

SUMMARY
Background
To assess activity and toxicity in newly diagnosed advanced stage epithelial ovarian cancer (EOC) patients receiving dose-intense paclitaxel, cyclophosphamide, cisplatin, and filgrastim delivered with a flexible dosing schedule.
Methods
Patients with Stage III/IV EOC received cyclophosphamide 750 mg/m2, followed by 24 hr infusion of paclitaxel 250 mg/m2, and cisplatin 75 mg/m2 on day 2. Filgrastim began on day 3 at 10 μg/kg/d × 9d. Patients received six cycles of all drugs. Those with pathologic complete response or microscopic residual disease at the conclusion of six cycles of therapy received an additional cycles two to four cycles of paclitaxel with cyclophosphamide. Patients with objective response continued cyclophosphamide and paclitaxel.
Results
62 patients were enrolled. Thirty-two of these 62 patients had stage IIIC disease, and 26 of 62 had stage IV disease. Using an intent to treat analysis, 55 (89%) experienced clinical complete remission (CCR). With a median potential follow-up of 11.4 years, the median progression free survival is 18.9 months and median survival is 5.4 years. The most serious toxicity was grade 3/4 neutropenic fever (35%). Although all participants developed peripheral neuropathy, improvement in neuropathic symptoms began with decrease or cessation of paclitaxel.
Conclusions
This regimen yielded a high response rate and encouraging overall survival. These data and those of the Japanese Gynecologic Oncology Group suggest that further study of dose dense or intense paclitaxel regimens in women with newly diagnosed advanced stage EOC is warranted.
Keywords: ovarian neoplasms, paclitaxel, cyclophosphamide, cisplatin, antineoplastic combined chemotherapy protocols, filgrastim, drug dose-response relationship
INTRODUCTION
Epithelial ovarian cancer (EOC) is the leading cause of death from gynecologic malignancies in the United States with 15,520 deaths expected in 2008 1. It continues to be associated with a poor prognosis and advanced stage at presentation 2. Most women with advanced stage disease can anticipate optimal surgical cytoreduction and subsequent response to platinum-based chemotherapy, yet most will experience tumor recurrence and eventual death from EOC. Sustained remission and cure remains rare in women with advanced stage disease.
Many approaches have been explored to optimize and improve initial treatment. Interval surgical cytoreduction has been tested without notable advantage in the paclitaxel era. Other approaches in initial systemic therapy include triple drug or sequential doublet regimens 3, 4, maintenance therapy 5, introduction of novel treatment regimens including molecularly targeted agents for initial treatment 6, and dose intensification 7-11. Limited advances, albeit statistically significant, have been observed. Recent reports indicate that dose dense taxane therapy benefits patients with breast cancer and EOC. Following standard adjuvant therapy, breast cancer patients treated with weekly paclitaxel showed improved disease-free (PFS) and overall survival (OS) compared to those receiving paclitaxel every 3 weeks 12. More relevant are recent results from the Japanese Gynecologic Oncology Group (JGOG) who have demonstrated improved PFS in women with Stage II-IV treated with carboplatin coupled with weekly paclitaxel 80mg/m2 as compared to standard schedule paclitaxel (180mg/m2). delivered every 3 weeks 11.
We now report the activity, toxicity, and 10 year survival outcome of women with EOC in a Phase II trial of a dose-intense paclitaxel regimen. Prospective goals were to define the surgical response rate, use of flexible filgrastim dosing schedule to maintain paclitaxel dose density, individualization of treatment duration, and portability through multi-institutional collaboration. Secondary objectives were characterization of OS and PFS and the kinetics of neurotoxicity.
Cisplatin and cyclophosphamide, with differing and potentially synergistic mechanisms of action, were combined with dose intensified paclitaxel 13-16. Paclitaxel stabilizes microtubule assembly 17 and slows repair of DNA lesions, including cisplatin adducts and cyclophosphamide cross-links 16, 18. We defined the maximally tolerated dose of paclitaxel as 250mg/m2 when given as a 24 hour infusion with flexible filgrastim support 19, and determined its safety when combined with cyclophosphamide 750mg/m2 and cisplatin 75mg/m2 20. Administration of the triplet to patients with advanced stage EOC led to an 89% clinical response and a 61% surgical response (microscopic and pathologic) in a single institution study of EOC patients 20. This Phase II study defines the activity and tolerability of this dose-intense paclitaxel combination.
