

Summary
Purpose
Clinically, perturbations in the semaphorin signaling system have been associated with autism and epilepsy. The semaphorins have been implicated in guidance, migration, differentiation, and synaptic plasticity of neurons. The semaphorin 3F (Sema3F) ligand and its receptor, neuropilin 2 (NPN2) are highly expressed within limbic areas. NPN2 signaling may intimately direct the apposition of presynaptic and postsynaptic locations, facilitating the development and maturity of hippocampal synaptic function. To further understand the role of NPN2 signaling in central nevous system (CNS) plasticity, structural and functional alterations were assessed in NPN2 deficient mice.
Methods
In NPN2 deficient mice, we measured seizure susceptibility after kainic acid or pentylenetetrazol, neuronal excitability and synaptic throughput in slice preparations, principal and interneuron cell counts with immunocytochemical protocols, synaptosomal protein levels with immunoblots, and dendritic morphology with Golgi-staining.
Results
NPN2 deficient mice had shorter seizure latencies, increased vulnerability to seizure-related death, were more likely to develop spontaneous recurrent seizure activity after chemical challenge, and had an increased slope on input/output curves. Principal cell counts were unchanged, but GABA, parvalbumin, and neuropeptide Y interneuron cell counts were significantly reduced. Synaptosomal NPN2 protein levels and total number of GABAergic synapses were decreased in a gene dose-dependent fashion. CA1 pyramidal cells showed reduced dendritic length and complexity, as well as an increased number of dendritic spines.
Discussion
These data suggest the novel hypothesis that the Sema 3F signaling system's role in appropriate placement of subsets of hippocampal interneurons has critical downstream consequences for hippocampal function, resulting in a more seizure susceptible phenotype.
Keywords: Semaphorin, Guidance, Electrophysiology, Immunocytochemistry, Morphometry, Excitability
Autism, a neurodevelopmental disorder affecting between 0.2% and 0.5% of the population, is characterized by deficits in communication and behavior. Although the cause of autism is unknown, studies strongly suggest a genetically heritable risk factor (reviewed in Geschwind & Levitt, 2007). Epilepsy, particularly seizure disorders of the frontal and temporal lobes (reviewed in Gedye, 1991; Salmond et al., 2005), is highly comorbid with autism (Clarke et al., 2005; Kagan-Kushnir et al., 2005), although the potential interactions between autistic symptoms and epileptiform activity are still under investigation (Deonna & Roulet, 2006). Mutations in a number of candidate genes have been suggested based on chromosomal linkage analysis, but causative roles have remained elusive (Maestrini et al., 1999; Newbury et al., 2002). Recently, however, single nucleotide polymorphisms in the autism susceptibility region 2q 34 of the human genome coding for neuropilin 2 (NPN2; also known as NRP2) have been identified and strongly associated with autism (Wu et al., 2007).
NPN2 is a receptor for the chemorepulsive axon guidance mediator semaphorin 3F (Sema3F; a member of the class III semaphorins) (Xiang et al., 1996) and is part of a family of axon guidance cues that are involved in responses to brain injury and environment changes (Tessier-Lavigne & Goodman, 1996; Song & Poo, 2001; Pasterkamp & Verhaagen, 2006), as well as during neuronal migration and differentiation in development (Marin et al., 2001; Pozas et al., 2001; Dent et al., 2003; Fenstermaker et al., 2004; Niell et al., 2004; Pascual et al., 2004). Further, this signaling system is highly expressed in adult hippocampus (Chen et al., 2000; Barnes & Slevin, 2003; Barnes et al., 2003; Sahay et al., 2003; Pascual et al., 2004; Sahay et al., 2005; Yang et al., 2005). In rodent studies, neuropilin expression levels are increased by exposure to enriched environments (Cao et al., 2004) and decreased by chemically induced status epilepticus (SE) and/or N-methyl D-aspartate (NMDA) receptor blockade (Barnes et al., 2003; de Wit & Verhaagen, 2003; Holtmaat et al., 2003; O'Donnell et al., 2003; Yang et al., 2005) (but see Shimakawa et al., 2002). Importantly, genetic lesioning studies knocking out different components of this signaling system (NPN2, Sema3F, or plexin A3) have resulted in animals with extended infrapyramidal mossy fiber axonal pathways and spontaneous seizures (Giger et al., 2000; Sahay et al., 2005), similar to those found in the epileptic brain (Cavazos & Cross, 2006; Holtkamp & Meierkord, 2007; Nadler et al., 2007). These data suggest that semaphorin signaling is normally associated with experience-dependent neuronal activity and that experimental manipulations decreasing this signaling pathway's function are closely allied with hyperexcitability and abnormal neuritic outgrowth in the hippocampus. However, NPN2 has pleiotropic roles in guidance, differentiation, and neuritogenesis, and it is not known to what degree developmental alterations contribute to these phenotypes.
Here, we used a NPN2 knockout murine model to assess the consequences of deficits in this signaling system to behavioral convulsions, hippocampal excitability, neuronal cell type/number, ultrastructural morphology (neurite length and complexity as well as dendritic spine type and number), and synaptic protein content. Our results strongly suggest that a deficit in NPN2 during development critically affects interneuron migration and differentiation in the hippocampus and that this has downstream consequences, negatively impacting seizure susceptibility and hippocampal network excitability. Given earlier work showing NPN2's association with autism, this model system that appears to recapitulate at least the seizure susceptibility aspects may be ideal for investigating NPN2's putative role in this neurodevelopmental disease and may aid in defining new therapeutic interventions for autism and autism-related epilepsy.
Methods
Generation of NPN2 knockout mice
The animal experiments were performed under protocols approved by the University of Kentucky Institutional Animal Care and Use Committee. We received NPN2 tau-green fluorescent protein male heterozygotes (+/−; one of the two NPN2 alleles knocked out) from Drs. Andreas Walz and Peter Mombaerts (Rockefeller University, New York, NY, U.S.A.) on a C57BL/6J X B129 mixed genetic background (Walz et al., 2002). These were backcrossed five generations; each time a heterozygous male was mated with a pure FVB/NJ wild-type female (Jackson Laboratory colony; The Jackson Laboratory, Bar Harbor, ME, U.S.A.). The progeny were genotyped according to the previously published polymerase chain reaction (PCR) protocol (Walz et al., 2002). The resulting 1,500 pups are approximately 40% wild-type (−/−), 50% heterozygous (+/−), and 10% homozygous (−/−). From the N1 to N5 generation, handling induced seizures, and SE was noticed only in mice either heterozygous or homozygous for NPN2 knockout (KO) allele.
Kainic acid-induced SE
Male, 25–30 g, >2 month old +/+ and +/− littermates (n = 9/group) were injected subcutaneously with vehicle or kainic acid (KA) (Yang et al. 2005). As noted in Results, −/− littermates were also injected with KA, but none survived beyond 1 week. In all experiments, the mice were observed for at least 6 h for evidence of SE, 4 h of which either continuous or intermittent class IV or V seizures were displayed (Racine, 1972; Pitkanen et al., 2002). Class IV or higher seizures were achieved after 2–3 doses (cumulative dose of 20–25 mg/kg with 15 mg/kg more common). The latency to class III or higher seizure onset, numbers of seizures, and duration of KA-induced SE (KA-SE) by genotype were recorded. Mice (n = 3) that did not develop KA-SE as noted by behavioral seizures were excluded from the analyses. SE was terminated after 6 h by subcutaneous injection (SC) injection of 40 mg/kg pentobarbital. Animals were anesthetized by injection of 100 mg/kg pentobarbital and decapitated. Brains were removed and frozen by slow immersion in −50°C dry ice/methanol/2-methylbutane bath to preserve morphology.
Quantification of KA-induced epileptogenesis
We investigated the long-term effects of KA-SE on cell death and epileptogenesis in the +/+ and +/− mice (n = 9/group). Systematic, treatment-blinded observations were made over 8 months via videotaping (10 h/month from 3 days to 240 days after KA-SE) to determine whether mice developed spontaneous recurrent behavioral seizures (>class II) indicating the presence of epilepsy (defined as >2 class IV or higher seizures in 80 h video period). The number of seizures/80 h of videotape/mouse was noted.
Penetylenetetrazol-induced seizures
Pentylenetretrazol (PTZ) dissolved in buffered saline was administered SC at a dose of 30 mg/kg. Animals were monitored for 30 min after injection. Behavioral responses were scored using the following scale: class 1, hypoactive; class 2, partial clonus (clonic seizure activity affecting face, head, and/or forelimb or forelimbs); class 3, generalized clonus (sudden loss of upright posture, whole body clonus involving all four limbs and tail, rearing, and autonomic signs); and class 4, tonic–clonic seizure (generalized seizure characterized by tonic hindlimb extension) (Ferraro et al., 1999). Latency to first seizure as well as the class of that seizure were analyzed [one-way analysis of variance (ANOVA) with Student-Newman-Keuls post hoc analysis; mice that did not develop seizures during the observation period were excluded from analysis].
Electrophysiology of NPN2 KO mice
Male +/+ and +/− mice (n = 3/group) were anesthetized in a CO2 chamber prior to rapid decapitation with a small animal guillotine. Brains were rapidly removed and transverse hippocampal slices (350 μm) were cut with a Vibratome 3000 (TPI, Saint Louis, MO, U.S.A.) in cold oxygenated artificial cerebrospinal fluid (ACSF) of the following composition: 128 mM NaCl, 1.25 mM KH2PO4, 10 mM glucose, 26 mM NaHCO3, 3 mM KCl, 0.1 mM CaCl2, and 2 mM MgCl2 (Thibault et al., 2001). Intact slices were placed in an interface-type chamber containing ACSF with 2 mM CaCl2 (Ca-ACSF) at 32°C and gassed with 95% O2/5% CO2 for at least 1 h. Individual slices were then transferred to an RC-22C perfusion chamber (Warner Instruments, Hamden, CT, U.S.A.) equipped with a bottom net for ACSF perfusion beneath the slice. The oxygenated Ca-ACSF was delivered at 1.5–2 ml/min and warmed to 33° ± 1°C using an TC2Bip inline heater (Cell Micro Controls, Norfolk, VA, U.S.A.).
Recordings
Electrophysiological data were acquired and analyzed using pCLAMP 8, a sharp-electrode amplifier (2A), and a DigiData 1320 digitization board (Molecular Devices, Union City, CA, U.S.A.). Extracellular recording electrodes were pulled from microhematocrit glass capillaries (Fisher Scientific, Pittsburgh, PA, U.S.A.) on a P80 pipet puller (Sutter Instruments, Novato, CA, U.S.A.). Electrodes had a 5–15 MΩ, tip resistance when filled with 2 M KCH3SO4 and 10 mM HEPES, pH 7.4. All experiments were conducted in current clamp mode. Voltage records were digitized at 2–20 kHz and low-pass-filtered at 1 kHz.
