

Abstract
The ability of a peptide hormone to affect many different intracellular targets is thought to be possible because of the modular organization of signal transducing molecules in the cell. Evidence for the presence of signaling modules in metazoan cells, however, is incomplete. Herein we show, with morphology and cell fractionation, that all the components of a mitogen-activated protein kinase pathway are concentrated in caveolae of unstimulated human fibroblasts. Addition of platelet-derived growth factor to either the intact cell or caveolae isolated from these cells stimulates tyrosine phosphorylation and activates mitogen-activated protein kinases in caveolae. The molecular machinery for kinase activation, therefore, is preorganized at the cell surface of quiescent cells.
The platelet-derived growth factor (PDGF) receptor belongs to a family of tyrosine kinases that modulate multiple cellular processes in response to ligand binding. Some of the cell activities affected by PDGF include cytoskeletal rearrangement and migration (1, 2), mitogenesis (3), differentiation (4), calcium mobilization (5), and apoptosis (6). The receptor appears to exert this control through multiple phosphorylation cascades, each of which begins with the phosphorylation of the receptor itself. Although many of the participating molecules and substrates have been identified and their sites of interaction have been mapped (7), exactly how the receptor is able to exert its pleiotropic effect is not known.
One way for a receptor to influence many targets is through modular intermediates (8–11). A module is a molecular cassette composed of transducer molecules, such as serine/threonine and tyrosine kinases, adapter molecules to control interactions among the transducers and scaffolding proteins that hold the adapter and transducer elements in the proper spatial relationship. There might be one module for turning on DNA synthesis, for example, and another for stimulating cell adhesion, etc. Each time a ligand binds its receptor, the ligand–receptor complex relays information by interacting with several different modules. Evidence for the existence of modules comes from two sources. Molecules with the properties of scaffolds (12), transducers (10), and adapters (7) have been identified. Some of these molecules are physically associated with scaffolding proteins during signal transduction in yeast (8).
It is still not clear whether signaling modules exist in metazoan cells. If they do, the scaffold, transducer, and adapter molecules making up a module should be physically associated and exist as a functional unit before a cell is stimulated. The activity of the module should also be detectable both in vivo and in vitro. Finally, the module is likely to have a specific location in the cell. A module activated by the PDGF receptor, for example, would be at the cell surface nearby the receptor, not randomly distributed in the membrane. We now present evidence that caveolae membrane fractions (13) from unstimulated human fibroblasts contain all of the molecular machinery required for PDGF to stimulate mitogen-activated protein (MAP) kinase activation, both in vivo and in vitro.
MATERIALS AND METHODS
Materials.
PDGF(BB), anti-PDGF β receptor IgG, anti-phosphotyrosine IgG, and anti-erk2 kinase IgG were all purchased from Upstate Biotechnology. Anti-caveolin IgG, anti-phosphatidylinositol 3-kinase (PI3 kinase) IgG, anti-Mek-1 IgG, and anti-Raf1 IgG were from Transduction Laboratories (Lexington, KY). Anti-activated MAP kinase IgG was purchased from Promega. Genistein and suramine were purchased from Biomol (Plymouth Meeting, PA). PVDF membrane was from Millipore. ECL Western blot detection reagents were from Amersham. OptiPrep was purchased from Accurate Chemicals.
Cell Culture.
Normal human fibroblasts (14) were cultured to confluence in MEM supplemented with penicillin and 10% fetal bovine serum and then incubated in MEM containing BSA (200 μg/ml) for 20 hr. Caveolae isolation was done on confluent cells cultured in 150-mm plates. Immunoelectron microscopy was carried out with either cells cultured on coverslips or membranes isolated from cells grown in dishes.
Caveolae Isolation.
Caveolae were isolated by the method of Smart et al. (13). Briefly, confluent normal human fibroblasts were collected in hypertonic buffer and homogenized with 20 passages in a Dounce homogenizer on ice. The plasma membrane and the cytosol were separated from the post-nuclear supernatant by using a 30% Percoll gradient (13). The plasma membrane fraction was sonicated and mixed with OptiPrep (final OptiPrep concentration, 23%) in a TH641 tube. A linear 20–10% OptiPrep gradient was overlaid on the sample and centrifuged at 52,000 × g for 90 min at 4°C. The top 5 ml was collected and mixed with 4 ml of 50% OptiPrep in a second TH641 tube. Two milliliters of 5% OptiPrep was overlaid and the sample was centrifuged at 52,000 × g for 90 min at 4°C. The gradient was fractionated in 0.7-ml fractions. Fractions 2 and 3 were designated the caveolae membrane fraction.