PATIENTS AND METHODS
Patient Eligibility and Pretreatment Evaluation
The study was approved by the Institutional Review Boards at the National Cancer Institute (NCI) and Massachusetts General Hospital (MGH). Eligibility criteria included women with untreated newly diagnosed FIGO stage III/IV EOC, initiation of therapy within 6 weeks of staging laparotomy, creatinine clearance ≥60 cc/min, good end organ function, ECOG performance status 0-2, and informed consent. Patients were excluded for a prior history of invasive cancer, myocardial infarction or unstable dysrhythmia within six months, or gastrointestinal bleeding within 1 month of entry.
Drug Administration
Agents were provided by the Cancer Therapy Evaluation Program, NCI. NCI patients received their first cycle in the hospital with subsequent cycles as outpatients, whereas MGH patients received all cycles in the hospital. The premedication, treatment, and dose modification schema are shown in Table I. Cisplatin was limited to 6 cycles to minimize risk of allergic reactions and cumulative neurotoxicity. Cyclophosphamide and paclitaxel were continued in patients requiring more than 6 cycles for best clinical response, and after surgical reassessment. Subsequent cycles were initiated if nonhematologic toxicity was less than grade 3 and the absolute granulocyte count was ≥1500 cells/mm3.
Table I.
| (a): Drug administration and dose modification schema | |||||||
|---|---|---|---|---|---|---|---|
| Administration | 21 Day Cycle | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7… | |
| Premedication* | XX | XX | |||||
| Hydration | XXXXXXXXXXXXXXX | ||||||
| Cyclophosphamide (C) 750 mg/m2 | X | ||||||
| Paclitaxel (T) 250 mg/m2 | 24 hour CI | ||||||
| Cisplatin (P) 75 mg/m2 | X | ||||||
| Filgrastim (G) 10 ug/kg/d** | Daily D3-13 | X | X | X | X | ||
| Antiemetics | |||||||
| - ondansetron*** | X | X | X | X | |||
| - Prochlorperazine**** | X | X | X | X | X | X | X |
| (b) Dose modification | |||||||
| Toxicity | 15t Occurrence | 2nd Occurrence | 3rd Occurrence | ||||
| Neutropenia >5d +/- F&N | G 20/ug/kg/d | T 200mg/m2 | C 500mg/m2 | ||||
| Gr 4 thrombocytopenia | T 200mg/m2 | P 50 mg/m2 | T 150mg/m2 | ||||
| Gr 3 neurotoxicity | T 200mg/m2 | T 175mg/m2 | T 150mg/m2 | ||||
| Gr 4 neurotoxicity | T 200mg/m2 P 50mg/m2 |
||||||
Premedication: Day 1, 20mg oral dexamethasone 14 and 7 hr prior, cimetidine 300mg and diphenhydramine 25mg 30 min prior to T; Day 2, dexamethasone 20 mg and mannitol diuresis.
Filgrastim, continued until the total granulocyte count was >30,000/dL, or for 9 days, whichever was longer
ondansetron 8mg IV prior to C then orally q 4-8 hours for 4-6 doses;
Prochlorperazine SR q 12 hours on 01-2 starting I hour prior to C followed by 30mg q 12 hours for 4 doses and 15mg q 12 hours for 4-6 doses on D5-6.
Toxicity Assessment and Dose Modification
Drug compliance and toxicity were assessed at each visit and graded using NCI Common Toxicity Criteria v1.0. Proprioception, vibration, and pinprick sensory modalities, deep tendon reflexes, Romberg’s maneuver, and tandem gait were evaluated each cycle. Paclitaxel dose was modified for objective clinical findings with functional impairment consistent with CTC v1.0 grade 3 neurologic toxicity.
Outcome Assessment
Patients underwent clinical assessment every cycle and radiographic assessment every 2 cycles. Patients with normal physical examination, CA125, and radiographic studies after 6 cycles (clinical complete response; CCR) underwent surgical reassessment. Therapy post surgical assessment was guided based on intraoperative findings. Two cycles of paclitaxel and cyclophosphamide were administered for patients with pathologically negative findings, while those women with microscopic residual disease triggered 2-4 additional cycles. Patients with unresectable gross residual tumor (pathologic partial response, PPR) were referred to other treatments. Patients not attaining a CCR after 6 cycles received up to 2-4 additional cycles of paclitaxel and cyclophosphamide if still responding. Continued persistent disease led to a change in therapy. All patients were followed for survival.