Synaptic stimulation was accomplished using a twisted bipolar stainless steel stimulation electrode (0.0045″ coated; A-M Systems, Everett, WA, U.S.A.) positioned in the Schaffer-collaterals/commissural fibers of stratum radiatum, approximately 500 μm from the recording electrode. Somatic field CA1 potentials were recorded (20 kHz sampling rate) from 4–6 slices per animal. Input-output (I/O) relationships were determined in every slice during baseline periods (0.2 Hz) prior to frequency facilitation (FF) runs. Data from these I/O analyses were used to determine the ratio of population spike (PS) over fiber potential at multiple stimulation intensities and were plotted and fit by linear regression. The slope and x-axis intercept of the linear fit from each slice were used for further analysis. First, the x-axis intercepts were used to determine the excitability threshold by estimating the fiber potential amplitude at which population spikes begin to appear. Second, the slope was used to measure the excitability across a range of stimulus intensities. Stimulus intensity during the FF paradigm was set to deliver pulses at 50% of the maximum PS amplitude measured during I/O and was delivered at 7 Hz for 18 s. Synaptic activation and stimulation protocols were accomplished using a SD9K stimulator (Astro Med, Grass Instrument, Warwick, RI, U.S.A.). Amplitude of the fiber volley was determined by comparison to prestimulus baseline. PS amplitudes were calculated as the difference between the maximum positive deflection of the field excitatory postsynaptic potential (EPSP) before the PS and the maximum negative deflection of the PS. The degree of FF was determined by calculating the ratio of difference to the first primary PS elicited in the FF stimulation period to all other PSs elicited throughout the stimulus train [(subsequent PS-1st PS)/1st PS]. Each second of ratios included seven measures that were then binned per second and reported as an average difference ratio per second. As FF stimulation progressed, a secondary PS gradually appeared. Again, the degree of facilitation of the secondary PS was determined by calculating the ratio increase compared to the first observed secondary PS.
Data was analyzed using Clampfit (v 8.0; Axon Instruments, Union City, CA, U.S.A.) and routines written in Igor Pro (v 5.0; Wavemetrics, Lake Oswego, OR, U.S.A.). Group comparisons were analyzed for main effects using ANOVA. Statistical analysis of the FF measures was performed using a ranked two-way repeated measures ANOVA. Means ± SEM were reported.
Cell counts
KA-SE-treated animals were killed 7 or 240 days after treatment, and their hippocampi was sectioned. For excitatory cells, the soma were counted in the cell layers of three major regions: Dentate gyrus/hilar (DG/H) region, CA3, and CA1 regions (see Supplementary Fig. 1 for demarcation) (Yang et al., 2005). Within these same demarcated regions, nissl cell counts (presumed interneurons) were quantified in molecular layer, stratum radiatum, and stratum oriens of each subregion. Five cresyl violet-stained hippocampal sections (at 200-μm intervals) per animal were counted using the Image ProPlus program (Media Cybernetics, Bethesda, MD, U.S.A.). Both hippocampi were counted. Experimenters were blinded to genotype and treatment group, and average measures per animal were used for statistical analysis (n = at least 5/group). The cell density was calculated for 200 μm2 of a given hippocampal region in each of five sections that were averaged together to establish a single value of cell density per hippocampal region for each mouse. For GABA, GAD-67, parvalbumin, calretinin, somatostatin, and neuropeptide Y cell counts, the total number of cells counted in each hippocampal subregion is noted instead of cell density (see Supplementary Fig. 1 for demarcation). The averages per region in Table 3 and Supplementary Table 3 are calculated from at least five mice. The statistical analysis was performed using ANOVA followed by a Student-Newman-Keuls post hoc analysis. Similar analyses were done for both pyramidal and interneuron cell counts of drug naive (+/+), (+/−), and (−/−) mice.
Table 3. Mean cell counts of interneurons in hippocampal subregions.
| +/+ n = 9 |
+/− n = 9 |
−/− n = 9 |
p-value | |
|---|---|---|---|---|
| GABA+ | ||||
| CA3 | 27 ± 4 | 11 ± 3 | 10 ± 2 | 0.006 |
| CA1 | 30 ± 4 | 14 ± 4 | 7 ± 1 | 0.009 |
| DG/hilus | 12 ± 2 | 5 ± 2 | 4 ± 1 | 0.009 |
| Neuropeptide Y+ | ||||
| CA3 | 28 ± 2 | 12 ± 2 | 8 ± 2 | 0.003 |
| CA1 | 25 ± 3 | 14 ± 2 | 11 ± 2 | 0.003 |
| DG/hilus | 20 ± 2 | 10 ± 2 | 6 ± 1 | 0.001 |
| Parvalbumin+ | ||||
| CA3 | 36 ± 4 | 27 ± 3 | 21 ± 1 | 0.020 |
| CA1 | 37 ± 3 | 28 ± 4 | 27 ± 2 | 0.050 |
| DG/hilus | 12 ± 1 | 9 ± 1 | 10 ± 1 | 0.150 |
| Calretinin+ | ||||
| CA3 | 32 ± 6 | 35 ± 7 | 41 ± 3 | 0.340 |
| CA1 | 41 ± 3 | 40 ± 5 | 38 ± 3 | 0.870 |
| DG/hilus | 25 ± 2 | 27 ± 3 | 22 ± 3 | 0.620 |
| Somatostatin+ | ||||
| CA3 | 26 ± 2 | 30 ± 4 | 32 ± 2 | 0.070 |
| CA1 | 32 ± 3 | 35 ± 4 | 40 ± 3 | 0.160 |
| DG/hilus | 15 ± 1 | 14 ± 3 | 15 ± 2 | 0.530 |
All values are mean ± SEM. The values are an average total number of interneurons per hippocampal subregion as described in the Methods section.
For counting of GABAergic synapses, hippocampal sections (at 200-μm intervals) per animal were counted using the Image ProPlus program. Confocal microscopy [Zeiss Laser Confocal Scanning Microscope (LSM); Carl Zeiss, Oberkochen, Germany] 0.5-μm sections (Z-series stacks) were taken along the z-axis in the CA3b region using the 100× oil immersion objective. The images taken from different sections were compared under constant conditions by adjusting the LSM parameters (pinhole size, laser power, detector voltage). Punctata below 0.1 μm2 were eliminated. Cells were counted only if the clearly defined largest diameter of the cell and clearly defined puntata could be visualized. The CA3b region of both hippocampi was counted. The numbers of Parv+ of GAD-65+ punctata around 50 pyramidal cells were counted, as well as the percent double labeling with both markers. Experimenters were blinded to genotype and treatment group, and average measures per animal were used for statistical analysis (n = 5/group). The Statistical analysis was performed using ANOVA followed by a Student-Newman-Keuls post hoc analysis.
Immunocytochemistry of mouse hippocampus
Drug naive adult mice (>P60) were perfused transcardially with 4% buffered paraformaldehyde. Free floating coronal sections (30 μm) were cut throughout the rostral to caudal extent of the hippocampus. Sections were washed with 1× phosphate-buffered saline (PBS), blocked with blocking buffer [1× PBS, 0.25% Triton X-100, 8% bovine serum albumin (BSA)] for 1 h, and incubated for 1–4 days at 4°C with 1:5000 GABA and 1:1000 parvalbumin (Sigma, St. Louis, MO, U.S.A.), 1:1000 calretinin, neuropeptide Y 1:2000, GAD-65 1:1000, GAD-67 1:500, 1:500 somatostatin (all from Chemicon, Millipore, Billerica, MA, U.S.A.), and 1:100 GABAA receptor α1 (Millipore, Billerica, MA, U.S.A.). Following incubation with primary antibody, sections were rinsed 6× with blocking buffer, then with biotinylated goat secondary antibody (1:1000; Jackson ImmunoResearch, West Grove, PA, U.S.A.) for 2 h at room temperature, and finally 1 h in 1% ABC reagent (Vector Laboratory, Burlingame, CA, U.S.A.). Antibodies were visualized with 3,3 diaminobenzidine (DAB; Sigma, St. Louis, MO, U.S.A.) as a peroxidase substrate with 0.01% H2O2 added as a catalyst. Immunocytochemical controls were performed using the same reaction procedure with nonspecific immunoglobulin G (IgG) or without the primary antibody. For neuropeptide Y experiments, a rhodamine-labeled donkey anti-sheep antibody (1:1000: Jackson ImmunoResearch) was used. For GAD-65/parvalbumin and GABAA receptor α1 double labeling experiments, a Texas Red goat anti-mouse or Cy5 goat anti-rabbit (1:1000; Jackson ImmunoResearch) was used. Cell counts were performed as described above.
To assess the distribution of various nerve fiber layers, four stained sections per mouse were photographed under bright field illumination with an Olympus BX-51 microscope (Olympus, Tubingen, Germany). Optical density (O.D.) measurements (four per subregion) were assessed based on average per subregion using the Image ProPlus analysis program. The O.D. of the fimbrae served as an internal constant control in each slide, and average measurement per subregion per animal was used for statistical analysis. Results were expressed as a mean ± SEM. Statistical analysis was performed using ANOVA followed by a Student-Newman-Keuls post hoc analysis. Sample sizes are noted in Table 3.
Quantitative analysis of synaptosomal proteins on immunoblots
Synaptosomes were prepared from +/+, +/−, and −/− adult mice (>P60, n = 3 per group) as previously described (Barnes et al., 1995). Briefly, 5 μg hippocampal synaptosomal protein were used on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels followed by transfer to Immobilon P membranes (Millipore). After blocking the membranes with 1× PBS, 0.1% Tween-20, and 5% milk, proteins were visualized by primary antibody [1:4000 NPN2 antiserum (gift of Alex Kolodkin, Johns Hopkins University), 1:1000 GABAA receptor β2/3 (Upstate, Danvers, MA, U.S.A.), 1:5000 GAD-67, GAT-1, GluR1, NMDA2A/2B, PSD-95 (all from Chemicon), 1:1000 Fyn (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), 1:5000 synapsin I and β tubulin (National Hybridoma Core Bank, Iowa City, IA, U.S.A.)] followed by secondary antibody detection with horseradish peroxidase (HRP)-linked goat anti-rabbit or anti-mouse IgG 1:5000 (Jackson ImmunoResearch) using the Supersignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, U.S.A.). Light emission was detected by BioMax film (Kodak, Rochester, NY, U.S.A.). O.D. of detected bands were expressed as a mean of the ratios of −/− versus +/+ mouse values ± SEM. Statistical analysis was performed using ANOVA and Newman-Keuls post hoc test.
Rapid Golgi staining and quantitative analysis
Brains isolated from +/+ and +/− mice (n = 7/group) were fixed with 10% formalin in PBS and processed using a modified rapid Golgi impregnation technique (Valverde, 1976). Briefly, coronal tissue blocks of cerebrum (2–3 mm thick) were impregnated using solutions containing osmium tetroxide plus potassium dichromate (for 5–7 days) followed by immersion in silver nitrate (for 36–42 h). After dehydration, blocks were embedded in nitrocellulose, and coronal sections were cut at 120 μm, cleared using α-terpineol and xylenes, and coverslipped under Permount (Fisher Scientific, Fair Lawn, NJ, U.S.A.). All slides were coded, and randomly selected neurons (6–8 per brain from each cell population; see below) meeting staining criteria were evaluated. Camera lucida drawings of individual neurons were used to quantitate the number and length of dendritic branches and the complexity of the dendritic tree. Only neurons that fulfilled all of the following criteria were analyzed: (1) Cell type must be identifiable; (2) neurons must be completely impregnated with the silver-chromate Golgi precipitate throughout all dendrites and spines; (3) soma of the selected neurons had to be located in the middle third of the thickness of the 120-μm section; and (4) all dendrites must be relatively free of neurites from nearby cells, from blood vessels, and from nondescript precipitate.
