoked by stimulation of the Schaffer collaterals with 0.2 ms pulses once every 15 s and were recorded in the CA1 stratum radiatum. L-LTP was induced by theta burst stimulation (TBS; 12 bursts, each of 4 pulses at 100 Hz). One train of high-frequency stimulation in 1 s at 100 Hz was used to induce E-LTP. AP20187 (final concentration at 5 μm) was applied to the bath 1 h before theta burst stimulation until recording was finished.
Behavioral paradigms
fPKR/Cre male mice (mutants) and their littermate controls (fPKR, T29-1 Cre, and C57BL/6N wild-type) from 8 to 12 weeks of age were used for the behavioral tasks. All subjects were maintained under a 12:12 h dark:light cycle with food and water ad libitum. All testing was conducted during the light phase of the cycle.
Elevated plus maze.
The anxiety of animals was evaluated via the elevated plus maze paradigm, conducted as previously described (Weisstaub et al., 2006) with some modifications. The maze (Mikes Machine Co.) consisted of two open arms (30 × 5 cm) with 3-mm-high ledges and two closed arms (30 × 5 cm) with 16-cm-high opaque walls. The floor of the arms and a central square (5 × 5 cm) were made of white plastic and were elevated 42 cm from the room floor. The maze was placed in the center of a room (10 × 10 feet) and was homogenously illuminated (∼100 lux across arms). Each mouse was place in the central square facing the open arm opposite the investigator. Mouse behavior was automatically video-recorded for 5 min and animal position was determined by automatic video tracking (ANY-maze, Stoelting).
One-trial contextual step-through active avoidance task.
Avoidance conditioning was conducted as previously described (Cravens et al., 2006), using a 0.18 mA, 2 s footshock. The test for avoidance memory was conducted 24 h later.
Contextual and auditory fear conditioning.
Experiments were conducted as described previously (Cravens et al., 2006) with some modifications. Training consisted of a 2 min period of acclimatization to the context, followed by 2 pairings of a tone (3000 Hz, 85 dB, 30 s) with a foot shock (0.75 mA, 2 s). Contextual retrieval tests were performed in the training chamber after retention delays of 30 min and 24 h. Tone tests for cued fear memory were performed in a new context 30 min after the 24-h-context test. Pretone freezing was monitored 2.5 min before the presentation of the same tone for 30 s.
Statistical analysis
Data are presented as means and SEMs. Three animals were used in RT-PCR and Western blotting, and 4 animals were used for immunohistochemistry and 35S-Met incorporation for each group. n values used in behavioral studies are stated in figure legends. Statistical significance between means of experimental and control groups was determined by Statistica 7 (StatSoft).
Results
Cell type-specific expression of PKR in mouse hippocampus
To manipulate translational machinery at the level of phospho-eIF2α, we generated floxed-mouse lines of PKR, a well characterized eIF2α kinase (Sadler and Williams, 2007). We first constructed the FKBP-PKR cDNA by fusing FKBP-12 in frame to the PKR kinase domain, to regulate eIF2α phosphorylation in an inducible manner (Fig. 1a) (Je et al., 2009). A loxP-nuclear localization signal (nls)LacZ-stop-loxP cassette was then placed upstream of the FKBP-PKR gene to serve as a reporter to monitor regional expression under the control of the CaMKIIα promoter. In floxed-PKR#6 (hereafter referred to as fPKR), one of the transgenic lines carrying the floxed-FKBP-PKR construct, expression of LacZ was robust in the dorsal hippocampal CA1 and dentate granule cell layer (Fig. 1b; supplemental Fig. 1a, available at www.jneurosci.org as supplemental material) in mice older than 8 weeks of age. No LacZ expression was detected in other brain areas including the olfactory bulbs, medial prefrontal cortex, neocortex, area CA3, or the amygdala, although LacZ-positive cells were sporadically detected in hypothalamic nuclei and presubiculum and postsubiculum (supplemental Fig. 1b, available at www.jneurosci.org as supplemental material). The region-specific expression pattern did not change until 12 weeks of age; scattered LacZ expression in the neocortex became apparent by 15 weeks of age (Fig. 1c). Minimal double-positive staining was observed with anti-β-galactosidase (LacZ product) and anti-GAD67 (a marker for interneurons); >90% of CaMKIIα (a marker for excitatory cells)-positive cells were also β-galactosidase immunoreactive in area CA1 (Fig. 1d) and in the dentate gyrus (data not shown). These results indicate that transgene expression was largely restricted to hippocampal CA1 pyramidal cells and the dentate granule cells (supplemental Fig. 2d, available at www.jneurosci.org as supplemental material). The restricted expression pattern of the transgene could be integration site-dependent, because only one line out of the 17 transgenic mice exhibited this pattern using the above construct.
Figure 1.

Cre-mediated PKR expression is largely confined to excitatory cells in area CA1. a, A DNA construct used for generating transgenic floxed-fPKR mice is depicted. A loxP nuclear localization signal(nls)-LacZ-stop-loxP cassette under the control of CaMKIIα promoter was placed before FKBP12-PKR fusion cDNA. Cell type-specific expression of FKBP-PKR fusion protein was achieved by the Cre/loxP-mediated deletion of the “nlsLacZ-stop” cassette. PKR was activated upon dimerization of the FKBP-fusion proteins by in vivo administration of a chemical cross-link inducer, AP20187. b, Region-restricted expression of LacZ reporter in 8-week-old fPKR mice was detected in hippocampal CA1 and dentate gyrus (DG) principal cells by X-Gal staining. No blue cells were observed in cortex, hippocampal CA3, or amygdala. c, LacZ expression was restricted in hippocampal CA1 and dentate gyrus until 12 weeks of age. Scattered blue cells were observed in the cortex at 15 weeks of age. d, LacZ was expressed in CA1 pyramidal cells and dentate granule cells, but not in interneurons. Anti-β-gal-positive cells (Cy3, red) did not overlap with anti-GAD67-positive cells (Alexa Fluor 488, green; indicated by white arrows). Conversely, >90% of anti-CaMKIIα-positive cells (Alexa Fluor 488, green) in CA1 were β-gal positive. e, X-Gal staining pattern of Cre/fPKR double hemizygous mutant mice (mutants; 8-week-old) compared to age-matched fPKR mice, showing Cre recombination was complete in the mutant CA1 pyramidal cells.