Disease outcome was measured using radiographic imaging and/or physical examination. Response was documented as a CCR or partial response (PR) lasting at least 4 weeks, or progressive disease (PD). Partial response (PR) required >50% reduction in the sum of the products of bi-directional measurements of tumor masses. Standard PD definitions were used. Disease response was further classified surgically. Patients with CCR who underwent surgical assessment were categorized as a pathologic complete response (PCR), microscopic residual disease (micro), or PPR. Off-study criteria included failure to achieve a CCR, PPR or disease progression as best response, grade 3 or greater non-hematologic toxicity, and voluntary withdrawal.
Statistical Analysis
The primary objectives were to identify the PCR rate and to determine regimen portability to another academic site; the study was defined to accrue up to 80 subjects. Assessment of OS and PFS were secondary objectives and were calculated from the on-study date until the date of death, progression, or last follow-up. OS and PFS probabilities were computed using the Kaplan-Meier method. Survival time was censored at the time of the most recent follow-up.
RESULTS
Patient Characteristics
This cohort had multiple indicators of poor prognosis (Table II). All patients underwent attempts at optimal debulking during surgical staging; 42% had pathologically documented stage IV disease. Residual disease was present at enrollment in 35 (44%), most of whom had hepatic parenchymal involvement; 93% had radiological or clinically measurable disease, with 7% having residual effusion only. Forty-three percent of patients were optimally debulked (<1cm; N = 27).
Table II.
Clinical Characteristics
| NCI (%) | MGH (%) | Total (%) | |
|---|---|---|---|
| Total patients accrued | 46 (75) | 16 (25) | 62 (100) |
| # evaluable for response | 45 | 16 | 61 |
| # evaluable for toxicity | 46 | 16 | 62 |
| Median age (yrs: range) | 50 (43-69) | 55 (40-69) | 53 (40-69) |
| Stage on study: | |||
| IIIA | 0 | 1 | 1 (1) |
| IIIB | 2 | 1 | 3 (5) |
| IIIC | 23 | 9 | 32 (52) |
| IV* | 21 | 5 | 26 (42) |
| Histology | |||
| Papillary serous | 24 | 13 | 37 (59) |
| Endometrioid/Transitional | 10 | 0 | 10 (16) |
| Undifferentiated | 4 | 0 | 4 (7) |
| Clear Cell | 2 | 1 | 3 (5) |
| Primary peritoneal | 6 | 2 | 8 (13) |
| Tumor Grade | |||
| I | 0 | 1 | 1 (1) |
| II | 9 | 2 | 11 (18) |
| III | 37 | 12 | 49 (79) |
| Sites of Disease | |||
| Liver | 13 | 6 | 19 (31) |
| Spleen | 3 | 1 | 4 (6) |
| Lung/chest parenchymal | 2 | 3 | 5 (8) |
| Mesenteric/omental | 22 | 6 | 28 (45) |
| Retroperitoneal LN | 17 | 2 | 19 (31) |
| Pelvis | 27 | 8 | 35 (56) |
| Pleural effusion | 18 | 4 | 22 (35) |
| Ascites | 21 | 3 | 24 (39) |
multiple sites within one organ were counted once.
Response and Duration
The PCR rate, the primary objective, is presented in Table III. CCRs occurred in 55 patients (89%) and 2 with PR (92% ORR; Table III (b)). All but one patient had an invasive reassessment. PCRs occurred in 26 (47%), with microscopic residual found in 20 (36%), yielding an 84% overall pathologic response rate. A patient who had microscopic disease at reassessment was subsequently found to have a PCR at surgery for an adhesion-induced small bowel obstruction. The median potential follow-up of the cohort is 11.4 years. The median PFS is 18.9 months (Figure I) and median OS 5.4 years (53% 5 year OS). When the results are restricted to Stage IV patients with parenchymal liver or splenic involvement, median PFS is 15.8 months and median OS 2.9 years, (42% 5 year OS).