Immunoelectron Microscopy.
Normal human fibroblasts were incubated in the absence of serum for 20 hr. Upper plasma membranes were attached to Formvar/carbon-coated grids (15). Localization of erk2 (1:25 dilution), PDGF receptor (1:25 dilution), and tyrosine phosphorylated proteins (1:25 dilution) on these membranes was carried out as described (16). All antibodies were mixed in buffer A (0.1 M sodium phosphate/0.15% BSA, pH 7.6) and incubated at room temperature. For colocalization experiments, the grids were incubated in a mixture of monoclonal anti-erk2 IgG and polyclonal anti-PDGF receptor IgG. They were washed in buffer A and incubated in a mixture of goat anti-mouse IgG (50 μg/ml) and sheep anti-rabbit IgG (50 μg/ml). Finally, samples were washed and incubated in a mixture of rabbit anti-goat IgG conjugated to 10-nm gold (1:30 dilution) and donkey anti-sheep IgG 5-nm gold (1:30 dilution). Immunogold localization of tyrosine phosphorylated proteins was carried out on isolated caveolae membranes as described (17).
Stimulation of Tyrosine Phosphorylation and MAP Kinase Activation in Vivo.
Confluent normal human fibroblasts were treated with PDGF, and caveolae membranes were isolated. Membrane proteins were solubilized in SDS sample buffer and separated by SDS/PAGE on 10% gels. Samples were immunoblotted with anti-phosphotyrosine and anti-activated MAP kinase antibodies by using an ECL method to detect reactive proteins.
Stimulation of Tyrosine Phosphorylation and MAP Kinase Activation in Vitro.
Four caveolae membrane preparations were combined (about 5.4 ml) and mixed with 0.6 ml of phosphorylation buffer [10× MEM, pH 7.4/BSA (800 μg/ml)/10 mM NaF/2 mM Na3VO4/leupeptin (100 μg/ml)/soybean trypsin inhibitor (100 μg/ml)/10 mM MgCl2/1 mM ATP]. Aliquots of the mixture were placed in 1-ml Eppendorf tubes and treated with the indicated factors. The samples were then incubated in a 37°C water bath for 5 min. The reaction was stopped by putting tubes into a salty ice bath. In some experiments BSA was removed by centrifuging the samples at 50,000 rpm in a SW60 rotor. Pellets were then dissolved into SDS sample buffer. In other experiments, 100 μl of 72% trichloroacetic acid was added to each sample to precipitate proteins. Proteins were separated by SDS/PAGE on 10% gels and blotted with either anti-phosphotyrosine or anti-activated MAP kinase IgG.
RESULTS
MAP Kinase Activation in Vivo.
This laboratory has found that the caveolae membrane fraction of unstimulated human fibroblasts is enriched in PDGF receptor, PI3 kinase, Shc, and erk2 (16) and that caveolae from rat-1 cells are enriched in both Ras and Grb2 (18). Fig. 1A shows by immunoblotting that caveolae fractions from the quiescent human fibroblasts used in this study contain PDGF receptor, Ras, Raf-1, Mek-1, and erk2. We verified the location of erk2 by using immunocytochemistry (Fig. 1 B and C). The upper plasma membrane of fibroblasts was attached to a solid substratum and processed for localization of erk2 by using immunogold (15). Anti-erk2 IgG gold labeling was found concentrated over invaginated membrane domains (Fig. 1B). Regions of membrane with this morphology are rich in the caveolae marker protein caveolin, as well as in PDGF receptors (16). When we colocalized PDGF receptors and erk2, both were clustered over the same region of membrane (Fig. 1C). Little gold label was found elsewhere on the plasma membrane, including coated pits (Fig. 1 B and C). Therefore, caveolae from unstimulated cells contain the components of a MAP kinase pathway.
Figure 1.