The fPKR mouse line was crossed with a forebrain-specific Cre mouse line (T29-1) (Tsien et al., 1996) to restrict PKR expression to the hippocampus, in particular to CA1 pyramidal cells. Heterozygous floxed, Cre-positive progeny (fPKR/Cre, hereafter referred to as mutants) were viable and fertile, and exhibited no gross developmental abnormalities. RT-PCR using total RNA extracted from the mutant hippocampi showed sustained and stable expression of FKBP-PKR by 8 weeks until at least 14 weeks of age (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material). To evaluate the regional pattern of Cre recombination producing the transgene expression, the X-gal staining pattern was compared between fPKR mice and mutant mice (Fig. 1e). LacZ expression in the CA1 pyramidal cell layer, which was observed in fPKR mice at 8 weeks of age, almost completely disappeared in the brains of mutant mice at the same age, indicating Cre recombination was complete in the mutant CA1 pyramidal cell nuclei. Although LacZ expression in the dentate granule cell layer was largely preserved in the mutant brains, in situ hybridization immunohistochemistry using human PKR-specific digoxigenin (DIG)-labeled riboprobes detected FKBP-PKR expression in the CA1 pyramidal cell layer and dentate granule cell layer at 12 weeks of age (supplemental Fig. 1d, available at www.jneurosci.org as supplemental material).
Activation of PKR in CA1 pyramidal cells with no effect on general translation
Because dimerization of the PKR kinase domain is required for kinase activation (Sadler and Williams, 2007) the activation of PKR in vivo was induced by intracerebroventricular injection of AP20187 (1 nmol), a chemical dimerizer of FKBP. PKR activation was determined by immunostaining of phospho-eIF2α and ATF4, whose translation is increased in response to eIF2α phosphorylation (Dever, 2002). Western blots using hippocampal lysates from the 12-week-old mutants revealed that phosphorylation of eIF2α was transiently increased 30 min after AP20187 infusion, sustained up to 1.5-fold over basal levels for at least 6 h, and returned back to normal levels by 18 h (Fig. 2a,b). Immunostaining using a phospho-eIF2α antibody conducted 2 h after the drug infusion showed increased expression of phospho-eIF2α-immunoreactivity (IR) in the CA1 pyramidal cell layer of the mutants compared to control mice (Fig. 2c,d; f,g). Densitometric analysis of phospho-eIF2α-IR in the CA1 pyramidal cell layer and dentate granule cell layer, which was normalized to the IR level of the CA3 cell layer, confirmed the significant increase in mutant CA1 phospho-eIF2α-IR compared to that of controls (Fig. 2i). Notably, phospho-eIF2α-IR levels were extremely low in the dentate granule cell layer, regardless of genotype and drug treatment (Fig. 2e,h; supplemental Fig. 2a, available at www.jneurosci.org as supplemental material), indicating that the increase in eIF2α phosphorylation was specific to CA1 pyramidal cells in our manipulation. Indeed, no obvious eIF2α phosphorylation in the dentate granule cell layer is observed after status epilepticus, during which extensive eIF2α phosphorylation is known to occur in many types of brain cells (Carnevalli et al., 2004). Here, low levels of phospho-eIF2α-IR in the dentate granule cell layer were probably due to the low level of eIF2α expression in this area (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material).
Figure 2.

Targeted activation of PKR in CA1 pyramidal cells. a, b, Phosphorylation of eIF2α was transiently increased up to 1.5-fold over basal levels after AP20187 injection in mutants (12 weeks old) but not in control mice. Animals intracerebroventricularly injected with AP20187 were killed at different time points and hippocampal lysates were stained with anti-phospho-eIF2α by Western blotting. Total protein loading of each time point was first normalized by anti-eIF2α immunoreactivity (IR). Relative phospho-eIF2α levels (percentage) upon AP20187 treatment at different time points were compared to the zero time point of controls or mutants. F(1,32) = 15.3 for genotype, p < 0.0005; F(7,32) = 4.2 for time, p < 0.005. post hoc Fisher's LSD test, *p < 0.05. c–i, Phospho-eIF2α-IR was increased selectively in the CA1 pyramidal cell layer. Control (c–e) and mutant (f–h) brains were fixed 2 h after AP20187 injection and brain sections were immunostained with anti-phospho-eIF2α. The boxed areas in the left panel are enlarged in the center and right panels (c–h). Hip, Hippocampus. i, Immunostaining was quantified in NIH ImageJ by measuring the mean gray value in hippocampal CA1, CA3, and dentate gyrus, respectively. Relative phospho-eIF2α values (means ± SEM) were normalized to the level of CA3 cell layer-IR. Mann–Whitney U test, *p < 0.05. Error bars represent SEM.
To evaluate whether the postulated decrease in ternary complex formation upon increased eIF2α phosphorylation resulted in suppressed de novo general translation in area CA1, we examined the degree of [35S]-methionine incorporation into newly synthesized proteins after AP20187 infusion in vivo. No obvious reduction of de novo general translation was observed in area CA1 2 or 8 h after the drug infusion (Fig. 3a,b), suggesting that the level of increased eIF2α phosphorylation in the mutant CA1 pyramidal cells following AP20187 infusion was insufficient to inhibit general translation. Moreover, no obvious change in the levels of CaMKIIα and β-tubulin, two major cytosolic proteins, was observed in hippocampal CA1 homogenates in mutants after AP20187 injection (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). We concluded that intracerebroventricular injection of AP20187 to the mutants activated PKR with little inhibition of general translation.