For dendritic branching analysis, using Zeiss bright-field microscopes equipped with drawing tubes, we first prepared camera lucida drawings of the basilar trees of randomly selected CA1 neurons at a magnification of 400×. A total of 5–6 CA1 neurons that met the above criteria were selected from each hippocampus. The camera lucida drawings were then quantified using Sholl analysis (Sholl, 1953). Using a template comprised of a series of enlarging concentric circles that originated at the soma, by quantifying the number of dendritic intersections with each circle, the Sholl analysis provides a profile of the amount and distribution of dendritic material measured at 10-μm intervals. We compared the branching and total length of the CAl dendrites as a function of genotype. An estimate of total dendritic length was obtained from the total number of dendritic intersections in the Sholl analysis to microns using a previously generated conversion factor. Dendritic complexity was assessed using a branch point analysis that ascertained the number, location, and branch order of dendritic branch bifurcations. Statistical significance was determined using the Wilcoxon rank-sign test. An adjusted α for significance for multiple comparisons was used.
Dendritic spines were quantified on the terminal tip segments of the CA1 basilar trees. At a magnification of 1,200×, using special long-working distance oil immersion lenses, dendritic spines were counted directly from the microscope along four segments of the 30-μm long terminal tip. These spine counts provide an accurate comparison of data between groups, but underestimate the actual total numbers of spines since only flanking spines were counted. (Spines directed either up toward the observer or away from the observer—on the underside of the dendritic branch segment—would be partially obscured by the Golgi stain within the branch).
In addition to total spine density, we also evaluated spine configurations. Spines may be designated by shape into three basic morphological categories: (1) L-spines, which look like a lollipop and have a well-defined spine head and a narrower definitive spine neck. These may be further subdivided into Ls spines (with a small spine head) or LL spines (with a large spine head). (The “L” type spine has also been designated by other investigators as an “M” spine for its “mushroom”-like appearance.) (2) “N” (nubby) spines, which lack a definitive spine head and typically have a thickened spine neck. (3) “D” (dimple) type spines, which are small and have neither a well-defined head nor neck. Spine data was evaluated for statistical significance using the unpaired Student's t-test.
Results
Spontaneous seizure activity in NPN2 KO mice
Initial experiments were observational and appreciated general activity and behavior in these animals. As reported previously, there were no appreciable differences in animal weight, grooming behavior, or ambulatory characteristics of adult mice. Approximately 5% of 500 NPN2 KO heterozygous (+/−) and homozygous (−/−) mice exhibited random handling-induced seizures (0 of 176 +/+ mice, 14 of 273 +/− mice, and 12 of 51 −/− mice) manifested as behavioral arrest and generalized myoclonus (including tail extension, clonic facial twitching, and clonus of the forelimbs). In some instances, NPN2 KO mice (+/− or −/−) were observed to die in spontaneous SE between P7 and P21. Mice 4 months or older tended to exhibit more spontaneous seizure activity, and some were observed to die in SE (data not shown).
Increased sensitivity to chemoconvulsants
After determining baseline seizure activity in this model, animals were challenged with two chemoconvulsant protocols, KA-SE and PTZ (see Methods). For the KA-SE experiment, 2-month-old (+/+) and (+/−), and (−/−) mice (n = 9/group) were given vehicle or KA (15 mg/kg) injections every hour until SE was induced. The average seizure class was IV ± one for both genotypes, although heterozygous mice showed a shorter latency to seize (Table 1). The average duration of KA-SE was 100 min ± 8 min in (+/+) and 107 min ± 10 min (+/−) mice. Mortality 10 h post first KA injection was 10% for both (+/+) and (+/−) genotypes, however, the KA-SE protocol was lethal to all nine (−/−) animals. Videotapes were analyzed of surviving mice up to 8 months post KA-SE (80 h/mouse) by an experimenter blinded to genotype and treatment. Interestingly, 7 of 9 KA-treated (+/−) mice developed at least two spontaneous seizures (>Class III), qualifying as epilepsy (Fig. 1). In contrast, KA-SE (+/+) littermates and vehicle-treated (+/+) and (+/−) mice did not develop observed spontaneous recurrent seizures during the period of study over 8 months of video recording. At 240 days post KA-SE, no cell loss was detected in KA- or vehicle-treated mice regardless of genotype (Supplementary Table 1). Stable hippocampal messenger RNA (mRNA) content of semaphorin/neuropilin gene family (NPN2, Sema3F, NPN1, Sema3A, Sema4C) was found in KA- versus vehicle-treated mice irrespective of genotypes (data not shown).
Table 1. Seizure induction by KA and PTZ.
| Experiment | Seizure type | Genotype | Latency to seizure |
|---|---|---|---|
| KA-SE | SE | +/+ | 47 ± 7 min |
| +/− | 23 ± 2a min | ||
| PTZ (wild-type) | Class I | +/+ | 240 ± 25 s |
| Class II | 180 ± 21 s | ||
| Class III | 400 ± 80 s | ||
| PTZ (NPN2 Het) | Class I | +/− | 140 ± 21b s |
| Class II | 180 ± 52 s | ||
| Class III | 268 ± 37b s |
p < 0.008 by Mann–Whitney test.
p < 0.0001 by ANOVA.
Figure 1.
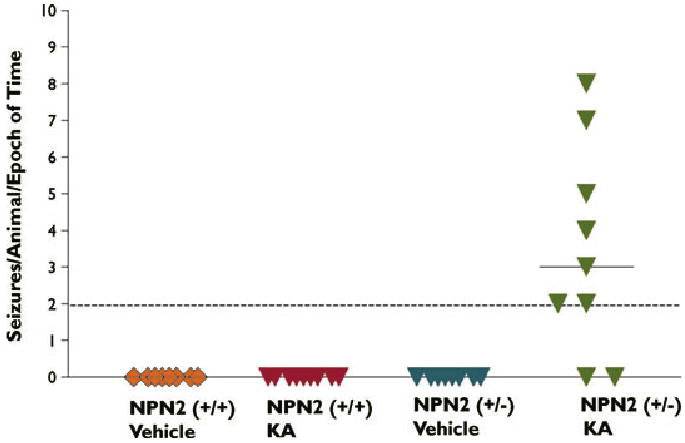
Facilitation of KA-induced epileptogenesis in NPN2 (+/−) mice. NPN2 (+/+) and (+/−) mice were subjected to equivalent duration and severity of KA-SE and then videotaped a consecutive 10 h (10 a.m. to 6 p.m.)/month for 8 months. Spontaneous seizures were only detected in the KA-treated (+/−) mice but not KA-treated (+/+) mice.
Epilepsia © ILAE
For the PTZ experiment, 2-month-old (+/−) and (+/+) littermates (n = 11/group) were challenged with vehicle or PTZ (50 mg/kg). The (+/−) and (+/+) mice had similar numbers of acute PTZ-induced seizures (data not shown). However, the latency to onset for PTZ seizures was significantly shorter in (+/−) animals (Table 1).
Enhanced excitability and altered short-term plasticity in the hippocampus of NPN2 KO mice
Our initial data suggested that NPN2 KO mice were more sensitive than their wild-type littermates to chemoconvulsant exposure. Therefore, we next investigated the impact of a deficit in NPN2 signaling on synaptic circuitry in the hippocampus. Population spikes elicited by synaptic stimulation in field CA1 of the hippocampus (Fig. 2A) in heterozygous and wild-type NPN2 KO animals (n = 3–4 animals/group and 3–4 slices per animal) using a slice recording preparation (see Methods). No significant differences in basic synaptic parameters were observed, although there was a trend towards lowered excitability thresholds in NPN2 KO animals (Table 2). I/O analysis (Fig. 2B) revealed that PS amplitude increased with fiber volley to a greater extent in (+/−) mice, with a significant effect of genotype (unpaired Student's t-test; p < 0.01) (Fig. 2C). Thus, the relative amount of excitation, as inferred by PS amplitude, was increased in the (+/−) animals.
Figure 2.
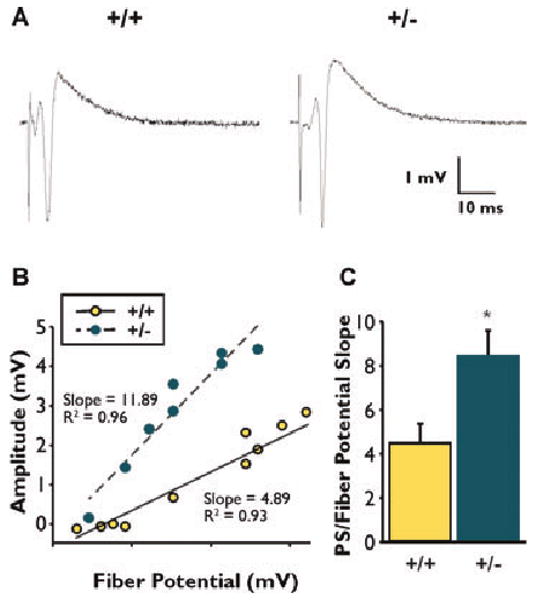
Measures of excitability obtained through synaptic stimulation of CA3-CA1 axonal pathway. (A) Typical examples of extracellular response recorded in the CA1 pyramidal layer at a stimulus intensity of 50% of maximal population spike (PS). (B) Slope measures derived from the linear fit of the fiber volley/PS relationship. (C) A significant difference (p < 0.01) was observed for (+/+) versus (+/−) mice. Averages ± SEM reported. Asterisks (*) indicates significant difference from respective wild-type control (p = 0.01).
Epilepsia © ILAE
Table 2. Basic synaptic parameters in CA1 neurons of all test groups.
| +/+ n = 12 |
+/− n = 9 |
p-value | |
|---|---|---|---|
| Stimulus intensity at 50% max (mA) | 13.20 ± 1.56 | 12.00 ± 0.99 | 0.20 |
| Fiber potential amplitude (mV) | 0.17 ± 0.05 | 0.15 ± 0.02 | 0.51 |
| Estimated minimum fiber potential (mV) | 0.17 ± 0.05 | 0.09 ± 0.02 | 0.11 |
Values are mean ± SEM. Stimulus intensity at 50% max (mA) required to elicit half-maximal population spikes (PS) were derived from stimulus voltage at the stimulator. Fiber potential amplitudes were measured as noted in the Methods section, and estimated minimum fiber potential was derived from the x-axis intercepts in the I/O analysis (see the Methods section).
In a second set of experiments performed on the same tissue, a short-term plasticity measure (FF; see Methods) (Thibault et al., 2001) was assessed in order to appreciate the consequences of a NPN2 deficit on the response of excitable cells to a train of action potentials. There was no difference between groups in the degree of primary PS facilitation during 7 Hz synaptic stimulation train (Fig. 3A). Surprisingly, facilitation of the secondary PS elicited during the 7 Hz stimulation train significantly decreased in (+/−) mice [one-ANOVA repeated measures; F(1,47) = 4.545; p < 0.05] (Fig. 3B). There was also no difference between groups with respect to the onset of the occurrence of the secondary PS. In summary, sensitivity to stimulation was greater in NPN2 KO animals, while short-term synaptic throughput, at least for the secondary PS in CA1 pyramidal cells, was markedly reduced.
Figure 3.