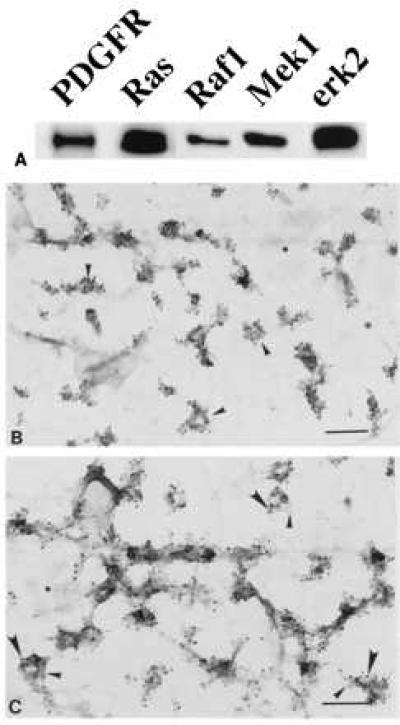
Components of MAP kinase module in caveolae. Normal human fibroblasts were grown as described. (A) Caveolae were purified and immunoblotted with the indicated antibody. (B and C) Membranes were attached to Formvar/carbon-coated electron microscope grids and labeled with either monoclonal anti-erk2 IgG (10-nm gold) alone (B) or monoclonal anti-erk2 IgG (10-nm gold) plus polyclonal anti-PDGF receptor IgG (5-nm gold) (C). In C, the small arrows point to caveolae containing PDGF receptors and the large arrows point to caveolae containing erk2. The asterisks indicate clathrin-coated pits. (Bars = 0.2 μm.)
To determine whether these components were functional, we measured the ability of PDGF to activate MAP kinase in vivo. Normal human fibroblasts were grown in the absence of serum for 20 hr before addition of PDGF (Fig. 2). Cells were homogenized after various times of incubation and caveolae were purified. Samples of caveolae (caveolae) and cytosol (cytosol) were separated by gel electrophoresis and immunoblotted with either an anti-activated MAP kinases IgG (AK) or an anti-MAP kinase IgG [MAPK(erk2)]. Small amounts of activated MAP kinase were initially present in both fractions (Fig. 2, lanes 1 and 7). Within 2 min of exposure to PDGF, the level of activated kinase increased in the caveolae fraction. The amount continued to increase until 5 min (Fig. 2, lane 3) and then declined. Little activated kinase was detected in the caveolae fraction after 120 min of incubation (Fig. 2, lane 6) even though the concentration of the enzyme changed little during this period (Fig. 2, lanes 1–6). Cytosolic MAP kinase was also activated by PDGF (Fig. 2, lanes 7–12) but the kinetics were slower. Peak activity occurred at 30 min of incubation (Fig. 2, lane 10). Because we did not detect any activated MAP kinase in noncaveolae membrane fractions (data not shown), nor does this fraction contain detectable Erk2 (16), caveolae appear to be a cell surface location where MAP kinase is functionally linked to the PDGF receptor. Furthermore, the activated MAP kinase appearing in cytosol after PDGF binding may originate in caveolae.
Figure 2.
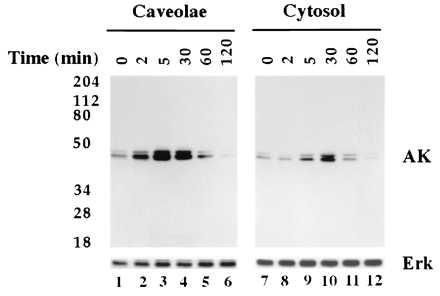
PDGF stimulates activation of caveolae MAP kinase in vivo. Normal human fibroblasts were incubated for the indicated time in the presence of PDGF(BB) at 30 ng/ml. At the end of the incubation, caveolae membranes and cytosol fractions were prepared. Caveolae membrane (5 μg per lane) and cytosol (25 μg per lane) were separated by SDS/PAGE on 10% gels and immunoblotted with either anti-activated MAP kinase IgG (AK) or anti-MAP kinase IgG (Erk).
MAP Kinase Activation in Vitro.