Figure 3.

CA1-specific-increase in ATF4 expression without general translation inhibition upon elevation of eIF2α phosphorylation. a, b, No general translation inhibition was detected upon activation of PKR induced by AP20187 infusion. [35S]-methionine was intraperitoneally injected into mutants and fPKR controls 2 or 8 h after AP20187 infusion. The images below are enlargements of the boxed area in the upper images. De novo protein synthesis was quantified in NIH ImageJ by measuring the mean gray value in hippocampal CA1 and normalized to the level of CA3. c, d, Increased ATF4 expression was observed in mutant CA1 area but not in dentate gyrus (DG). Sections from fPKR control and mutant animals were double-immunostained with anti-ATF4 (red signal) and anti-MAP2 (green signal) and were image-collected by confocal microscope. The boxed areas from the left panel are enlarged in the right panel. The numbers of red dots for ATF4 signal in somatic area of CA1, dentate granule cells, and CA3 were counted using NIH ImageJ. The relative ATF4 values were normalized to the level of CA3 cell layer-IR. Mann–Whitney U test, *p < 0.05. Error bars represent SEM.
Increased ATF4 translation and concomitant decrease in CREB-dependent transcription/translation
Increased ATF4 translation is tightly associated with elevated eIF2α phosphorylation (Hinnebusch, 2005). Indeed, we found that ATF4 protein levels in the hippocampal homogenates obtained 2 h after AP20187 infusion were robustly increased in the mutants than in control fPKR mice on the Western blots with ATF4 antibody (relative ATF4 protein level, 234.9 ± 23.3% for mutants compared to those from control animals as 100%; Mann–Whitney U test, p < 0.05). Furthermore, a significant increase in ATF4-IR was observed in the CA1 pyramidal cell layer, but not in the dentate granule cell layer, in mutants 2 h after drug infusion (Fig. 3c,d). Increased ATF4 translation could result in suppressed CREB-dependent transcription in the CA1 pyramidal cells. We therefore monitored changes in mRNAs of several CREB-dependent genes in the CA1 cell layer after AP20187 treatment, including BDNF from promoter IV (Shieh et al., 1998; Tao et al., 1998), c-fos (Berkowitz et al., 1989), zif268 (Sakamoto et al., 1994), and c/EBPβ (Alberini et al., 1994). We found a transient decrease in all of the mRNAs after AP20187 infusion in the CA1 area of the mutant as compared to control mice (Fig. 4a). Furthermore, we detected a transient decrease in BDNF protein levels in mutant CA1 homogenates (Fig. 4b). In contrast, mRNA levels of ATF4 (Fig. 4a), AMPA-type glutamate receptor subunit 1 (GluR1), NMDA receptor subunit NR2A, and PSD95 (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material), whose transcriptions seem to be CREB-independent, were unaffected by the AP20187 treatment. These results suggest that AP20187-induced eIF2α phosphorylation significantly suppressed subsequent CREB-dependent transcriptions, including that of BDNF, in the mutant CA1 cells by increasing ATF4 translation.
Figure 4.

Downregulation of CREB-dependent gene expression. a, Detection of CREB-dependent gene transcription with RT-PCR. Total RNA from area CA1 of control and mutant animals at different time points after AP20187 injection were reverse transcribed into single-stranded cDNA and amplified by PCR with gene-specific primers. The PCR products were separated on a polyacrylamide gel and visualized by PhosphorImager analysis. GAPDH transcripts were used as a control for RNA input and reverse transcription efficiency. Relative mRNA level (percentage) for each gene at different time points after AP20187 treatment were compared to those of zero time point of controls or mutants. F(1,24) = 43.9 for genotype effect on BDNF, p < 0.000005; F(1,24) = 63.6 for genotype effect on c-fos, p < 0.000001; F(1,24) = 17.9 for genotype effect on zif268, p < 0.0003; F(1,24) = 6.7 for genotype effect on c/EBPβ, p < 0.02. Post hoc Fisher's LSD test, *p < 0.05. b, BDNF protein levels in the mutant hippocampi were transiently decreased from 2 to 6 h after AP20187 injection. Total protein loading was normalized by Western blots with anti-eIF2α Ab. Relative BDNF protein levels (percentage) upon AP20187 treatment at different time points were compared to those of zero time point of controls or mutants. post hoc Fisher's LSD test, *p < 0.05. Error bars represent SEM.
Levels of BACE1, another target protein of eIF2α phosphorylation-dependent translational control, were also significantly elevated in the CA1 area of the mutants upon eIF2α phosphorylation (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). Therefore, it is possible that the proteins using eIF2α phosphorylation-dependent translation mechanism were not limited to ATF4 (see Discussion). Excess activation of PKR could also lead to a nontranslational process, namely, induction of apoptotic cell death (Barber, 2005). However, Nissl and Fluoro-Jade B staining revealed no induction of apoptotic processes in the mutant area CA1 following AP20187 infusion (supplemental Fig. 2c,d, available at www.jneurosci.org as supplemental material), suggesting that the eIF2α phosphorylation-dependent nontranslational process, if any, was negligible.
Activation of PKR blocked L-LTP at CA1 synapses
Sal003, an eIF2α phosphatase(s) inhibitor, prevents the induction of L-LTP at CA1 synapses (Costa-Mattioli et al., 2007). To test whether mutant mice with AP20187 exhibit a similar phenotype, acute hippocampal slices were treated with AP20187 or vehicle and stimulated with theta burst stimulation (12 bursts, each of 4 pulses at 100 Hz) at the Schaffer collateral axons; field EPSP was recorded in the stratum radiatum of area CA1. L-LTP was inhibited when slices from mutant mice were treated with AP20187, but not the vehicle control (Fig. 5b). AP20187 had no effect on L-LTP in fPKR control littermates (Fig. 5a). In both fPKR and mutant mice, no difference was observed in basal synaptic transmission (supplemental Fig. 4a, available at www.jneurosci.org as supplemental material) or paired-pulse facilitation (supplemental Fig. 4b, available at www.jneurosci.org as supplemental material) between animals treated with AP20187 or vehicle. These results indicated that impaired L-LTP in mutants was due to postsynaptic rather than presynaptic processes, because FKBP-PKR was selectively expressed in the postsynaptic CA1 pyramidal cells of the Schaffer collateral pathway. Furthermore, early-phase LTP (E-LTP), which is independent of protein synthesis (Huang and Kandel, 1994), was normal in the mutant mice treated with AP20187 (Fig. 5c). Together, these results suggest that postsynaptic control of eIF2α phosphorylation at CA1 synapses was critical for the induction of L-LTP, but not for E-LTP.