Table III.
| (a): Pathologic response after surgical assessment of CCR patients (N=55) | |||
|---|---|---|---|
| Type of response | Number of patients (%) | ||
| NCI | MGH | Total | |
| Pathologic CR (PCR) | 16 | 10 | 26 (47) |
| Microscopic residual (M)* | 18 | 2 | 20 (36) |
| Gross Residual (PPR) | 6 | 3 | 9 (16) |
| PCR + M (eligible for surgery) | 34/40 (85) | 12/15 (80) | 46/55 (84) |
| (b): Clinical response: intent-to-treat (N=62) | |||
| Type of response | Number of patients (%) | ||
| NCI | MGH | Total | |
| CCR | 40 | 15 | 55 (89) |
| PR | 2 | 0 | 2 (3) |
| SD | 3 | 0 | 3 (5) |
| NE | 1* | 1 | 1 |
| Overall Clinical RR | 42 (91) | 15 (94) | 57 (92) |
includes a patient who did not undergo second look but was found to have microscropic residual on a procedure performed to relieve a small bowel obstruction.
pt withdrew after 2 cycles with unconfirmed PR and tinnitus.
Figure l. Kaplan-Meier plots.
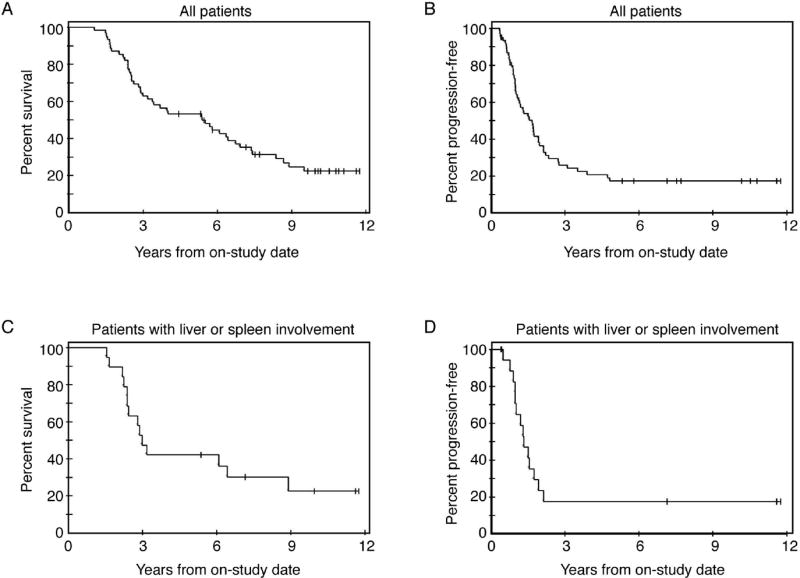
A: Progression-free survival n=62. B: Overall survival n=62. C: Progression-free survival for NCI patients with liver/splenic involvement (n=19). D: Overall survival for NCI patients with liver/splenic involvement (n=19).
Drug Administration and Toxicity
A secondary objective was maintenance of dose density in the first 6 cycles. A median of 10 and 7 courses were given to patients at NCI and MGH, respectively. Details of dose modifications are shown in Table IV. Administered dose was maintained in 89% of patients during the first 6 cycles, with 10 patients (16%) requiring increased filgrastim doses. Toxicity is presented in Table V. Seventeen (27%) patients had grade 3 neutropenic fever without a documented source and five had grade 4 neutropenic fever requiring aggressive management. Filgrastim induced bone pain was grade 1/2 in 19% of patients, and was reduced by premedication with acetaminophen and diphenhydramine. The most common adverse events were grade 2: fatigue (37%), peripheral neuropathy (37%), nausea (27%), and diarrhea (24%). One patient withdrew from study with grade 2 tinnitus after 2 cycles. No deaths and no severe anaphylactoid reactions occurred on study. There have been no secondary malignancies.
Table IV.