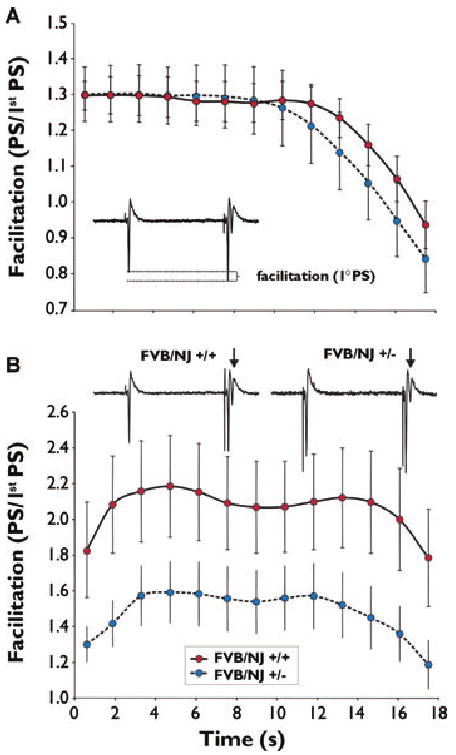
Comparison of frequency facilitation (FF) measures from wild-type and NPN2 (+/−) mice. (A) No significant difference was found in FF of primary (1°) population spike (PS) between wild-type and (+/−) mice. (B) Secondary (2°) PS (arrows) during FF was significantly decreased at all time points (p < 0.05). Averages ± SEM reported. Ranked 2-way ANOVA on repeated measures was used for statistical tests (see Methods section).
Epilepsia © ILAE
Selective loss of GABAergic cells in the hippocampus of NPN2 KO mice
After determining behavioral activity, response to chemical challenge, and electrophysiologic properties of hippocampal circuits, we examined the microanatomy of the hippocampus. To ascertain the morphologic effects of genetic NPN2 lesioning, we analyzed the major cellular populations of the mouse hippocampus. Because the entire hippocampus was not sampled, stereological procedures were not used, and the cell counting method used here (see Methods) is considered semiquantitative. The principal cell numbers counted in the cell layers of cresyl violet-stained sections [DG region, NPN2 (+/+) 185 ± 25 of 200 μm2 versus NPN2 (−/−) 174 ± 32 of 200 μm2; CA3 region, NPN2 (+/+) 123 ± 17 of 200 μm2 versus NPN2 (−/−) 113 ± 22 of 200 μm2; CA1 region, NPN2 (+/+) 115 ± 14/200 μm2 versus NPN2 (−/−) 109 ± 19 of 200 μm2] of hippocampus were not significantly different (Fig. 4A versus 4B and Supplementary Table 1). However, using immunocytochemical protocols (see Methods), a major decrease in GABA+ interneuron populations (67% in DG/H region, 63% in CA3 region, and 77% in CA1 region) of (−/−) mice compared to wild-type littermates (Fig. 4C versus 4D, and Table 3) was revealed. On closer inspection of cresyl violet-stained sections, a major decrease (40%–53%) in nissl-stained large nuclei outside the principal cell layers in hippocampal subregions (Supplementary Table 2) was observed in (−/−) mice compared to wild-type littermates.
Figure 4.
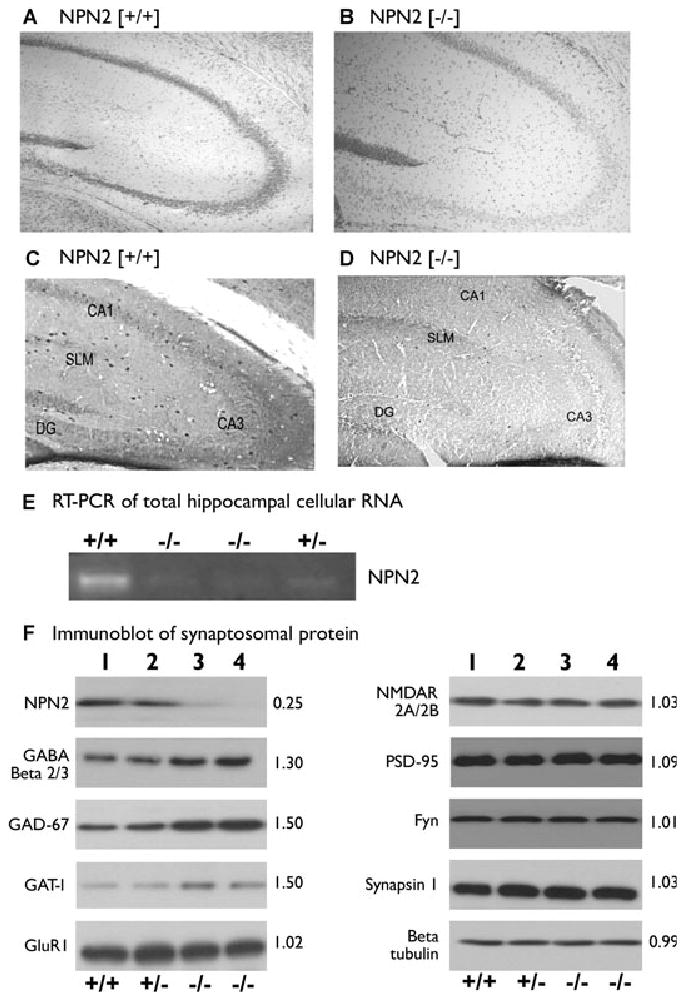
Deletion of NPN2 signaling leads to hippocampal GABAergic deficiency. The initial overall look of the cytoarchitecture of hippocampus was very similar using cresyl violet staining in both (−/−) and (+/+) littermates (panel A versus B). However, analysis of the GABA+ interneuron populations revealed a major decrease of in the DG/H region, CA3 region, and in CA1 region of (−/−) mice compared to wild-type littermates (panel C versus D). In panel E, RT-PCR of total cellular hippocampal RNA indicated a 90% reduction in NPN2 mRNA content in (−/−) mice compared to (+/+) mice. In panel F, hippocampal synaptosomes of (−/−) mice contained 75% less NPN2 protein (lane 3 and 4) compared to (+/+) mice (lane 1). Synaptosomes from these same mice (lanes 3 and 4) contained a 50% increase in GAD-67 and GAT-1 plus a 30% increase in GABAA receptor β 2/3 immunoreactive protein relative to (+/+) mice (lane 1) without any differences in excitatory synaptic protein content. The number to the right of each blot is the optical density (O.D.) measurements (n = 3 experiments), which is the O.D. ratio of NPN2 (−/−) to NPN2 (+/+) protein.
Epilepsia © ILAE
Altered protein profile in NPN2 KO synapses and synaptosomes
In the NPN2 (−/−) mice, reverse transcription PCR (RT-PCR) analysis (Yang et al. 2005) revealed a 90% decrease in total hippocampal NPN2 mRNA (Fig. 4E), and this change was validated at the protein level in synaptosomal preparations. We counted the number of GAB-Aergic synapses around CA3b pyramidal cells in sections. The number of Parv+ [NPN2 (+/+) 30 ± 2/cell versus NPN2 (−/−) 17 ± 2/cell, p < 0.007] or GAD-65+ punctata [NPN2 (+/+) 42 ± 2/cell versus NPN2 (−/−) 23 ± 2/cell, p < 001] was significantly decreased (Supplementary Fig. 3A–3F) as well as the percent co-labeling [NPN2 (+/+) 65% ± 4% versus NPN2 (−/−) 29% ± 3%, p < 0.006] in the NPN2 KO mouse. The percent colabeling between parvalbumin and the GABAA receptor α1 protein [NPN2 (+/+) 66% ± 5% versus NPN2 (−/−) 31% ± 3%] on presumed interneurons in the CA3b region was also significant decreased (p < 0.001, Supplementary Fig. 3G–3L) in the NPN2 KO mouse. We investigated the synaptic protein content in biochemical experiments on synaptosomes isolated from (+/+), (+/−), and (−/−) mice using immunoblots. Hippocampal synaptosomes of (−/−) mice contained approximately 75% less NPN2 protein (Fig. 4F, lanes 3 and 4) as compared to (+/+) mice (lane 1). Interestingly, despite the decrease in total number of hippocampal GABA+ interneurons and GAD-65+ punctata/pyramidal cell (presumed synapses), synaptosomes from these same mice (Fig. 4F) showed a 50% increase in GAD-67 and GAT-1 plus a 30% increase in GABAA receptor β 2/3 immunoreactive protein, but did not reveal differences in other proteins (GluR1, PSD-95, NMDAR 2A/2B, Fyn, synapsin I, and β-tubulin) (Fig. 4F, lanes 1–4). However, as expected, the total number of GAD-67+ cells had a 30%–67% decrease in the NPN2 KO mouse, depending on the hippocampal subregion (Supplementary Fig. 4 and Supplementary Table 3).
Other interneurons subtypes are selectively reduced in NPN2 KO animals
After determining that GABAergic neuron numbers are reduced in association with deficits in NPN2 expression (Fig. 4), we examined other interneuron populations in the hippocampus, including parvalbumin (Parv+), calretinin (CR+), neuropeptide Y (NPY+), and somatostatin (SS+) positive cells (Danglot et al., 2006). Parv+ neurons in the CA3 region of dorsal hippocampus in (−/−) mice were decreased by 42% (p = 0.02) (Fig. 5C versus 5B, black arrows, and Table 3). In CA3, there was a 45% reduction (p = 0.008) in staining in stratum lucidum and stratum oriens (Fig. 5C versus 5D, and Table 4). However, Parv+ neurons but not immunoreactive Parv+ fibers were decreased by 28% [NPN2 (+/+), 37 ± 3 versus NPN2 (−/−), 27 ± 2] within all CA1 subregions (Fig. 6E versus 6F, and Tables 3 and 4).
Figure 5.
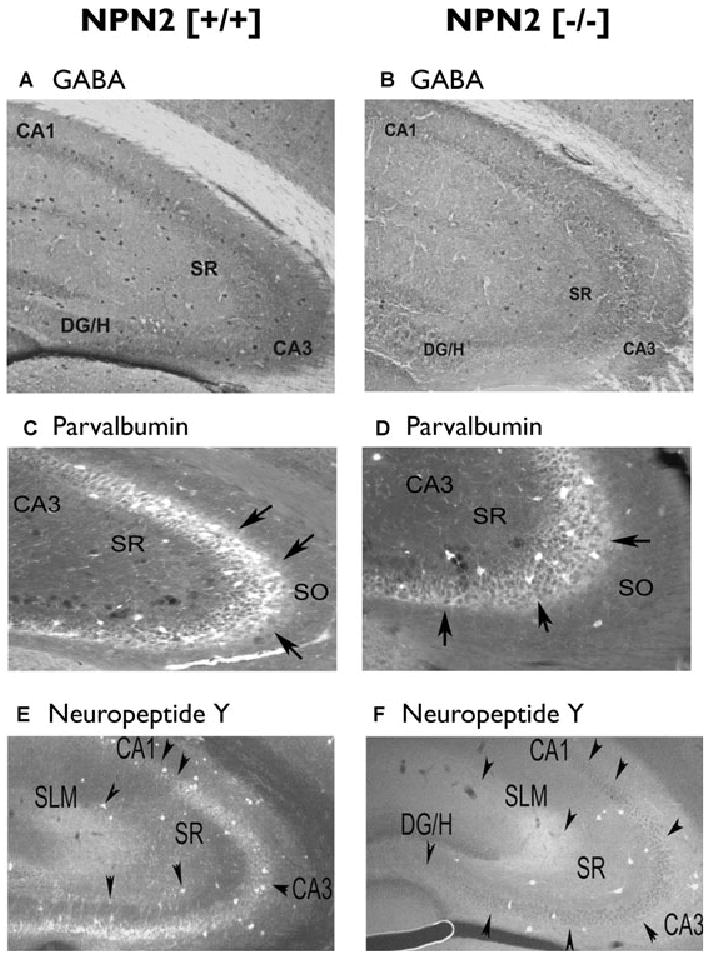
Decreased parvalbumin and neuropeptide Y neurons in the CA3 regions of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (n = 9) or NPN2 (−/−) (n = 9) mice were subjected to immunocytochemistry using the GABA (panel A versus B), Parvalbumin (panel C versus D), neuropeptide Y (panel E versus F) antibodies. The number of GABA+ neurons was decreased by 63% in both subregions of CA3 (panel A versus B). Cell counts of Parv+ neurons in the CA3 region were decreased by 42% (panel C versus D, black arrows). There was a remarkable 71% decrease in NPY+ neurons in the CA3 region of (−/−) mice (panel E versus F).