Figure 5.
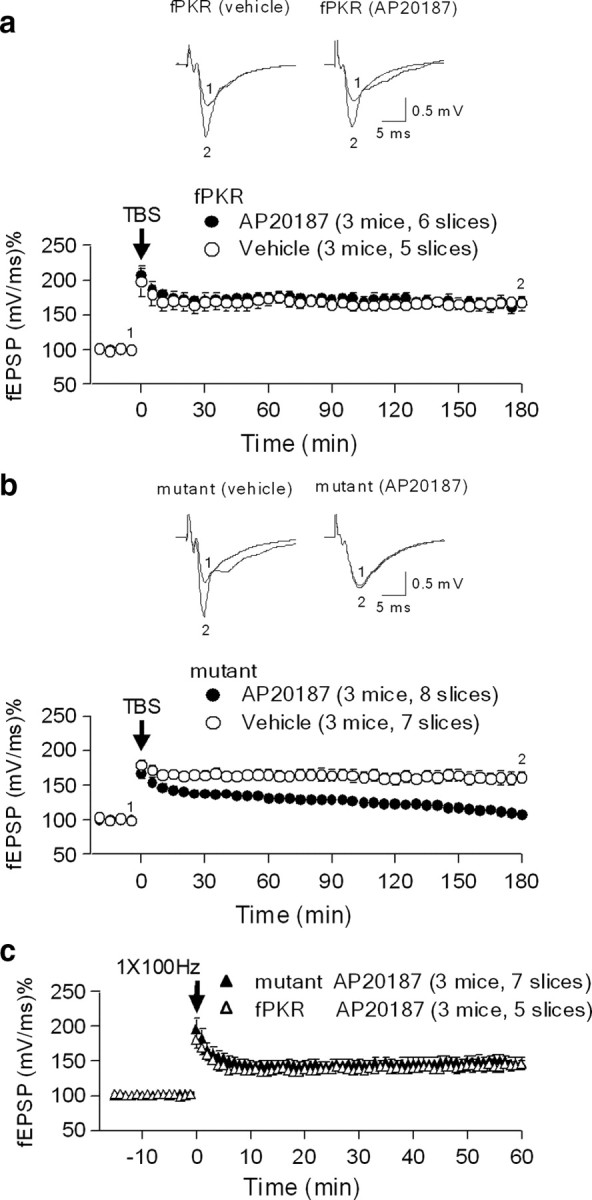
L-LTP but not E-LTP was impaired in mutants with AP20187. a, b, L-LTP was induced by 12 theta burst stimulation (TBS) at 100 Hz (arrows) in the CA1 area of acutely prepared hippocampal slices from 12-week-old mutants (b) and littermate-matched fPKR control mice (a). Examples of fEPSP recordings before (1) and 180 min after (2) LTP induction are shown in the upper panel. L-LTP in control mice was comparable with (filled circle) and without (open circle) application of the dimerizer AP20187. However, L-LTP was nearly absent in mutant mice with 5 μm AP20187 (filled circles) compared to vehicle control (open circles; p < 0.0002, Student's t test). c, E-LTP was induced by a single train of high-frequency stimulation (100 Hz for 1 s) in the Schaffer collateral pathway of hippocampal slices from 12-week-old mutants and littermate-matched fPKR control mice. Both control (open triangle) and mutant (filled triangle) slices showed normal E-LTP induction with AP20187. All results are means ± SEM.
Impaired contextual memory upon activation of PKR in CA1 pyramidal cells
To test whether the increase in eIF2α phosphorylation, which occurs specifically in CA1 pyramidal cells, was sufficient to impair long-term memory formation, mutants and their control littermates (8 to 12 weeks of age), which received vehicle or AP20187 2 h before, underwent to a context discrimination test using a one-trial step-through active avoidance paradigm to evaluate a context-specific memory whose formation was hippocampus-dependent (Olton and Isaacson, 1968). Before conditioning, all mice were observed to have similar initial cross latencies from the light to the dark compartment A of the chamber regardless of the injection with vehicle or AP20187 (supplemental Fig. 4c, available at www.jneurosci.org as supplemental material), suggesting no difference in anxiety or activity levels among the genotypes. We also confirmed no difference in exploratory activity or anxiety levels between genotypes by conducting the elevated-plus maze task (supplemental Fig. 4f, available at www.jneurosci.org as supplemental material). Context discrimination was tested 24 h after one trial conditioning by placing the mice in either the shocking compartment A, or a contextually different, non-shock-associated compartment B, and measuring the latency to cross from the dark to the light compartment (D–L latency) (Fig. 6a). Control mice with AP20187 were able to distinguish compartments A and B as demonstrated by a significantly shorter latency to escape from the shock-associated compartment A. In contrast, mutant mice with AP20187 crossed with similar latencies from either compartment. These latencies were significantly longer than the mean escape latency of control animals from the conditioning-compartment A, suggesting a deficit in long-term context-specific memory. Memory impairments appeared to be induced by PKR activation after AP20187 infusion, because both mutant and control mice with vehicle infusion exhibited significantly shorter escape latencies from the dark compartment A to the light compartment, compared to the AP20187-treated mutant mice (supplemental Fig. 4d, available at www.jneurosci.org as supplemental material; Fig. 6a) (drug × genotype interaction, p < 0.02, post hoc Tukey HSD test, p < 0.05 for drug-treated mutant compared to other groups).