Treatment and dose modifications all cycles for all pts (N = 62)
| Dose Changes | # pts | Median # cycles (range) | Reason for dose modification | ||||
|---|---|---|---|---|---|---|---|
| ANC | PN | F&N | Hearing | Unknown | |||
| Full Dose | 55 (89%) | ||||||
| GCSF 20 mcg/kg/d | |||||||
| cycle 1-6 | 10 (16%) | 2 (2-6) | 10 | 0 | 0 | 0 | 0 |
| cycles > 6 | 3 (5%) | 8 | 3 | 0 | 0 | 0 | 0 |
| T 200 mg/m2 | |||||||
| cycle 1-6 | 4 (6%) | 3 (3-4) | 1 | 2 | 1 | 0 | 0 |
| cycles > 6 | 12 (19%) | 8 (7-10) | 2 | 9 | 4 | 0 | 0 |
| T 175 mg/m2 | |||||||
| cycle 1-6 | 2 (5%) | 5 (4-6) | 0 | 0 | 1 | 0 | 1 |
| cycles > 6 | 6 (15%) | 8 (8-10) | 1 | 5 | 1 | 0 | 1 |
| T 150 mg/m2 | |||||||
| cycle 1-6 | 1 | 6 | 0 | 1 | 0 | 0 | 0 |
| cycles > 6 | 1 | 9 | 0 | 1 | 0 | 0 | 0 |
| C 500 mg/m2 | |||||||
| cycle 1-6 | 1 | 3 | 1 | 0 | 0 | 0 | 0 |
| cycles > 6 | 3 (5%) | 9 (7-9) | 2 | 0 | 0 | 0 | 1 |
| P 50 mg/m2 | |||||||
| cycle 1-6 | 3** | 6 | 0 | 0 | 0 | 1 | 1 |
more than one reason for dose modification in some patients.
for delayed thrombocytopenia
Table V.
Toxicity
| Toxicity | Grade 1(%) | Grade 2 (%) | Grade 3 (%) | Grade 4 (%) |
|---|---|---|---|---|
| Alopecia | 4 (6) | 48 (77) | 0 | 0 |
| Allergic Reaction | 2 (3) | 1 (1) | 0 | 0 |
| Anorexia | 27 (44) | 11 (17) | 0 | 0 |
| Arthralgia/myalgia | 19 (31) | 12 (19) | 0 | 0 |
| Bone pain | 29 (47) | 12 (19) | 0 | 0 |
| Constipation | 14 (23) | 5 (8) | 1 (1) | 0 |
| Dehydration/IV fluids | 5 (8) | 1 (1) | 0 | 0 |
| Depression | 10 (16) | 8 (13) | 0 | 0 |
| Diarrhea | 18 (29) | 15 (24) | 8 (13) | 0 |
| Dizziness | 30 (48) | 2 (3) | 0 | 0 |
| Fatigue | 19 (31) | 23 (37) | 8 (13) | 0 |
| Fever | 11 (17) | 12 (19) | 2 (3) | 0 |
| Edema | 15 (24) | 3 (5) | 0 | 0 |
| Headache | 15 (24) | 2 (3) | 0 | 0 |
| Hypotension | 1 (1) | 3 (5) | 0 | 0 |
| Hematologic | ||||
| Neutropenia | 1 (1) | 2 (3) | 17 (27)* | 5 (8)* |
| Neutropenic fever | 0 | 0 | 17 (27) | 5 (8) |
| Thrombocytopenia | 12 (26) | 18 (39) | 16 (35) | 0 |
| Laboratory | ||||
| Abnormalities** | 0 | 26 (46) | 20 (43) | 0 |
| Anemia | 33 (72) | 3 (6) | 6 (13) | 0 |
| Hypomagnesemia | 0 | 1 (2) | 0 | 0 |
| Elevated creatinine | 21 (46) | 5 (11) | 2 (4) | 2 (4) |
| Transaminitis | 0 | 2 (4) | 0 | 1 (2)*** |
| Hyperbilirubinemia | ||||
| Mucositis | 17 (27) | 5 (8) | 0 | 0 |
| Nausea | 26 (42) | 17 (27) | 3 (5) | 1 (1) |
| Neuropathy | 23 (37) | 19 (31) | 12 (19) | 0 |
| Rash | 7 (11) | 1 (1) | 0 | 0 |
| Tinnitus | 2 (3) | 3 (5) | 0 | 0 |
| Vomiting | 24 (39) | 11 (17) | 5 (8) | 1 (1) |
| Weight Loss | 3 (5) | 2 (3) | 1 (1) | 0 |
does not include patients with neutropenic fever
NCI cohort, n=46
Gilbert’s syndrome
The natural history of treatment-induced neuropathy was a secondary objective analyzed at NCI (Table VI). Cisplatin dose was not modified for early peripheral neuropathy; but was reduced for one patient who developed impaired hearing during cycle 2. Gabapentin, oxycodone, sertraline, and amitriptyline provided symptomatic relief. Neurologic improvement began with paclitaxel dose reduction or treatment discontinuation. Nineteen patients (41%) had complete resolution of neuropathy with mean time to resolution of 14 months (median 11; range 3-36).