Epilepsia © ILAE
Table 4. O.D. measurements of immunoreactive fibers in hippocampal interneuron subpopulations.
| +/+ n = 9 |
−/− n = 9 |
p-value | |
|---|---|---|---|
| Neuropeptide Y+ | |||
| CA1 | 1.62 ± 0.12 | 0.76 ± 0.24 | 0.007 |
| CA3 | 1.34 ± 0.05 | 0.80 ± 0.15 | 0.008 |
| DG/hilus | 1.45 ± 0.10 | 0.80 ± 0.24 | 0.006 |
| Parvalbumin+ | |||
| CA1 | 0.78 ± 0.05 | 0.68 ± 0.06 | 0.170 |
| CA3 | 1.32 ± 0.10 | 0.73 ± 0.13 | 0.008 |
| DG/hilus | 0.67 ± 0.10 | 0.78 ± 0.06 | 0.690 |
Values are mean ± SEM.
Figure 6.
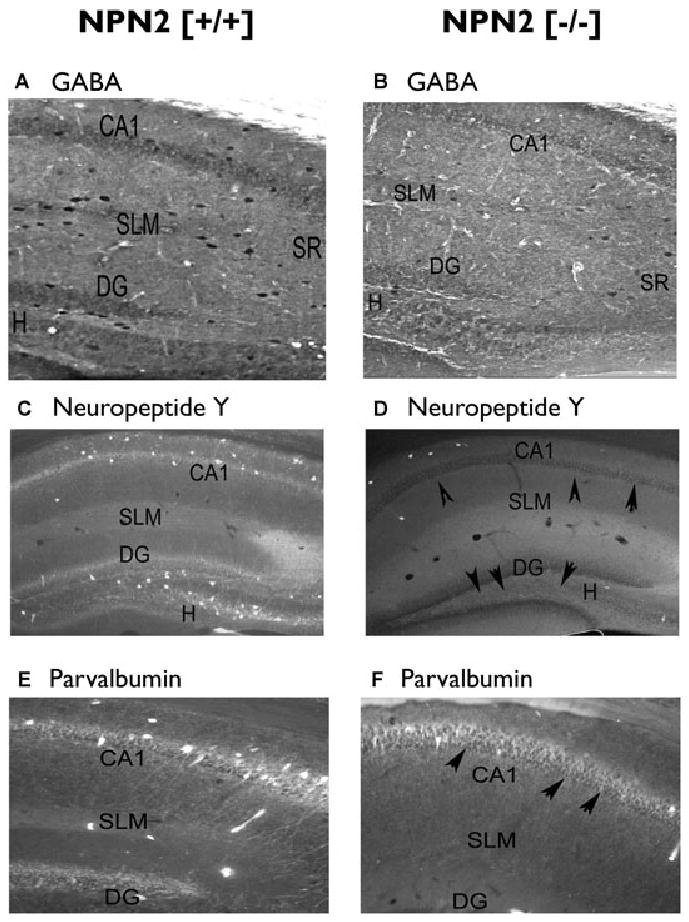
Decreased neuropeptide Y neurons in the CA1 and dentate gyrus/hilar regions of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (n = 9) or NPN2 (−/−) (n = 9) mice were subjected to immunocytochemistry using the GABA (panel A versus B), neuropeptide Y (panel C versus D), or parvalbumin (panel E versus F) antibodies. The number of GABA+ neurons was decreased by 77% in the CA1 and 67% in the DG/H regions of the (−/−) mice (panel A versus B). Cell counts of NPY+ neurons in the CA1 and H/DG regions were decreased by 56% and 71%, respectively compared to (+/+) mice (panel C versus D, black arrows). Cell counts of Parv+ cells decreased by 28% (panel E versus F) in the CA1 of (−/−) mice but no changes were noted in the H/DG regions of both genotypes.
Epilepsia © ILAE
NPY+ neurons were located in stratum lucidum moleculaire (SLM), stratum rafiatum (SR), and stratum oriens (SO) of the CA3 region in (+/+) mice (Fig. 5E). Immunoreactive NPY fibers were also noted in the pyramidal cell layer (Fig. 5E, black arrows). In contrast, there was a marked 71% decrease (p = 0.003) in NPY+ neurons (via cell counts) as well as immunoreactive NPY fibers (34% decrease, p = 0.008) in the CA3 regions of (−/−) mice (Fig. 5E versus 5F, and Tables 3 and 4). Cell counts of NPY+ neurons in the CA1 region of dorsal hippocampus in (−/−) mice were decreased by 56% (p = 0.003) in SR/SO/SLM (Fig. 6C versus 6D, black arrows, and Table 3). O.D. analyses in CA1 revealed there was a 54% reduction (p = 0.007) in SL and SO of CA1, as well as a similar reduction in the SLM (Fig. 6C versus 6D, and Table 4) in (−/−) mice compared to (+/+) mice.
SS+ neurons as well as immunoreactive SS fibers were present in similar numbers [NPN2 (+/+), 26 ± 2 versus NPN2 (−/−), 32 ± 2] within all CA3 subregions in both (+/+) and (−/−) mice (Supplementary Fig. 2A versus 2B, and Tables 3 and 4). SS+ neurons as well as immunoreactive SS fibers were present in similar numbers [NPN2 (+/+), 32 ± 3 versus NPN2 (−/−), 40 ± 3] within all CA1 subregions in both (+/+) and (−/−) mice (Supplementary Fig. 2C versus 2D, and Table 3). CR+ neurons as well as immunoreactive CR fibers were present in similar numbers [NPN2 (+/+), 30 ± 3 versus NPN2 (−/−), 41 ± 3] within all regions (Supplementary Fig. 2E versus 2F, and Tables 3 and 4).
In the DG/H region of (−/−) mice (Fig. 6A versus 6B, and Tables 3 and 4), cell counts of NPY+ neurons in the DG/H region of were decreased by 70% (p < 0.001) (Fig. 6C versus 6D, black arrows, and Table 3). O.D. analyses in DG/H revealed there was a 45% reduction of NPY+ staining fibers (p = 0.006) in molecular layer (ML) and hilar regions (Fig. 6C versus 6D, and Table 4) in NPN2. All other interneuron subtypes and fibers were similar across genotype (Tables 3 and 4 and Supplementary Fig. 2). Taken together, these data indicate that a deficit in NPN2 signaling is associated with a loss of subpopulations of interneurons in the hippocampus.
Dendrite and spine analyses of CA1 pyramidal neurons in the NPN2 KO mice
Given the apparent increased excitability of CA3-CA1 circuitry and the apparent decrease in GABAergic interneurons seen with NPN2 KO allele, we further examined the potential ultrastructural alterations of CA1 pyramidal cells. Previous experiments have already established that there is an increased length of the infrapyramidal bundle of mossy fiber axons in this transgenic strain of NPN2 (−/−) mice (Walz et al., 2002). Therefore, we analyzed the dendritic morphology/structure within field CA1 of the hippocampus in NPN2 KO mice. Brains isolated from 12-week-old naive (+/+) mice and (+/−) mice (n = 7/genotype) underwent the rapid Golgi staining protocol (Valverde, 1976), performed in a manner blinded to genotype. Using camera lucida drawings of the hippocampal CA1 dendrites, dendritic branching was evaluated for estimated total dendritic length and complexity. A moderate decrease in dendritic branching (p < 0.001) and total dendritic length of CA1 pyramidal cells (18% decrease, p < 0.01) was detected in the (+/−) mice (Fig. 7). Further, N type and D type spines in NPN2 (+/−) mice were significantly increased 11% (p-values of p < 0.01 and p < 0.05, respectively) compared to CA1 neurons from (+/+) mice (Table 5). The total spine density of NPN2 (+/−) CA1 pyramidal cells showed a trend towards increased density (11%, p = 0.10). Whether the synapses on the excess spines are excitatory or inhibitory in nature is unknown.
Figure 7.
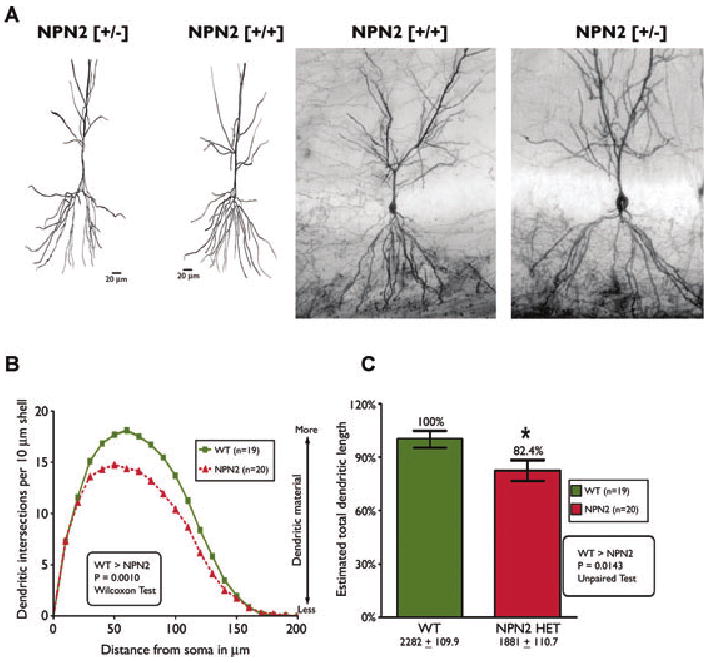
Decreased dendritic complexity of CA1 pyramidal cells in hippocampus of NPN2 KO mice. Rapid Golgi staining of brains from (+/+) and (+/−) mice revealed consistent differences in dendritic complexity of CA1 pyramidal cells from (+/+) versus (+/−) mice. As shown in panels A and B, dendritic branching of CA1 pyramidal cells was less in (+/−) mice compared to (+/+) littermates (p = 0.001). Consistent with the Sholl's analyses, the total dendritic length in (+/−) mice was 18% less than (+/+) littermates (panel C, p = 0.01).
Epilepsia © ILAE
Table 5. Spine density analyses of wild-type and NPN2 KO mice.
| Parameter | +/+ | +/− | p-value |
|---|---|---|---|
| Total spines | 42.6 ± 2.6 | 46.8 ± 1.0 | 0.10 |
| LL type spines | 13.8 ± 1.1 | 15.3 ± 0.6 | 0.20 |
| LS type spines | 12.6 ± 0.8 | 12.9 ± 0.5 | 0.98 |
| D type spines | 3.87 ± 0.2 | 4.77 ± 0.2 | 0.01a |
| N type spines | 12.2 ± 0.7 | 13.8 ± 0.4 | 0.05a |
Significant by unpaired Student's t-test.