We next determined whether MAP kinase could be activated in isolated caveolae. First, we tested whether PDGF could stimulate tyrosine kinase activity. Caveolae were isolated and incubated in the presence or absence of PDGF(BB) for 30 min (Fig. 3, lanes 1 and 2). The samples were then separated by gel electrophoresis and immunoblotted with anti-PDGF receptor IgG (PDGFR), anti-phosphotyrosine IgG (PY), anti-erk2 IgG [MAPK(erk2)], or anti-PI3 kinase IgG [PI3K (85-kDa subunit)]. We detected two phosphotyrosine (PY) bands in caveolae fractions before incubation with PDGF. By contrast, PDGF caused the appearance of many phosphorylated proteins, indicating the receptor mediated the phosphorylation of multiple substrates. These results indicate the PDGF receptor and downstream substrates share a common membrane domain.
Figure 3.
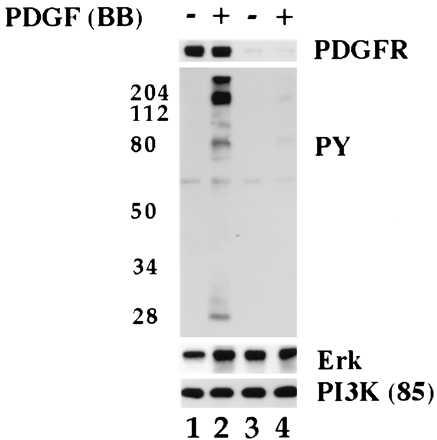
PDGF stimulates tyrosine phosphorylation of caveolae proteins in vitro. Normal human fibroblasts were grown as described. Caveolae membranes were isolated from cells that had been incubated for 2 hr in the presence (lanes 3 and 4) or absence (lanes 1 and 2) of PDGF(BB) (40 ng/ml) and transferred to phosphorylation buffer. Aliquots of caveolae membranes (5 μg per tube) were incubated in the presence (lanes 2 and 4) or absence (lanes 1 and 3) of PDGF(BB) (30 ng/ml) for 30 min on ice followed by 5 min at 37°C. The reaction was stopped by adding ice-cold PBS. The samples were pelleted, dissolved in SDS sample buffer, and immunoblotted with the indicated antibody.
Caveolae from cells that had been incubated in the presence of PDGF no longer responded to ligand binding in vitro (Fig. 3). Cells were incubated in the presence of PDGF for 2 hr prior to preparing caveolae fractions. Immunoblots showed these fractions were depleted of receptor (Fig. 3, lanes 3 and 4) but retained erk2 (MAP kinase) and PI3 kinase (PI3K). As indicated by the absence of anti-phosphotyrosine IgG positive bands (Fig. 3, compare lane 2 and 4), tyrosine kinase activity did not increase when these caveolae were incubated in the presence of PDGF (Fig. 3, compare lane 4 with lane 2).
We used immunogold labeling (19) to confirm the localization of tyrosine-phosphorylated proteins. Whole plasma membranes from cells incubated in the presence (Fig. 4B) or absence (Fig. 4A) of PDGF for 15 min were prepared for immunogold labeling. PDGF-treated cells had clusters of gold particles over regions of membrane (Fig. 4B) that had the same morphology as PDGF receptor containing membrane (compare with Fig. 1C). Many fewer gold particles were associated with these structures in membranes from unstimulated cells (Fig. 4A). Similar results were obtained with isolated caveolae. Anti-phosphotyrosine IgG labeled isolated caveolae after exposure to PDGF for 5 min in vitro (Fig. 4D). The gold particles were generally concentrated at one region of the vesicle-like profiles or decorated small fragments of membrane. Very few gold particles were associated with caveolae that had been incubated in buffer alone (Fig. 4, compare C with D).
Figure 4.
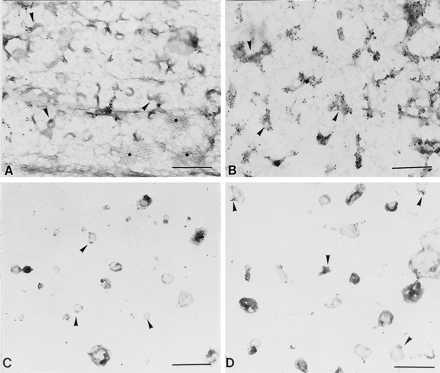
Immunogold localization of tyrosine-phosphorylated proteins. Normal human fibroblasts were grown. (A and B) One set of cells was incubated in the presence (B) or absence (A) of PDGF(BB) (30 ng/ml) for 15 min. Membranes were attached to grids and incubated with anti-phosphotyrosine IgG followed by anti-IgG conjugated to 10-nm gold. Small arrows point to caveolae membrane. Asterisks mark clathrin coated pits. (C and D) A second set of cells was used to isolate caveolae. Isolated caveolae were incubated in the presence (D) or absence (C) of PDGF(BB) (30 ng/ml) for 5 min before labeling with the anti-phosphotyrosine IgG followed by gold (10 nm)-conjugated second antibody. Small arrows point to isolated membrane. [Bars = 0.3 μm (A and B) and 0.4 μM (C and D).]