Table VI.
Follow up of peripheral neuropathy (NCI pts, N= 46)*
| Grade | # | Median cycle of onset (range) | Clinical manifestation | Physical examination | Duration (weeks)** | Mean | # pts taking relief meds (%) |
|---|---|---|---|---|---|---|---|
| 1 | 42 | 3 (1-10) | Subjective numbness and tingling; gait dysfunction | Loss of DTRs with time. | 4 (0.1-3400) | 19.5 | 4 (9) |
| 2 | 26 | 7 (2-10) | Subjective functional limitations; pain in extremities; increased tingling and numbness | Abnormal exam: decreased light touch, proprioception or vibration; wide based gait | 16 (0.1.-144) | 33 | 6 (23) |
| 3 | 12 | 7 (3-10) | Functional limitations; ambulatory difficulty; painful neuropathy; loss of balance; loss of fine motor skills | Decreased/loss of proprioception, light touch, pin prick, vibration; abnormal cerebellar exam; fine motor skill dysfunction | 15 (0.1-28) | 9 | 1 (8) |
| 4 | 0 | 0 | - | - | - | - | - |
Each grade manifest per pt is presented.
Duration implies either time to resolution or improvement to a lower grade; excludes patients whose condition was not resolved.
DISCUSSION
We hypothesized that dose intense paclitaxel in combination with cyclophosphamide and cisplatin and flexible filgrastim dosing would yield improved pathologic complete or microscopic response rate compared to historical controls, and that this might translate into improved long term outcomes. We individualized the number of administered treatment cycles, gave consolidation therapy after reassessment surgery, and maintained dose intensity with flexible filgrastim dosing. Portability of this regimen was demonstrated through a multi-institutional collaboration between the NCI and MGH. Administration of this three drug regimen resulted in 92% CCR and 84% PCR or microscopic residual disease. The median OS is 5.4 years and median PFS, 18.9 months. The median PFS is 15.8 months for patients with stage IV parenchymal organ involvement. While direct comparison is neither feasible nor appropriate, outcome is at least comparable and may be superior to institutional historical controls and to those reported for phase III carboplatin/paclitaxel control arms for optimally debulked patients 4, 20, 21.
Since the initiation of this trial in 1995, several reports have suggested a benefit of dose-dense paclitaxel in breast and ovarian cancers 7, 8, 11, 12. Breast cancer patients who received weekly paclitaxel 80mg/m2 over 1 hour for 12 doses following standard adjuvant therapy experienced improved PFS and OS compared to those randomized to 175mg/m2 over 3 hours every 3 weeks for four doses 12. A Phase II trial of Stage IIB-IV EOC patients who received weekly paclitaxel 100mg/m2 followed by carboplatin (AUC2) for a total of 18 cycles, interrupted by treatment breaks each 6 cycles, had a median PFS of 21 months, and OS of 43 months 22. The JGOG recently reported their randomized trial of Stage II-IV EOC patients treated with carboplatin (AUC6) with either dose-dense paclitaxel (80mg/m2 weekly) or paclitaxel 180mg/m2 every 3weeks 11. Twenty percent of the patients had stage II disease, and 45% were optimally debulked. PFS was significantly improved in the dose-dense paclitaxel arm (median, 27.9 months v. 17.1 months; p=0.0014). OS at 2 years favored the dose-dense cohort (83.6% v. 77.7%; p=0.05). Our median PFS of 18.9 months in a cohort of stage IIIC/IV patients suggests potential of our regimen.
Randomized clinical trials to date have failed to demonstrate superiority of a triplet regimen using pegylated liposomal doxorubicin or gemcitabine against standard carboplatin and paclitaxel, (PFS, 15.4 -16.4 months) 4. No triplets have incorporated dose-dense or dose-intense paclitaxel. In a randomized trial, addition of epirubicin to paclitaxel and carboplatin (AUC5) increased toxicity and did not improve activity 23. In the absence of a randomized trial, it is impossible to determine the contribution of each component in the triplet to the observed activity and toxicity.