Figure 6.

Consolidation of context-specific memory was impaired after PKR activation. a, Mutant mice, but not fPKR controls, were impaired in context-specific memory formation in the step-through active avoidance paradigm 2 h after AP20187 infusion. F(1,36) = 5.98 for genotype × context interaction, p < 0.02, post hoc Fisher's LSD test, *p < 0.05. Twelve controls and 11 mutants were used for the context A test; 8 controls and 9 mutants were used for the context B test. b, Mutant mice and control mice underwent fear conditioning 2 h after AP20187 infusion. Contextual freezing during the conditioning and 30 min after the training was similar between mutant (n = 8) and control mice (n = 8), suggesting acquisition and retrieval of memory was not affected by the drug infusion. c, Mutant mice (n = 20) were impaired in contextual memory 24 h after the training compared to controls (n = 21) (left panel). Both genotypes showed normal levels of freezing during the tone presentation (right panel). Student's t test, *p < 0.05. d, e, Anti-phospho-eIF2α staining was performed using mutant and fPKR brains obtained 2 h after AP20187 infusion but immediately before fear conditioning (pre) or 2 h after fear conditioning (post). Both genotypes showed postconditioning decrease in phospho-eIF2α levels in CA1 pyramidal cells. Levels in mutants were higher than those in controls both before and after conditioning. Immunostaining was quantified by measuring mean intensity values of the CA1 cell layer using NIH ImageJ. F(1,12) = 20.4 for genotype effect, p < 0.001; F(1,12) = 21.8 for pre–post effect, p < 0.001; post hoc Fisher's LSD test, *p < 0.05. f, g, Mutant ATF4-IR in CA1 pyramidal cells was higher than controls both before and 2 h after conditioning. Brain sections from mutants and fPKR controls were double stained with anti-MAP2 (Alexa Fluor 488, green) and anti-ATF4 (Cy3, red). Images were obtained by confocal microscope using ×63 oil objective. The ATF4 expression level in area CA1 was quantified by counting the number of red dots in the CA1 cell layer using NIH ImageJ. F(1,12) = 17.3 for genotype effect, p < 0.002; F(1,12) = 3.2 for pre–post effect, p = 0.1; post hoc Fisher's LSD test, *p < 0.05. Error bars represent SEM.
Consolidation deficit with impaired training-induced decrease in eIF2α phosphorylation
To evaluate whether the memory deficits of drug-treated mutants were attributable to consolidation impairments, animals underwent contextual fear conditioning 2 h after AP20187 infusion and tested for context- and cued-fear memory retrieval after the conditioning. We first conducted the contextual fear retrieval test 30 min after conditioning to assess the acquisition and retrieval of protein synthesis-independent memory. Both control and mutant mice exhibited equivalent levels of freezing immediately after the training and during the retrieval test (Fig. 6b), suggesting that drug infusion had no effect on memory acquisition or retrieval. Next, we tested contextual fear retrieval 24 h after conditioning using different cohorts of animals, to evaluate memory consolidation. In this paradigm, mutant mice with AP20187 exhibited a significantly reduced level of freezing after a retention delay of 24 h (Fig. 6c, left panel). Furthermore, when tone-dependent fear retrieval was assessed in a distinct context, the same mutants exhibited normal associative memory for the tone (Fig. 6c, right panel), suggesting that decreased freezing in the conditioning context was not caused by nonspecific impairment of the animal's ability to perform the task. The result was also consistent with X-gal staining data showing no transgene expression in the amygdala (Fig. 1b). In a different cohort of animals, the mutant mice infused with vehicle showed robust long-term associative memories for context and tone, which were equivalent to the levels of the drug-treated controls (supplemental Fig. 4e, available at www.jneurosci.org as supplemental material; Fig. 6c, left panel) (post hoc Tukey HSD test, p < 0.05 for drug-treated mutant compared to other groups). Together, these results suggest that our drug manipulation, which activated PKR and subsequent increased eIF2α phosphorylation, selectively impaired hippocampus-dependent memory consolidation without affecting acquisition and retrieval of memory processes.
eIF2α phosphorylation levels are known to decrease following fear conditioning (Costa-Mattioli et al., 2007), presumably due to the increased demand of de novo translation by memory acquisition. We assessed whether a deficit in context memory consolidation could be linked to impaired training-induced decrease in eIF2α phosphorylation. First, we confirmed in the control fPKR mice that levels of phospho-eIF2α decreased to approximately half in CA1 pyramidal cells 2 h after the fear conditioning (Fig. 6d,e). We then found that levels of phospho-eIF2α in CA1 pyramidal cells were much higher in mutants than in control fPKR mice before, and 2 h after fear conditioning. However, such an increase in phospho-eIF2α was unlikely to inhibit general translation, because the inhibition of general protein synthesis was not observed 2 or 8 h after AP20187 treatment in mutants (Fig. 3a,b). In contrast, ATF4-IR was higher in area CA1 compared to that of the controls both before and after the conditioning (Fig. 6f,g), possibly resulting in the suppression of CREB-dependent transcription in the mutants, as observed after AP20187 treatment. Together, these results suggest that increased eIF2α phosphorylation-dependent translations in CA1 pyramidal cells caused consolidation deficits of hippocampal memories without suppressing general translation.