PDGF receptor and MAP kinase appear to be in the same caveola (Fig. 1B) and PDGF activates caveolae MAP kinase in vivo (Fig. 2). Fig. 5 shows that PDGF also activates MAP kinase in isolated caveolae. Caveolae were prepared from fibroblasts and incubated in the presence of various concentrations of PDGF. Samples were separated by gel electrophoresis and immunoblotted with either anti-activated MAP kinase IgG (AK) or anti-PY IgG (PY). In the absence of PDGF (Fig. 5A, lane 1), very little activated MAP kinase was detected and few tyrosine phosphorylated proteins were present. With the addition of increasing amounts of PDGF, however, there was an increase in the amount of activated MAP kinases (5–60 ng/ml, AK) in parallel with the appearance of tyrosine-phosphorylated proteins (5–60 ng/ml, PY). Suramine, which prevents PDGF binding to receptor (20), inhibited both PDGF activation of MAP kinase (Fig. 5B, compare lanes 2 and 4) and tyrosine phosphorylation. Genistein also blocked tyrosine phosphorylation (Fig. 5B, compare lane 2 with lane 5) and MAP kinase activation (Fig. 5B, lane 5).
Figure 5.
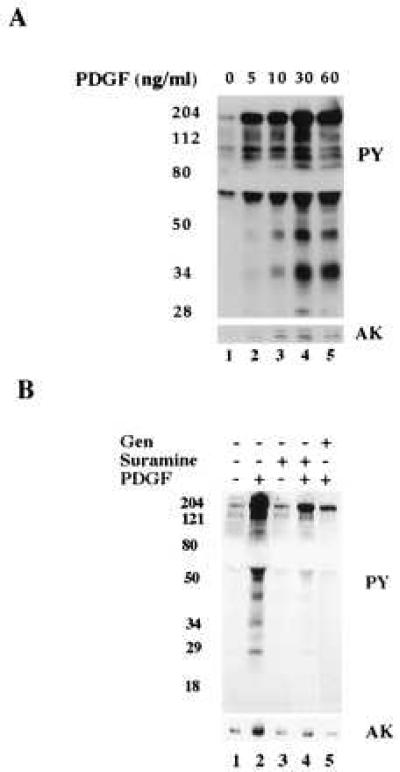
PDGF stimulates tyrosine phosphorylation and MAP kinase activation in vitro. Caveolae were isolated from normal human fibroblasts grown overnight in the absence of serum. (A) Aliquots of caveolae membranes (5 μg per tube) were incubated on ice for 30 min in the presence of the indicated concentration of PDGF(BB) followed by 5 min at 37°C. (B) Aliquots of caveolae membranes (5 μg per tube) were pretreated with either genistein (100 μg/ml; lane 5) or suramine (100 μg/ml; lanes 3 and 4) for 30 min on ice. Samples were then incubated in the presence (lanes 2, 4, and 5) or absence (lanes 1 and 3) of PDGF(BB) (100 ng/ml) for another 30 min on ice followed by 5 min at 37°C. All reactions were stopped by adding 7% trichloroacetic acid and then immunoblotted with either anti-phosphotyrosine IgG (PY) or anti-activated MAP kinase IgG (AK). The distortion in the gel (blank space) is due to the high concentration of BSA present in the phosphorylation buffer.