Biasing against overall PFS may be the nonstandard and aggressive follow up approach used at the NCI: every other month CT, physical examination, and CA125 measurement for the first year. This may have resulted in earlier relapse detection, often prior to notable increases in CA125. This early recurrence detection was followed by prompt therapeutic intervention and thus may account for the relatively short median PFS. However, in view of the JGOG data, it is also possible that the dose-intense paclitaxel played a role. The JGOG regimen administered paclitaxel 240mg/m2 over 3 weeks, comparable to our 250mg/m2 every three weeks. A previous GOG study failed to demonstrate improved survival in patients randomized to paclitaxel 200mg/m2 or cisplatin 100mg/m2 every three weeks compared to those receiving paclitaxel 135mg/m2 and cisplatin 75mg/m2 however, interpretation of this trial was complicated due to imbalance in the arms 24. Our dose-intense regimen and the JGOG dose-dense regimen employed higher net doses of paclitaxel in combination with a platinum compound. Unlike regimens containing carboplatin, there was no evidence of long-term marrow injury.
All patients experienced some peripheral neuropathy, a major reason for discontinuation of cisplatin use in EOC, but it was ≤ grade 2 in the majority of patients. Despite the presumption that cisplatin was the cause of neuropathy in this regimen, neuropathy began to resolve after decrease or discontinuation of paclitaxel. We previously described neuropathy as the dose-limiting toxicity of high dose single agent paclitaxel and demonstrated that toxicity was not augmented by the addition of cisplatin 19, 20. Early dose reduction of paclitaxel for neuropathy in this study occurred in only three patients, whereas 9 patients required late dose modification after six cycles, consistent with a late and cumulative process. Resolution of neuropathy was observed in 41% of patients with median time to resolution 11months (3 –36).
In summary, this triplet dose-intense paclitaxel-based regimen resulted in a high pathologic response rate, and encouraging overall survival. Outcome analysis of this generally poor prognosis group of advanced-stage patients is generally poor, making these findings potentially of interest. The JGOG results and ours reported herein suggest that combination of dose-dense/intense paclitaxel and a platinum may warrant further study for the treatment of advanced stage EOC patients.
Acknowledgments
We thank P. Davis RN, A. Jones-Wells RN, M. Raggio RN and C. Annunziata MD PhD for their assistance, and A. Bicher MD for participation. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research; patient care support was provided to the MGH investigators from Amgen Corporation.
Footnotes
Financial disclosures: Sarosy, Hussain, Minasian, Steinberg, Kohn--None
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18287387. [DOI] [PubMed]
- 2.Bukowski RM, Ozols RF, Markman M. The management of recurrent ovarian cancer. Semin Oncol. 2007;34(2 Suppl 2):S1–15. doi: 10.1053/j.seminoncol.2007.03.012. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17512352. [DOI] [PubMed]
- 3.Thigpen T. The role of gemcitabine in first-line treatment of advanced ovarian carcinoma. Semin Oncol. 2006;33(2 Suppl 6):S26–32. doi: 10.1053/j.seminoncol.2006.03.015. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16716800. [DOI] [PubMed]
- 4.Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M, et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J Clin Oncol. 2009;27(9):1419–25. doi: 10.1200/JCO.2008.19.1684. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19224846. [DOI] [PMC free article] [PubMed]
- 5.Herzog TJ, Coleman RL, Markman M, Cella D, Thigpen JT. The role of maintenance therapy and novel taxanes in ovarian cancer. Gynecol Oncol. 2006;102(2):218–25. doi: 10.1016/j.ygyno.2005.12.001. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16460787. [DOI] [PubMed]
- 6.Ozols RF. Systemic therapy for ovarian cancer: current status and new treatments. Semin Oncol. 2006;33(2 Suppl 6):S3–11. doi: 10.1053/j.seminoncol.2006.03.011. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16716797. [DOI] [PubMed]
- 7.Cadron I, Leunen K, Amant F, Van Gorp T, Neven P, Vergote I. The “Leuven” dose-dense paclitaxel/carboplatin regimen in patients with recurrent ovarian cancer. Gynecol Oncol. 2007;106(2):354–61. doi: 10.1016/j.ygyno.2007.04.003. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17499348. [DOI] [PubMed]