Discussion
The present work provides a comprehensive overview of hippocampal structure and function in mice with a deficit in NPN2 expression. Although it is tempting to conclude that these findings are a direct result of NPN2 manipulation, it is important to keep in mind, particularly when dealing with molecules such as NPN2 that are so heavily implicated in neurodevelopment, that indirect compensatory responses by the biological system may also contribute importantly to these findings. Results from this animal model may contribute importantly to our understanding of NPN2's role in development and maintenance of a normal excitatory/inhibitory network in the hippocampus, and how perturbations in this signaling system might affect susceptibility to epilepsy. Further, based on recent work, this model may be highly relevant for the study of autism (Wu et al., 2007). Here, we show for the first time: (1) gene-dose-dependent expression of hippocampal NPN2 mRNA and synaptosomal NPN2 protein, validating this model for studies of NPN2 deficits in signaling; (2) heightened sensitivity to chemical challenge with KA and PTZ, suggesting a lower seizure threshold in NPN2-deficient mice; (3) increased excitability in field CA1, manifested as an increased slope in the I/O curve, along with an apparent reduction in FF-induced short-term synaptic throughput (Landfield et al., 1978, 1986; Thibault et al., 2001); (4) a dramatic and subtype selective decrease in interneuron cell counts in multiple hippocampal regions, while principal cell numbers in the CA and DG layers are preserved; and (5) reduced dendritic length, complexity, and spine numbers in CA1 neurons of NPN2 deficient animals.
Protein expression in synaptosomes of NPN2 deficient animals
Loss of NPN2 expression was gene-dose-dependent, with heterozygous animals showing an intermediate level of expression. Further, mRNA and protein levels showed highly correlated changes in expression. Interestingly, mRNA expression levels for the homozygous KO animals were reduced by 90% compared to their wild-type littermates. Ostensibly, detection of NPN2 mRNA in homozygous KO animals should have been reduced 100%. Thus, this 10% difference between what was expected and what was measured may indicate that, at least with the mRNA quantification system used, there is some uncompensated background noise, cross-hybridization, or lack of specificity contributing to these measures.
Other proteins measured in the synaptosomal preparation, including those related to structure and synaptic elements (β-tubulin, synapsin I, PSD-95), as well as those related to excitatory neurotransmission (NMDAR 2A/2B, Glur1) were apparently unaffected by NPN2 deletion, suggesting that synapse number as well as excitatory protein content were maintained.
Surprisingly, there was an increase in synaptic protein levels for gene products related to GABAergic function, and this occurred in the face of a robust decrease in the number of GABAergic interneurons and GABAergic synapses. GABAA receptors increased significantly, and this increase may reflect a “denervation supersensitivity” adaptation in which postsynaptic excitatory neurons, deprived of their normal inhibitory tone, upregulate expression in an attempt to enhance inhibitory tone. If true, this mechanism could have important consequences regarding the treatment of seizure disorders in which increased GABAergic receptor expression is present (Scheffer & Berkovic, 2003). Further, GAD-67 (the rate-limiting enzyme in the synthesis of GABA) and GAT-1 (the presynaptic GABA reuptake transporter that plays a role in terminating GABA signaling) were both highly upregulated. These results suggest the intriguing possibility that despite a loss of GABAergic interneurons, there is an increase in presynaptic GABAergic terminals, which would further suggest an increase in branching and complexity of the remaining GABAergic neurons in NPN2 deficient animals. However, at least on CA3b pyramidal cells, the number of presynaptic GABAergic terminals is decreased in NPN2 deficient animals. It is also possible that, at least in the case of GAT-1, glial cells increased expression in order to scavenge GABA for the few remaining overworked GABA interneurons. Intriguingly, these findings also may indicate that the GABAergic interneurons are exhibiting an immature phenotype (Powell et al., 2003; Levitt et al., 2004). These possibilities suggest future studies and hint at new approaches to therapeutic intervention.
Sensitivity to chemical challenge
As reported in the Methods section, the homozygous KOs represented 10% of the 2-month-old population, suggesting that the gene deletion was not 100% lethal. However, our dose of KA across all animals was selectively lethal for the homozygous KO animals. While this rendered any downstream analysis moot, it did strongly suggest that the homozygous KO mice were more sensitive to the chemical challenge. Further, the heterozygous mice also showed a facilitated response with ensuing apparent epileptogenesis. This could be due genomic elements or spontaneous seizures of hetereozygous mice “primed” cells for subsequent KA-induced epileptogenesis. Further, a separate cohort of heterozygous mice also showed a decreased latency to PTZ-induced seizures, supporting the idea that NPN2 deficiency during development leads to a lowered seizure threshold.
Electrophysiology
The chemical challenge studies pointed to lowered seizure thresholds in NPN2 KO animals. To take a closer look at the performance of brain circuitry in drug naive animals with NPN2 deficits, we used the hippocampal slice preparation to examine I/O relationships in the field CA1 (Schaeffer collateral) pathway. The steeper I/O curves indicated that the CA1 somatic layer of NPN2 deficient animals yielded a greater response, suggesting increased excitability. In the frequency facilitation model of short-term synaptic plasticity (Landfield et al., 1978, 1986; Thibault et al., 2001), NPN2 KO animals exhibited deficient synaptic throughput with fewer maintained second spikes. This could reflect a compensatory mechanism whereby in the face of increased excitability (I/O data), cells try to dampen signaling by decreasing synaptic throughput. However, changes in excitatory, as well as inhibitory pathways could mediate this phenotype either alone or in concert, and further experiments with pharmacological GABA blockade and/or selective stimulation (i.e., antidromic versus orthodromic) protocols are needed to address these issues.
Loss of interneurons
The behavioral increase in susceptibility to systemically applied drugs could have been due to a number of factors, including altered metabolism or blood flow. However, subsequent electrophysiologic studies did support a local perturbation in at least one of the excitatory pathways of the hippocampus. Here again, multiple hypotheses could account for these changes, including facilitated excitatory or reduced inhibitory activity. Thus, we used immunocytochemical approaches to label and count, using a semiquantitative approach, different excitatory and inhibitory neuronal populations within the hippocampus. While this approach may over- or underestimate actual changes as assessed stereologically, the direction and relative magnitude of change across different cell subtypes should be more reliable. We found no differences in excitatory cell body counts within the principal cell body layers (CA and DG). However, one of the more robust findings reported here was a profound loss of interneurons. Neuropeptide Y (Npy+) interneurons were significantly reduced in all regions, and changes in Npy expression levels have recently been associated with seizure activity (Kharlamov et al., 2007). In mouse hippocampus, NPY+ interneurons and SS+ interneurons accounted for 33% and 17% of the immunoreactive GABAergic neurons (Jinno & Kosaka, 2003), respectively. Interestingly, NPY+ and SST+ positive interneurons have a highly overlapping (52%) distribution (Jinno & Kosaka, 2003), yet we observed a relatively selective loss of NPY+/SST- interneurons. NPY+/SST- interneurons are 48% of the NPY subpopulations based on stereological measurements in mouse hippocampus. This suggests that the SST expressing interneurons are relatively spared (and in fact show a trend towards increasing cell numbers), with NPN2 KO. It may be interesting to examine in future studies whether the NPY+/SST+ interneurons are spared themselves, or if this represents an increase in NPY-/SST+ interneurons. Subclasses of GABA+ interneurons originate from distinct parts of the ganglionic eminence (GE). Parv+, SS+, and NPY+ interneurons develop and migrate from the median GE (MGE), whereas Calretinin (CR+) cells develop and migrate from the caudal GE (CGE). Our data suggest defects in NPN2 signaling affects interneurons derived from MGE but not CGE (Pleasure et al., 2000).
Alternatively, decreases in cells immunoreactive for parvalbumin, calretinin, calbindin, and somatostatin have been reported in human epileptic tissue (temporal lobe) and animal models of epilepsy (Gruber et al., 1994; Spreafico et al., 2000; Andre et al., 2001; Sundstrom et al., 2001), and seizure-induced release of neuropeptide Y and somatostatin (reviewed in Sperk et al., 2007; Tallent & Qiu, 2007) has been reported. However, mice null for these proteins do not have spontaneous seizures or increased vulnerability to KA-induced seizures. The spontaneous seizures may have affected the neuropeptide profile in interneurons (Andre et al., 2001). Further, neuropeptide Y expression is well known to increase in response to seizure activity, albeit this work usually focuses on excitatory cell layers (e.g., Kharlamov et al., 2007; Sperk et al., 2007).
Numbers of GABAergic interneurons were significantly reduced in all areas of the hippocampus (Supplementary Table 3), supporting earlier work implicating the semaphorin-neuropilin signaling system in GABAergic migration and differentiation (Marin et al., 2001) as well as formation, stability, and function of excitatory and GABAergic synapses (Sahay et al., 2005). Regarding migration, NPN2 is expressed in migrating GABAergic neurons (Tamamaki et al., 2003), its deficiency impairs GABAergic migration (Marin et al., 2001), and its ligand (Sema3F), is expressed during development along the cortical/hippocampal GABAergic neuron migratory pathway (Pleasure et al., 2000). Further, Sema3F's ectopic expression can alter GABAergic neuron migration (Tamamaki et al., 2003). Thus, cell migration during development may at least in part account for the reduced number of GABAergic interneurons seen in NPN2 deficient mice.
GABAergic differentiation is mediated by a variety of signaling mechanisms including Sema3A/NPN1, fibroblast growth factor 2 (FGF-2), brain derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and hepatocyte growth factor/scatter factor (HGF/SF) (Pappas & Parnavelas, 1998; Fiumelli et al., 2000; Korhonen et al., 2000; Marin et al., 2001; Powell et al., 2001). Similar to Slit/Robo signaling in cultured hippocampal neurons (Sang et al., 2002), reductions of Parv+ and NPY+ fibers along the apical and basilar dendritic layers of CA3 and CA1 regions in NPN2 KO mice could imply that proper GABAergic neuritic extension is altered (Figs. 5 and 6 and Tables 3 and 4), consistent with the deletion of NPN2 signaling in excitatory pyramidal cells and DG neurons leading to improper extension of spines on dendrites (Fig. 7) and extension of axons (Sahay et al., 2005). A similar hypothesis has been proposed for hippocampal parvalbumin+ interneurons in mice deficient of C-Met oncogene (MET) hepatocyte growth factor (HGF) signaling (Martins et al., 2007). Several mechanisms are possible, including (1) abnormal extension of neurites or (2) mistargeting to wrong cell types or incorrect subcellular localization. However, the lack of GABA+ neurons alone could account for these observations.
Morphometry
Based on the altered excitability of field CA1 and the loss of innervating inhibitory interneurons, we examined the structure of CA1 pyramidal cells, especially dendritic arbors and synaptic spines. Decreased dendritic complexity, multisynaptic inputs, filopodia, and increased density of immature spines were observed (Liu et al., 2005; Sahay et al., 2005; Faulkner et al., 2007) (Fig. 7 and Table 5), suggesting that synapse formation is immature in NPN2 deficient animals. In development, interneurons receive synaptic input first from other GABAergic neurons and then pyramidal cells (Ben-Ari et al., 2004). Only interneurons of sufficient morphological maturity receive functional synaptic input, suggesting that GABAergic synaptogenesis is governed by the degree of maturation of the postsynaptic neuron (Danglot et al., 2006).