DISCUSSION
The interaction of as many as 11 different molecules (peptide hormone, tyrosine kinase receptor, nonreceptor tyrosine kinase, SOS, Ras, Raf-1, Grb2, SHC, 14-3-3, Mek, and MAP kinase) may be required for a receptor tyrosine kinase to stimulate activation of a MAP kinase (11, 21). We have found that the entire pathway is functional in isolated caveolae. Other investigators have reconstituted portions of the MAP kinase cascade from crude membrane preparations (22, 23). Our isolated caveolae, however, contained only discrete pieces of membrane (Fig. 4 C and D) making up 5–10% of the total plasma membrane protein. Nothing was added to the incubation mixtures other than ATP, phosphatase inhibitors, and PDGF. The functionality we observed in vitro appears to reflect the natural way these molecules are organized in quiescent cells because PDGF activation of MAP kinases in isolated caveolae (Fig. 4) was comparable to what is seen in the whole cell (Fig. 3). Therefore, the molecules that link the PDGF receptor to MAP kinase were functionally organized prior to isolating the caveolae, which suggests they operate independently of molecular traffic from other cellular compartments.
Several different membrane (18, 24, 25) and cytosolic (16, 18) signaling molecules have been found to move in and out of caveolae in response to hormonal stimuli. Molecular movement of this type probably is a reflection of the feedback loops that exist between proteins in caveolae and specific targets within the cell. The information carried by these molecules remains to be determined. Nevertheless, these molecules form pathways of communication between caveolae and other cellular compartments. The pattern of molecular movement is probably an important determinant of cell function.
We first proposed that caveolae were involved in cell signaling because morphological and biochemical studies had shown they house many different types of signaling molecules and they are able to sequester molecules at the cell surface (26, 27). Since then, additional signaling molecules (for reviews, see refs. 28 and 29) have been localized to this domain, including growth factor receptors (16, 30), endothelial nitric oxide synthase (31, 32), calmodulin (31), and polyphosphoinositides (33, 34). Four different signaling events that originate from this domain have also been identified, (i) epidermal growth factor-stimulated Raf-1 recruitment (18), (ii) PDGF-dependent activation of its receptor (16), (iii) interleukin 1β-stimulated ceramide production (35, 36), and (iv) histamine regulation of caveolae internalization (24). Now we have found that a MAP kinase pathway is functionally intact in isolated caveolae. Caveolae, therefore, must play a major role in signal transduction. The next step is to identify the forces responsible for attracting all of these molecules to caveolae and determine what regulates their activity.
Before the modern era of molecular research, there was no reason to suspect cells might have membrane structures with preorganized sets of signaling molecules. The identification of multiple signaling pathways has led to the realization that the molecules involved create information networks that control cell function (9, 21). There are sound reasons why the molecules that make up these networks should naturally organize at specific locations in the cell (37, 38), as they appear to do in caveolae membrane. The organization we have detected is a reflection of the molecular arrangement existing in caveolae at the time they were isolated from the cell. The transducers, adaptors, and scaffolding proteins in caveolae appear to create molecular circuits that can be detected by using a combination of in vivo and in vitro techniques. Detection of the circuit does not require knowledge of the participating molecules. Once the circuit is identified, however, isolated caveolae become useful tools for determining the molecules involved and how they are functionally connected.
Acknowledgments
We thank William Donzell and Ann-Sofi Horton for their valuable technical assistance and Stephanie Baldock for administrative assistance. We also thank Dr. Michael White for many helpful conversations. This work was supported by Grants HL 20948 and GM 43169 from the National Institutes of Health and the Perot Family Foundation.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: PDGF, platelet derived growth factor; MAP kinase, mitogen-activated protein kinase; PI3 kinase, phosphatidylinositol 3-kinase.