Memory consolidation occurred despite a ∼70% reduction in general translational capacity
To address whether de novo general translation is required for memory consolidation, we examined the effects of systemic infusion of various doses of the most widely used protein synthesis inhibitor, anisomycin, on de novo general translation and consolidation of contextual fear memory. A pronounced decrease in [35S]-methionine incorporation into newly synthesized proteins was observed in the entire brain 1 h after animals were subcutaneously injected with anisomycin at doses of either 5 mg/kg, 10 mg/kg, or 100 mg/kg (Fig. 7a–e). We then evaluated whether the same doses impaired consolidation of contextual fear memory (Fig. 7f,g). Significant impairments in the consolidation of both context memory and tone (cued-fear) memory were observed 24 h after fear conditioning in mice that received a subcutaneous anisomycin injection at a dose of 100 mg/kg 10 min before conditioning. This result was consistent with extensive evidence showing consolidation impairment by anisomycin at a dose of at least 30 mg/kg body weight in rodents (Flood et al., 1973; Davis and Rosenzweig, 1978). Remarkably, a single preconditioning subcutaneous injection of anisomycin at the 5 mg/kg or 10 mg/kg doses, which clearly suppressed [35S]-methionine incorporation to ∼50% or ∼30% of the levels present in vehicle-treated animals, respectively (Fig. 7b,c), did not elicit freezing deficits in either context- or cued-retrieval tests on the following day (Fig. 7f,g). Similar results were obtained when anisomycin was injected immediately after the conditioning (supplemental Fig. 4g,h, available at www.jneurosci.org as supplemental material).
Figure 7.

Low-dose anisomycin inhibits general translation with intact fear memory and normal c-Fos induction. a–e, De novo general translation was significantly inhibited when animals were injected with anisomycin. C57BL/6N mice (2–4 months old) were intraperitoneally infused with [35S]-methionine 1 h after they were subcutaneously injected with saline (a) or anisomycin at a dose of 5 mg/kg (b), 10 mg/kg (c), or 100 mg/kg (d). The mean gray value of radioactivity signals of areas CA1 and CA3 were quantified by NIH ImageJ and compared to the saline-injected group (e). Mann–Whitney U test compared to saline-treated group, *p < 0.05. Ani 5, Anisomycin 5 mg/kg; Ani 10, anisomycin 10 mg/kg; Ani 100, anisomycin 100 mg/kg. f, g, Mice were subjected to fear-conditioning 10 min after saline or anisomycin injection. Contextual freezing during the conditioning was similar among groups (f). However, 24 h after training, contextual (f) and cued-fear memory (g) were impaired with injection of 100 mg/kg anisomycin (n = 9), but were not impaired with 5 mg/kg (n = 12) or 10 mg/kg (n = 10) of anisomycin. One-way ANOVA, post hoc Dunnett test compared to saline-injected group (n = 9). *p < 0.05. h–m, C57BL/6N mice (2–4 months old) were kept in home cage (h) or underwent contextual fear conditioning (i–l). Ten minutes before training, animals were injected with saline (h, i), 5 mg/kg anisomycin (j), 10 mg/kg anisomycin (k) or 100 mg/kg anisomycin (l). The number of c-Fos-positive cells in the CA1 cell layer 1 h after conditioning was assessed compared to the number of the controls (saline injected with fear conditioning). Conditioning-induced c-Fos expression was attenuated by 100 mg/kg anisomycin, but not by 5 mg/kg or 10 mg/kg anisomycin (m). Mann–Whitney U test compared between saline-injected with fear conditioning group and other groups. *p < 0.05. w/o FC, Without fear conditioning; w/FC, with fear conditioning. Error bars represent SEM.
To assess the relationship between anisomycin dose and consolidation impairment, we further monitored the expression of c-Fos protein in the CA1 pyramidal cell layer; c-Fos is a member of the immediate early gene family of transcription factors, and its expression is an index of long-term memory formation (Guzowski, 2002) (Fig. 7h–m). Animals treated with 5 mg/kg anisomycin 10 min before fear conditioning showed a robust increase in the number of c-Fos-positive cells in the CA1 cell layer 1 h after training, and this increase was equivalent to that achieved in animals following saline injection. However, this increase was significantly attenuated by 100 mg/kg anisomycin treatment.
Together, the results suggest that memory consolidation occurs despite the fact that the rate of peptide bond formation during the translation is diminished to ∼30% of the steady-state levels.
Discussion
An important but unresolved issue in memory research is whether memory consolidation requires gene-specific transcription/translation changes or whether it requires the changes in general or global translation regulated by basic translational machinery. To address this issue, we focused on behavioral training-induced Ser51 dephosphorylation of eIF2α, which could result in both a decrease in ATF4 translation and an increase in the translation initiation rate of the basic translational machinery. To the end, we developed a CA1 pyramidal cell-restricted conditional transgenic mouse strain in which the level of eIF2α phosphorylation is chemically augmented to the level which elicits the ATF4 translation increase, but without affecting the rate of general translation initiation. Using this mutant as well as the general protein synthesis inhibitor anisomycin, we made several interesting discoveries. Namely, (1) PKR-mediated increase in eIF2α phosphorylation in CA1 pyramidal cells was sufficient to impair CA1 L-LTP and consolidation of hippocampal memories, despite no decreases in de novo general translation in the CA1; (2) consequently, we observed ATF4 translation increase selectively in the CA1, which suppressed CREB-dependent transcription/translation, including that of BDNF, a key protein for L-LTP and memory consolidation (Bekinschtein et al., 2008); and (3) conversely, inhibition of de novo general translation by low-dose anisomycin did not impair hippocampal-dependent memory or c-Fos induction in the CA1 cell layer. Cumulatively, these results suggest that consolidation of hippocampal memories through eIF2α dephosphorylation efficiently recruits transcription/translation of particular genes, including ATF4 and BDNF, in CA1 pyramidal cells rather than inducing an overall increase in the activity of basic translational machinery.