Summary
NPN2 deficient animals show an apparent lower seizure threshold, enhanced excitability, and altered short-term synaptic throughput. Further, they have reduced numbers of interneurons and altered dendritic branching and dendritic spine counts in excitatory neurons of the CA1 field. These findings could be accounted for by pleiotropic effects on cell migration, synaptic localization, and assembly or stability of synaptic content, thereby affecting synaptic physiology, plasticity, and neuronal excitability (Contractor et al., 2002; Henkemeyer et al., 2003). Interestingly, polymorphic alleles of the human NPN2 gene are associated with the clinical expression of autism (a disease of GABAergic deficiency associated with seizures) in Chinese populations (Wu et al., 2007). Thus, this animal model may have broader applications as a model for some aspects of autism, just as Reeler mice can serve as models for both epilepsy and psychosis (Patrylo et al., 2006). Conditional deletions of Sema3F signaling, either via inducible KOs, pharmacologic agents, or other approaches (e.g., small interfering RNA or siRNA) will provide an experimental basis to dissect the molecular pathways responsible for interneuron migration, excitatory and inhibitory neuritogenesis, and subsequent synaptic connectivity.
Supplementary Material
Figure S1. Demarcation boundaries for counting immunoreactive or nissl-stained positive neurons in the hippocampus of NPN2 (−/−) and NPN2 (+/+) mice. The hippocampal subregions for principal cell and interneuron counting are shown in distinct opaque boxes. All cells whose full nuclei were within this region were counted (see Methods section). CA1, stratum radiatum, stratum oriens, and stratum pyramidale of the CA1 region; CA3, stratum radiatum, stratum oriens, and stratum pyramidale of the CA3 region; DG/H, cell layers and molecular layers of the dentate gyrus and hilar regions.
Figure S2. Equivalent somatostatin and calretinin positive neurons in the hippocampus of NPN2 (−/−) and NPN2 (+/+) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 1A or 1C) or NPN2 (−/−) (Supplementary Fig. 2B and 2D mice were subjected to immunocytochemistry using the somatostatin antibodies. There were no differences in somatostatin cell counts from the CA3 (Supplementary Fig. 2A and 2B), CA1, or H/DG regions (Supplementary Fig. 2C and 2D) of either genotype. There were also no differences in somatostatin cell counts from the CA3 (Supplementary Fig. 2E and 2F), CA1, or H/DG regions (Supplementary Fig. 2G and 2H) of either genotype.
Figure S3. Decreased GABAergic synapses on CA3b pyramidal cells in the hippocampus of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 3D–3F and 3J–3L) or NPN2 (−/−) (Supplementary Fig. 3A–3C and 3G–3I) mice were subjected to immunocytochemistry using the parvalbumin (Parv), glutamate decarboxylase 65 (GAD-65), GABAA receptor α1 subunit (GABARA1) antibodies. The number of Parv+ and GAD-65+ punctata was decreased by 44% and 46% around CA3b pyramidal cells in NPN2 (−/−) mice (Supplementary Fig. 3A–3C). The percent colabeling between Parv+ and GABARA1+ cells decreased from 65% in NPN2 (+/+) mice (Supplementary Fig. 3J–3L) to 29% in NPN2 (−/−) mice (Supplementary Fig. 3G–3I).
Figure S4. Decreased cell numbers of GAD-67+ interneurons in the hippocampus of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 4A and 4C) or NPN2 (−/−) (Supplementary Fig. 4B and 4D) mice were subjected to immunocytochemistry using the parvalbumin (Parv) and glutamate decarboxylase 67 (GAD-67) antibodies. There were substantial reduction of numbers of GAD-67+ in the CA3 stratum oriens (67%), CA1 stratum radiatum (43%), and CA1 stratum oriens (50%) of NPN2 (−/−) mice (Supplementary Fig. 4A–4D). The number of GAD-67+ cells was decreased by 65% in the molecular layer (ML) of NPN2 (−/−) mice (Supplementary Fig. 4C and 4D). There was a moderate reduction of numbers of GAD-67+ in the CA3 stratum radiatum (30%) of NPN2 (−/−) mice (Supplementary Fig. 4A and 4B).
Table S1. Excitatory cell counts in murine hippocampus 240 days after KA-SE.
Table S2. Nissl cell counts in hippocampus from NPN2 KO mice.
Table S3. GAD-67 cell counts in hippocampus from NPN2 KO mice.
Acknowledgments
This study was supported in part by a Partnership for Pediatric Epilepsy Research grant and Kentucky Spinal Cord & Brain Injury Trust grant (G.N.B.) and also by National Institutes of Health (NIH) grant no. AG-029268 (J.G. and O.T.).
Footnotes
Conflict of interest: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. The authors have no conflicts of interest to disclose.
Supporting Information: Additional Supporting Information may be found in the online version of this article
Please note: Wiley Periodicals is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Andre V, Marescaux C, Nehlig A, Fritschy JM. Alterations of hippocampal GABAergic system contribute to development of spontaneous recurrent seizures in the rat lithium-pilocarpine model of temporal lobe epilepsy. Hippocampus. 2001;11:452–468. doi: 10.1002/hipo.1060. [DOI] [PubMed] [Google Scholar]
- Barnes GN, Slevin JT, Vanaman TC. Rat brain protein phosphatase 2A: an enzyme that may regulate autophosphorylated protein kinases. J Neurochem. 1995;64:340–353. doi: 10.1046/j.1471-4159.1995.64010340.x. [DOI] [PubMed] [Google Scholar]
- Barnes GN, Slevin JT. Ionotropic glutamate receptor biology: effect on synaptic connectivity and function in neurological disease. Curr Med Chem. 2003;10:2059–2072. doi: 10.2174/0929867033456800. [DOI] [PubMed] [Google Scholar]
- Barnes G, Puranam RS, Luo Y, McNamara JO. Temporal specific patterns of semaphorin gene expression in rat brain after kainic acid-induced status epilepticus. Hippocampus. 2003;13:1–20. doi: 10.1002/hipo.10041. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Represa A, Gozlan H. Interneurons set the tune of developing networks. Trends Neurosci. 2004;27:422–427. doi: 10.1016/j.tins.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36:827–835. doi: 10.1038/ng1395. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Cross DJ. The role of synaptic reorganization in mesial temporal lobe epilepsy. Epilepsy Behav. 2006;8:483–493. doi: 10.1016/j.yebeh.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, Lowenstein DH, Skarnes WC, Chedotal A, Tessier-Lavigne M. Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron. 2000;25:43–56. doi: 10.1016/s0896-6273(00)80870-3. [DOI] [PubMed] [Google Scholar]
- Clarke DF, Roberts W, Daraksan M, Dupuis A, McCabe J, Wood H, Snead OC, 3rd, Weiss SK. The prevalence of autistic spectrum disorder in children surveyed in a tertiary care epilepsy clinic. Epilepsia. 2005;46:1970–1977. doi: 10.1111/j.1528-1167.2005.00343.x. [DOI] [PubMed] [Google Scholar]
- Contractor A, Rogers C, Maron C, Henkemeyer M, Swanson GT, Heinemann SF. Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science. 2002;296:1864–1869. doi: 10.1126/science.1069081. [DOI] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus. 2006;16:1032–1060. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- de Wit J, Verhaagen J. Role of semaphorins in the adult nervous system. Prog Neurobiol. 2003;71:249–267. doi: 10.1016/j.pneurobio.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Dent EW, Tang F, Kalil K. Axon guidance by growth cones and branches: common cytoskeletal and signaling mechanisms. Neuroscientist. 2003;9:343–353. doi: 10.1177/1073858403252683. [DOI] [PubMed] [Google Scholar]
- Deonna T, Roulet E. Autistic spectrum disorder: evaluating a possible contributing or causal role of epilepsy. Epilepsia. 2006;47(Suppl 2):79–82. doi: 10.1111/j.1528-1167.2006.00697.x. [DOI] [PubMed] [Google Scholar]
- Faulkner RL, Low LK, Cheng HJ. Axon pruning in the developing vertebrate hippocampus. Dev Neurosci. 2007;29:6–13. doi: 10.1159/000096207. [DOI] [PubMed] [Google Scholar]
- Fenstermaker V, Chen Y, Ghosh A, Yuste R. Regulation of dendritic length and branching by semaphorin 3A. J Neurobiol. 2004;58:403–412. doi: 10.1002/neu.10304. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, Ballas C, Schill J, Buono RJ, Berrettini WH. Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. J Neurosci. 1999;19:6733–6739. doi: 10.1523/JNEUROSCI.19-16-06733.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Kiraly M, Ambrus A, Magistretti PJ, Martin JL. Opposite regulation of calbindin and calretinin expression by brain-derived neurotrophic factor in cortical neurons. J Neurochem. 2000;74:1870–1877. doi: 10.1046/j.1471-4159.2000.0741870.x. [DOI] [PubMed] [Google Scholar]
- Gedye A. Frontal lobe seizures in autism. Med Hypotheses. 1991;34:174–182. doi: 10.1016/0306-9877(91)90189-6. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- Gruber B, Greber S, Rupp E, Sperk G. Differential NPY mRNA expression in granule cells and interneurons of the rat dentate gyrus after kainic acid injection. Hippocampus. 1994;4:474–482. doi: 10.1002/hipo.450040409. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163:1313–1326. doi: 10.1083/jcb.200306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp M, Meierkord H. Anticonvulsant, antiepileptogenic, and antiictogenic pharmacostrategies. Cell Mol Life Sci. 2007;64:2023–2041. doi: 10.1007/s00018-007-7021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Gorter JA, De Wit J, Tolner EA, Spijker S, Giger RJ, Lopes da Silva FH, Verhaagen J. Transient downregulation of Sema3A mRNA in a rat model for temporal lobe epilepsy. A novel molecular event potentially contributing to mossy fiber sprouting. Exp Neurol. 2003;182:142–150. doi: 10.1016/s0014-4886(03)00035-9. [DOI] [PubMed] [Google Scholar]
- Jinno S, Kosaka T. Patterns of expression of neuropeptides in GABAergic nonprincipal neurons in the mouse hippocampus: quantitative analysis with optical disector. J Comp Neurol. 2003;461:333–349. doi: 10.1002/cne.10700. [DOI] [PubMed] [Google Scholar]
- Kagan-Kushnir T, Roberts SW, Snead OC., 3rd Screening electroencephalograms in autism spectrum disorders: evidence-based guideline. J Child Neurol. 2005;20:197–206. doi: 10.1177/08830738050200030601. [DOI] [PubMed] [Google Scholar]
- Kharlamov EA, Kharlamov A, Kelly KM. Changes in neuropeptide Y protein expression following photothrombotic brain infarction and epileptogenesis. Brain Res. 2007;1127:151–162. doi: 10.1016/j.brainres.2006.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen L, Sjoholm U, Takei N, Kern MA, Schirmacher P, Castren E, Lindholm D. Expression of c-Met in developing rat hippocampus: evidence for HGF as a neurotrophic factor for calbindin D-expressing neurons. Eur J Neurosci. 2000;12:3453–3461. doi: 10.1046/j.1460-9568.2000.00260.x. [DOI] [PubMed] [Google Scholar]