References
- 1.Seppa H, Grotendorst G, Seppa S, Schiffmann E, Martin G R. J Cell Biol. 1982;92:584–588. doi: 10.1083/jcb.92.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mellstrom K, Heldin C-H, Westermark B. Exp Cell Res. 1988;177:347–359. doi: 10.1016/0014-4827(88)90468-5. [DOI] [PubMed] [Google Scholar]
- 3.Cochran B H, Reffel A C, Stiles C D. Cell. 1983;33:939–947. doi: 10.1016/0092-8674(83)90037-5. [DOI] [PubMed] [Google Scholar]
- 4.Noble M, Murray K, Stroobant P, Waterfield M D, Riddle P. Nature (London) 1988;333:560–562. doi: 10.1038/333560a0. [DOI] [PubMed] [Google Scholar]
- 5.Moolenaar W H, Tertooler L G J, de Last S W. J Biol Chem. 1984;259:8066–8069. [PubMed] [Google Scholar]
- 6.Kim H R, Upadhyay S, Li G, Palmer K C, Deuel T F. Proc Natl Acad Sci USA. 1995;92:9500–9504. doi: 10.1073/pnas.92.21.9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claesson-Welsh L. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- 8.Herskowitz I. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- 9.Pawson T. Nature (London) 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 10.Cohen G B, Ren R, Baltimore D. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 11.Marshall C J. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 12.Faux M C, Scott J D. Cell. 1996;85:9–12. doi: 10.1016/s0092-8674(00)81075-2. [DOI] [PubMed] [Google Scholar]
- 13.Smart E J, Ying Y-S, Mineo C, Anderson R G W. Proc Natl Acad Sci USA. 1995;92:10104–10108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein J L, Basu S K, Brown M S. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 15.Sanan D A, Anderson R G W. J Histochem Cytochem. 1991;39:1017–1024. doi: 10.1177/39.8.1906908. [DOI] [PubMed] [Google Scholar]
- 16.Liu P, Ying Y-S, Ko Y-G, Anderson R G W. J Biol Chem. 1996;271:10299–10303. doi: 10.1074/jbc.271.17.10299. [DOI] [PubMed] [Google Scholar]
- 17.Chang W-J, Ying Y-S, Rothberg K G, Hooper N M, Turner A J, Gambliel H A, DeGunzburg J, Mumby S M, Gilman A G, Anderson R G W. J Cell Biol. 1994;126:127–138. doi: 10.1083/jcb.126.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mineo C, James G L, Smart E J, Anderson R G W. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- 19.Carpentier J L, White M F, Orci L, Kahn R C. J Cell Biol. 1987;105:2751–2762. doi: 10.1083/jcb.105.6.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosang M. J Cell Biochem. 1985;29:265–273. doi: 10.1002/jcb.240290310. [DOI] [PubMed] [Google Scholar]
- 21.Hunter T. Cell. 1997;88:333–346. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- 22.Dent P, Romero G, Castle D, Sturgill T W. Methods Enzymol. 1995;255:265–273. doi: 10.1016/s0076-6879(95)55029-1. [DOI] [PubMed] [Google Scholar]
- 23.Dent P, Jelinek T, Morrison D K, Weber M J, Sturgill T W. Science. 1995;268:1902–1906. doi: 10.1126/science.7604263. [DOI] [PubMed] [Google Scholar]
- 24.Smart E J, Ying Y-S, Anderson R G W. J Cell Biol. 1995;131:929–938. doi: 10.1083/jcb.131.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smart E J, Mineo C, Anderson R G W. J Cell Biol. 1996;134:1169–1177. doi: 10.1083/jcb.134.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson R G W. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson R G W, Kamen B A, Rothberg K G, Lacey S W. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- 28.Couet J, Li S W, Okamoto T, Scherer P E, Lisanti M P. Trends Cardiovascular Med. 1997;7:103–110. doi: 10.1016/S1050-1738(97)00001-7. [DOI] [PubMed] [Google Scholar]
- 29.Nishizuka Y. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 30.Jaken S. Curr Opin Cell Biol. 1996;8:168–173. doi: 10.1016/s0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- 31.Shaul P W, Smart E J, Robinson L J, German Z, Ying Y, Anderson R G W, Michel T. J Biol Chem. 1996;271:6518–6522. doi: 10.1074/jbc.271.11.6518. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Cardena G, Oh P, Liu J, Schnitzer J E, Sessa W C. Proc Natl Acad Sci USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pike L J, Casey L. J Biol Chem. 1996;271:26453–26456. doi: 10.1074/jbc.271.43.26453. [DOI] [PubMed] [Google Scholar]
- 34.Hope H R, Pike L J. Mol Biol Cell. 1996;7:843–851. doi: 10.1091/mbc.7.6.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu P, Anderson R G W. J Biol Chem. 1995;45:27179–27185. doi: 10.1074/jbc.270.45.27179. [DOI] [PubMed] [Google Scholar]
- 36.Mochly-Rosen D. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 37.Morowitz H J. Energy Flow in Biology. Woodbridge, CT: Ox Bow; 1979. [Google Scholar]
- 38.Nicolis G, Prigogine I. Exploring Complexity: An Introduction. New York: Freeman; 1989. [Google Scholar]