Hippocampal CA1 pyramidal cells as a site of memory consolidation
CA1 pyramidal cells have substantially more extrinsic projections than CA3 or dentate granule cells and project to a number of subcortical and cortical areas (Cenquizca and Swanson, 2007). While this anatomical feature supports the idea that selective genetic manipulation in CA1 pyramidal cells would alter or disrupt hippocampal memory consolidation, it remained unclear whether CA1 pyramidal cell-restricted genetic manipulation was sufficient to impair consolidation of hippocampal-dependent memories. For instance, Pittenger et al. (2002) generated two tetracycline-dependent inducible transgenic lines expressing KCREB, a dominant-negative inhibitor of the CREB family of transcription factors. In one line (line #1), KCREB transcription was induced in area CA1 of the dorsal hippocampus, striatum, and piriform cortex, while another line (line #2) showed a similar expression pattern but with no detectable expression in the hippocampus. Because performance in the Morris water maze was impaired in line #1 but not in line #2, the authors extrapolated that KCREB expressed in dorsal CA1 was responsible for spatial long-term memory formation. The present findings expanded this work by directly and selectively targeting the induction of eIF2α phosphorylation to CA1 pyramidal cells, preferentially in the dorsal hippocampus. We found that this increased ATF4 translation in these cells, and subsequently suppressed CREB-dependent transcription. This CA1-restricted manipulation resulted in impaired hippocampal memory consolidation. Because acquisition and retrieval of short-term fear memory was not affected by the induction of eIF2α phosphorylation, our results demonstrated that translational process in CA1 pyramidal cells plays a pivotal role in hippocampal memory consolidation.
The level of eIF2α phosphorylation leading to ATF4 translation increase and consolidation deficits
Costa-Mattioli et al. (2007) demonstrated that intrahippocampal injection of Sal003 immediately after behavioral training in rats increases the eIF2α phosphorylation level ∼2.8-fold, which impaired memory consolidation. This treatment was also shown to result in a significant decrease in general translation in vitro. Conversely, in our mutant, infusion of AP20187 elicited eIF2α phosphorylation to 1.5-fold over the basal level and induced a subsequent increase in ATF4 translation in CA1 pyramidal cells. These effects were sufficient to impair consolidation of the learned behavior. Notably, no decrease in the initiation rate of basic translational machinery was detected with this level of phospho-eIF2α by the [35S]-methionine incorporation method. Furthermore, we did not observe the protein level changes of CaMKIIα or β-tubulin following AP20187 treatment. These results suggest that our manipulation was well controlled with regards to eIF2α phosphorylation levels and maintained the high ATF4 level needed to impair behavioral consolidation without affecting general translation. While it has been shown that the increases in the mRNA translation of GCN4, a yeast homolog of ATF4, are more sensitive toward ternary complex formation levels than general protein synthesis inhibition (Dever et al., 1993; Hinnebusch, 2005), the present study clearly demonstrated this phenomenon in mammalian neuronal cells. Even more importantly, our results suggested that gene-specific transcription/translation following a slight increase in eIF2α phosphorylation is sufficient to elicit behavioral consolidation impairments.
On the other hand, low doses of anisomycin did not result in any observable consolidation deficits, despite a clear suppression of de novo general translation up to ∼70% across the entire brain. This suggests that the remaining translational capacity of the neurons is sufficient for memory consolidation. It is also suggested that levels of particular key proteins rather than general translation are critical for consolidation.
Memory consolidation was mediated through a specific set of translation/transcription events
The present study focused on the levels of ATF4 following a slight increase in eIF2α phosphorylation, because a deficit in L-LTP by eIF2α phosphatase inhibitor Sal003 is known to be mediated entirely by ATF4 (Costa-Mattioli et al., 2007). Moreover, ATF4 is a key protein regulating CREB-dependent gene transcription. Indeed, a large body of evidence has already suggested that the CREB pathway is essential for L-LTP and long-term memory formation (Silva et al., 1998; Shaywitz and Greenberg, 1999; Lonze and Ginty, 2002; Carlezon et al., 2005). ATF4 functions as a transcriptional antagonist of CREB-mediated gene expression (Bartsch et al., 1995; Abel and Kandel, 1998). Our results further support a critical role for the CREB pathway in plasticity and memory consolidation. Interestingly, we also observed a translational increase in BACE1 protein following increased eIF2α phosphorylation, as recently reported (O'Connor et al., 2008). However, the involvement of increased BACE1 translation in consolidation is unclear, because BACE1-mediated cleavage of APP in the brain facilitates memory and synaptic plasticity (Ma et al., 2007). Nonetheless, we do not exclude the possibility that other target proteins of eIF2α phosphorylation-dependent translation, or even potential targets of PKR per se if any, could also contribute to these consolidation deficits.
In summary, our data add evidence to the de novo general protein synthesis hypothesis (Davis and Squire, 1984) positing that controlled translation of a subset of mRNAs, rather than boosting general translation rates, during an early phase after learning in CA1 pyramidal cells is crucial for the consolidation of long-lasting plasticity and hippocampal memory. Consistent with our data, an mTOR inhibitor rapamycin, which blocks both long-lasting plasticity and memory consolidation in mammals in many behavioral tasks (Costa-Mattioli et al., 2009), preferentially represses translation of a subset of mRNAs rather than general translation in Jurkat T cells (Grolleau et al., 2002). However, it is difficult to directly address the roles of “general” translation in memory consolidation without comprehensively analyzing overall changes of thousands of proteins at the transcription/translation levels. Indeed, there is substantial evidence that LTP- and LTD-inducing protocols activate the signaling pathways that may globally control translation (Tsokas et al., 2005). Further studies are necessary to determine how many proteins are activated by this learning process, and which proteins are essential for consolidation.
Footnotes
This research was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health. We are grateful to ARIAD Pharmaceuticals, Inc. (www.ariad.com/regulationkits), and Dr. Thomas Dever at National Institute of Child Health and Human Development for providing us with AP20187 and pC939 plasmid. We thank Drs. Beatriz Castilho, Jinsheng Dong, Alan Hinnebusch, Pamela Maher, Hiroaki Matsunami, and Kim Christian and Ioline Henter for their technical advice and critical reading of the manuscript.