- Landfield PW, McGaugh JL, Lynch G. Impaired synaptic potentiation processes in the hippocampus of aged, memory-deficient rats. Brain Res. 1978;150:85–101. doi: 10.1016/0006-8993(78)90655-8. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA, Applegate MD. The effects of high Mg2+-to-Ca2+ ratios on frequency potentiation in hippocampal slices of young and aged rats. J Neurophysiol. 1986;56:797–811. doi: 10.1152/jn.1986.56.3.797. [DOI] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Liu XB, Low LK, Jones EG, Cheng HJ. Stereotyped axon pruning via plexin signaling is associated with synaptic complex elimination in the hippocampus. J Neurosci. 2005;25:9124–9134. doi: 10.1523/JNEUROSCI.2648-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestrini E, Lai C, Marlow A, Matthews N, Wallace S, Bailey A, Cook EH, Weeks DE, Monaco AP. Serotonin transporter (5-HTT) and gamma-aminobutyric acid receptor subunit beta3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. The International Molecular Genetic Study of Autism Consortium. Am J Med Genet. 1999;88:492–496. doi: 10.1002/(sici)1096-8628(19991015)88:5<492::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Marin O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science. 2001;293:872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- Martins GJ, Plachez C, Powell EM. Loss of embryonic MET signaling alters profiles of hippocampal interneurons. Dev Neurosci. 2007;29:143–158. doi: 10.1159/000096219. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Tu B, Timofeeva O, Jiao Y, Herzog H. Neuropeptide Y in the recurrent mossy fiber pathway. Peptides. 2007;28:357–364. doi: 10.1016/j.peptides.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbury DF, Bonora E, Lamb JA, Fisher SE, Lai CS, Baird G, Jannoun L, Slonims V, Stott CM, Merricks MJ, Bolton PF, Bailey AJ, Monaco AP. FOXP2 is not a major susceptibility gene for autism or specific language impairment. Am J Hum Genet. 2002;70:1318–1327. doi: 10.1086/339931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, Smith SJ. In vivo imaging of synapse formation on a growing dendritic arbor. Nat Neurosci. 2004;7:254–260. doi: 10.1038/nn1191. [DOI] [PubMed] [Google Scholar]
- O'Donnell J, Stemmelin J, Nitta A, Brouillette J, Quirion R. Gene expression profiling following chronic NMDA receptor blockade-induced learning deficits in rats. Synapse. 2003;50:171–180. doi: 10.1002/syn.10258. [DOI] [PubMed] [Google Scholar]
- Pappas IS, Parnavelas JG. Basic fibroblast growth factor promotes the generation and differentiation of calretinin neurons in the rat cerebral cortex in vitro. Eur J Neurosci. 1998;10:1436–1445. doi: 10.1046/j.1460-9568.1998.00147.x. [DOI] [PubMed] [Google Scholar]
- Pascual M, Pozas E, Barallobre MJ, Tessier-Lavigne M, Soriano E. Coordinated functions of netrin-1 and class 3 secreted semaphorins in the guidance of reciprocal septohippocampal connections. Mol Cell Neurosci. 2004;26:24–33. doi: 10.1016/j.mcn.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Verhaagen J. Semaphorins in axon regeneration: developmental guidance molecules gone wrong? Philos Trans R Soc Lond B Biol Sci. 2006;361:1499–1511. doi: 10.1098/rstb.2006.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrylo PR, Browning RA, Cranick S. Reeler homozygous mice exhibit enhanced susceptibility to epileptiform activity. Epilepsia. 2006;47:257–266. doi: 10.1111/j.1528-1167.2006.00417.x. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Nissinen J, Nairismagi J, Lukasiuk K, Grohn OH, Miettinen R, Kauppinen R. Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res. 2002;135:67–83. doi: 10.1016/S0079-6123(02)35008-8. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Anderson S, Hevner R, Bagri A, Marin O, Lowenstein DH, Rubenstein JL. Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron. 2000;28:727–740. doi: 10.1016/s0896-6273(00)00149-5. [DOI] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozas E, Pascual M, Nguyen Ba-Charvet KT, Guijarro P, Sotelo C, Chedotal A, Del Rio JA, Soriano E. Age-dependent effects of secreted semaphorins 3A, 3F, and 3E on developing hippocampal axons: in vitro effects and phenotype of semaphorin 3A (-/-) mice. Mol Cell Neurosci. 2001;18:26–43. doi: 10.1006/mcne.2001.0999. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Sahay A, Molliver ME, Ginty DD, Kolodkin AL. Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci. 2003;23:6671–6680. doi: 10.1523/JNEUROSCI.23-17-06671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond CH, Ashburner J, Connelly A, Friston KJ, Gadian DG, Vargha-Khadem F. The role of the medial temporal lobe in autistic spectrum disorders. Eur J Neurosci. 2005;22:764–772. doi: 10.1111/j.1460-9568.2005.04217.x. [DOI] [PubMed] [Google Scholar]
- Sang Q, Wu J, Rao Y, Hsueh YP, Tan SS. Slit promotes branching and elongation of neurites of interneurons but not projection neurons from the developing telencephalon. Mol Cell Neurosci. 2002;21:250–265. doi: 10.1006/mcne.2002.1156. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. The genetics of human epilepsy. Trends Pharmacol Sci. 2003;24:428–433. doi: 10.1016/S0165-6147(03)00194-9. [DOI] [PubMed] [Google Scholar]
- Shimakawa S, Suzuki S, Miyamoto R, Takitani K, Tanaka K, Tanabe T, Wakamiya E, Nakamura F, Kuno M, Matsuura S, Watanabe Y, Tamai H. Neuropilin-2 is overexpressed in the rat brain after limbic seizures. Brain Res. 2002;956:67–73. doi: 10.1016/s0006-8993(02)03482-0. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Song H, Poo M. The cell biology of neuronal navigation. Nat Cell Biol. 2001;3:E81–88. doi: 10.1038/35060164. [DOI] [PubMed] [Google Scholar]
- Sperk G, Hamilton T, Colmers WF. Neuropeptide Y in the dentate gyrus. Prog Brain Res. 2007;163:285–297. doi: 10.1016/S0079-6123(07)63017-9. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Tassi L, Colombo N, Bramerio M, Galli C, Garbelli R, Ferrario A, Lo Russo G, Munari C. Inhibitory circuits in human dysplastic tissue. Epilepsia. 2000;41(Suppl 6):S168–173. doi: 10.1111/j.1528-1157.2000.tb01576.x. [DOI] [PubMed] [Google Scholar]
- Sundstrom LE, Brana C, Gatherer M, Mepham J, Rougier A. Somatostatin- and neuropeptide Y-synthesizing neurones in the fascia dentata of humans with temporal lobe epilepsy. Brain. 2001;124:688–697. doi: 10.1093/brain/124.4.688. [DOI] [PubMed] [Google Scholar]
- Tallent MK, Qiu C. Somatostatin: an endogenous antiepileptic. Mol Cell Endocrinol. 2008;286(12):96–103. doi: 10.1016/j.mce.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Fujimori K, Nojyo Y, Kaneko T, Takauji R. Evidence that Sema3A and Sema3F regulate the migration of GABAergic neurons in the developing neocortex. J Comp Neurol. 2003;455:238–248. doi: 10.1002/cne.10476. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde F. Aspects of cortical organization related to the geometry of neurons with intra-cortical axons. J Neurocytol. 1976;5:509–529. doi: 10.1007/BF01175566. [DOI] [PubMed] [Google Scholar]
- Walz A, Rodriguez I, Mombaerts P. Aberrant sensory innervation of the olfactory bulb in neuropilin-2 mutant mice. J Neurosci. 2002;22:4025–4035. doi: 10.1523/JNEUROSCI.22-10-04025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Yue W, Jia M, Ruan Y, Lu T, Gong X, Shuang M, Liu J, Yang X, Zhang D. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2007;144:492–495. doi: 10.1002/ajmg.b.30495. [DOI] [PubMed] [Google Scholar]
- Xiang RH, Hensel CH, Garcia DK, Carlson HC, Kok K, Daly MC, Kerbacher K, Van Den Berg A, Veldhuis P, Buys CH, Naylor SL. Isolation of the human semaphorin III/F gene (SEMA3F) at chromosome 3p21, a region deleted in lung cancer. Genomics. 1996;32:39–48. doi: 10.1006/geno.1996.0074. [DOI] [PubMed] [Google Scholar]
- Yang J, Houk B, Shah J, Hauser KF, Luo Y, Smith G, Schauwecker E, Barnes GN. Genetic background regulates semaphorin gene expression and epileptogenesis in mouse brain after kainic acid status epilepticus. Neuroscience. 2005;131:853–869. doi: 10.1016/j.neuroscience.2004.09.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Demarcation boundaries for counting immunoreactive or nissl-stained positive neurons in the hippocampus of NPN2 (−/−) and NPN2 (+/+) mice. The hippocampal subregions for principal cell and interneuron counting are shown in distinct opaque boxes. All cells whose full nuclei were within this region were counted (see Methods section). CA1, stratum radiatum, stratum oriens, and stratum pyramidale of the CA1 region; CA3, stratum radiatum, stratum oriens, and stratum pyramidale of the CA3 region; DG/H, cell layers and molecular layers of the dentate gyrus and hilar regions.
Figure S2. Equivalent somatostatin and calretinin positive neurons in the hippocampus of NPN2 (−/−) and NPN2 (+/+) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 1A or 1C) or NPN2 (−/−) (Supplementary Fig. 2B and 2D mice were subjected to immunocytochemistry using the somatostatin antibodies. There were no differences in somatostatin cell counts from the CA3 (Supplementary Fig. 2A and 2B), CA1, or H/DG regions (Supplementary Fig. 2C and 2D) of either genotype. There were also no differences in somatostatin cell counts from the CA3 (Supplementary Fig. 2E and 2F), CA1, or H/DG regions (Supplementary Fig. 2G and 2H) of either genotype.
Figure S3. Decreased GABAergic synapses on CA3b pyramidal cells in the hippocampus of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 3D–3F and 3J–3L) or NPN2 (−/−) (Supplementary Fig. 3A–3C and 3G–3I) mice were subjected to immunocytochemistry using the parvalbumin (Parv), glutamate decarboxylase 65 (GAD-65), GABAA receptor α1 subunit (GABARA1) antibodies. The number of Parv+ and GAD-65+ punctata was decreased by 44% and 46% around CA3b pyramidal cells in NPN2 (−/−) mice (Supplementary Fig. 3A–3C). The percent colabeling between Parv+ and GABARA1+ cells decreased from 65% in NPN2 (+/+) mice (Supplementary Fig. 3J–3L) to 29% in NPN2 (−/−) mice (Supplementary Fig. 3G–3I).
Figure S4. Decreased cell numbers of GAD-67+ interneurons in the hippocampus of NPN2 (−/−) mice. Forty-micron sections from NPN2 (+/+) (Supplementary Fig. 4A and 4C) or NPN2 (−/−) (Supplementary Fig. 4B and 4D) mice were subjected to immunocytochemistry using the parvalbumin (Parv) and glutamate decarboxylase 67 (GAD-67) antibodies. There were substantial reduction of numbers of GAD-67+ in the CA3 stratum oriens (67%), CA1 stratum radiatum (43%), and CA1 stratum oriens (50%) of NPN2 (−/−) mice (Supplementary Fig. 4A–4D). The number of GAD-67+ cells was decreased by 65% in the molecular layer (ML) of NPN2 (−/−) mice (Supplementary Fig. 4C and 4D). There was a moderate reduction of numbers of GAD-67+ in the CA3 stratum radiatum (30%) of NPN2 (−/−) mice (Supplementary Fig. 4A and 4B).
Table S1. Excitatory cell counts in murine hippocampus 240 days after KA-SE.
Table S2. Nissl cell counts in hippocampus from NPN2 KO mice.
Table S3. GAD-67 cell counts in hippocampus from NPN2 KO mice.