References
- Abel T, Kandel E. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Barber GN. The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ. 2005;12:563–570. doi: 10.1038/sj.cdd.4401643. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A. 2008;105:2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz LA, Riabowol KT, Gilman MZ. Multiple sequence elements of a single functional class are required for cyclic AMP responsiveness of the mouse c-fos promoter. Mol Cell Biol. 1989;9:4272–4281. doi: 10.1128/mcb.9.10.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse F, Ephrussi A. Translational control of localized mRNAs restricting protein synthesis in space and time. Nat Rev Mol Cell Biol. 2008;9:971–980. doi: 10.1038/nrm2548. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Carnevalli LS, Pereira CM, Longo BM, Jaqueta CB, Avedissian M, Mello LE, Castilho BA. Phosphorylation of translation initiation factor eIF2α in the brain during pilocarpine-induced status epilepticus in mice. Neurosci Lett. 2004;357:191–194. doi: 10.1016/j.neulet.2003.12.093. [DOI] [PubMed] [Google Scholar]
- Cenquizca LA, Swanson LW. Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res Rev. 2007;56:1–26. doi: 10.1016/j.brainresrev.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, Lu X, Cerasoli F, Jr, Gilman M, Holt DA. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, Sonenberg N. Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjević K, Lacaille JC, Nader K, Sonenberg N. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravens CJ, Vargas-Pinto N, Christian KM, Nakazawa K. CA3 NMDA receptors are crucial for rapid and automatic representation of context memory. Eur J Neurosci. 2006;24:1771–1780. doi: 10.1111/j.1460-9568.2006.05044.x. [DOI] [PubMed] [Google Scholar]
- Davis HP, Rosenzweig MR. Recovery as a function of the degree of amnesia due to protein synthesis inhibition. Pharmacol Biochem Behav. 1978;8:701–710. doi: 10.1016/0091-3057(78)90269-1. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- Dever TE, Chen JJ, Barber GN, Cigan AM, Feng L, Donahue TF, London IM, Katze MG, Hinnebusch AG. Mammalian eukaryotic initiation factor 2 alpha kinases functionally substitute for GCN2 protein kinase in the GCN4 translational control mechanism of yeast. Proc Natl Acad Sci U S A. 1993;90:4616–4620. doi: 10.1073/pnas.90.10.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Rosenzweig MR, Bennett EL, Orme AE. The influence of duration of protein synthesis inhibition on memory. Physiol Behav. 1973;10:555–562. doi: 10.1016/0031-9384(73)90221-7. [DOI] [PubMed] [Google Scholar]
- Grolleau A, Bowman J, Pradet-Balade B, Puravs E, Hanash S, Garcia-Sanz JA, Beretta L. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J Biol Chem. 2002;277:22175–22184. doi: 10.1074/jbc.M202014200. [DOI] [PubMed] [Google Scholar]
- Guzowski JF. Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fluorescent imaging approaches. Hippocampus. 2002;12:86–104. doi: 10.1002/hipo.10010. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. In: Translational control of gene expression. Sonenberg N, Hershey JWB, Mathews MB, editors. New York: Cold Spring Harbor Laboratory; 2000. pp. 185–243. [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- Je HS, Lu Y, Yang F, Nagappan G, Zhou J, Jiang Z, Nakazawa K, Lu B. Chemically inducible inactivation of protein synthesis in genetically targeted neurons. J Neurosci. 2009;29:6761–6766. doi: 10.1523/JNEUROSCI.1280-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Zheng X, Rich KM. Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J Neurochem. 2003;84:273–281. doi: 10.1046/j.1471-4159.2003.01522.x. [DOI] [PubMed] [Google Scholar]
- Karpinski BA, Morle GD, Huggenvik J, Uhler MD, Leiden JM. Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc Natl Acad Sci U S A. 1992;89:4820–4824. doi: 10.1073/pnas.89.11.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova AY, Tervo DG, Gray NW, Svoboda K. Rapid and reversible chemical inactivation of synaptic transmission in genetically targeted neurons. Neuron. 2005;48:727–735. doi: 10.1016/j.neuron.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, Younkin S, Forster C, Sergeant N, Delacourte A, Vassar R, Citron M, Kofuji P, Boland LM, Ashe KH. Involvement of β-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–8172. doi: 10.1073/pnas.0609521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami H, Buck LB. A multigene family encoding a diverse array of putative pheromone receptors in mammals. Cell. 1997;90:775–784. doi: 10.1016/s0092-8674(00)80537-1. [DOI] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olton DS, Isaacson RL. Importance of spatial location in active avoidance tasks. J Comp Physiol Psychol. 1968;65:535–539. doi: 10.1037/h0025830. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25:5087–5095. doi: 10.1523/JNEUROSCI.0093-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Williams BRG. Structure and function of the protein kinase R. Curr Top Microbiol Immunol. 2007;316:253–292. doi: 10.1007/978-3-540-71329-6_13. [DOI] [PubMed] [Google Scholar]
- Sakamoto KM, Fraser JK, Lee HJ, Lehman E, Gasson JC. Granulocyte-macrophage colony-stimulating factor and interleukin-3 signaling pathways converge on the CREB-binding site in the human egr-1 promoter. Mol Cell Biol. 1994;14:5975–5985. doi: 10.1128/mcb.14.9.5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Scheel JR, Garrett LJ, Allen DM, Carter TA, Randolph-Moore L, Gambello MJ, Gage FH, Wynshaw-Boris A, Barlow C. An inbred 129SvEv GFPCre transgenic mouse that deletes loxP-flanked genes in all tissues. Nucleic Acids Res. 2003;31:e57. doi: 10.1093/nar/gng057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Dever TE. Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisstaub NV, Zhou M, Lira A, Lambe E, González-Maeso J, Hornung JP, Sibille E, Underwood M, Itohara S, Dauer WT, Ansorge MS, Morelli E, Mann JJ, Toth M, Aghajanian G, Sealfon SC, Hen R, Gingrich JA. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science. 2006;313:536–540. doi: 10.1126/science.1123432. [DOI] [PubMed] [Google Scholar]