- 8.Vasey PA. “Dose dense” chemotherapy in ovarian cancer. Int J Gynecol Cancer. 2005;15(Suppl 3):226–32. doi: 10.1111/j.1525-1438.2005.00438.x. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16343237. [DOI] [PubMed]
- 9.van der Burg ME, van der Gaast A, Vergote I, Burger CW, van Doorn HC, de Wit R, et al. What is the role of dose-dense therapy? Int J Gynecol Cancer. 2005;15(Suppl 3):233–40. doi: 10.1111/j.1525-1438.2005.00432.x. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16343238. [DOI] [PubMed]
- 10.McGuire WP, Hoskins WJ, Brady MF, Homesley HD, Creasman WT, Berman ML, et al. Assessment of dose-intensive therapy in suboptimally debulked ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 1995;13(7):1589–99. doi: 10.1200/JCO.1995.13.7.1589. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7602348. [DOI] [PubMed]
- 11.Isonishi S, Yasuda M, Takahashi F, Katsumata N, Kimura E, Aoki D, et al. Randomized phase III trial of conventional paclitaxel and carboplatin (c-TC) versus dose dense weekly paclitaxel and carboplatin (dd-TC) in women with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer: Japanese Gynecologic Oncology. J Clin Oncol. 2008;26(Proceedings ASCO suppl) abstract 5506. [Google Scholar]
- 12.Sparano JA, Wang M, Martino S, Jones V, Perez EA, Saphner T, et al. Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med. 2008;358(16):1663–71. doi: 10.1056/NEJMoa0707056. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18420499. [DOI] [PMC free article] [PubMed]
- 13.Parker RJ, Dabholkar MD, Lee KB, Bostick-Bruton F, Reed E. Taxol effect on cisplatin sensitivity and cisplatin cellular accumulation in human ovarian cancer cells. J Natl Cancer Inst Monogr. 1993;15:83–8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7912534. [PubMed]
- 14.Ragazzi E, D’Ancona S, Berti E, Carrara M. Cytotoxicity of paclitaxel in combination with cisplatin and a new Pt-mercaptopyridine complex. Anticancer Res. 2002;22(5):2783–8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12529997. [PubMed]
- 15.Engblom P, Rantanen V, Kulmala J, Helenius H, Grenman S. Additive and supra-additive cytotoxicity of cisplatin-taxane combinations in ovarian carcinoma cell lines. Br J Cancer. 1999;79(2):286–92. doi: 10.1038/sj.bjc.6690046. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9888470. [DOI] [PMC free article] [PubMed]
- 16.Reed E, Kohn EC, Sarosy G, Dabholkar M, Davis P, Jacob J, et al. Paclitaxel, cisplatin, and cyclophosphamide in human ovarian cancer: molecular rationale and early clinical results. Semin Oncol. 1995;22(3 Suppl 6):90–6. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7541159. [PubMed]
- 17.Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994;5(Suppl 6):S3–6. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7865431. [PubMed]
- 18.Vanhoefer U, Harstrick A, Wilke H, Schleucher N, Walles H, Schroder J, et al. Schedule-dependent antagonism of paclitaxel and cisplatin in human gastric and ovarian carcinoma cell lines in vitro. Eur J Cancer. 1995;31A(1):92–7. doi: 10.1016/0959-8049(94)00440-g. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7695986. [DOI] [PubMed]
- 19.Sarosy G, Kohn E, Stone DA, Rothenberg M, Jacob J, Adamo DO, et al. Phase I study of taxol and granulocyte colony-stimulating factor in patients with refractory ovarian cancer. J Clin Oncol. 1992;10(7):1165–70. doi: 10.1200/JCO.1992.10.7.1165. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1376773. [DOI] [PubMed]
- 20.Kohn EC, Sarosy GA, Davis P, Christian M, Link CE, Ognibene FP, et al. A phase I/II study of dose-intense paclitaxel with cisplatin and cyclophosphamide as initial therapy of poor-prognosis advanced-stage epithelial ovarian cancer. Gynecol Oncol. 1996;62(2):181–91. doi: 10.1006/gyno.1996.0213. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8751547. [DOI] [PubMed]
- 21.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7494563. [DOI] [PubMed]
- 22.Sehouli J, Stengel D, Mustea A, Camara O, Keil E, Elling D, et al. Weekly paclitaxel and carboplatin (PC-W) for patients with primary advanced ovarian cancer: results of a multicenter phase-II study of the NOGGO. Cancer Chemother Pharmacol. 2008;61(2):243–50. doi: 10.1007/s00280-007-0466-z. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17393164. [DOI] [PubMed]
- 23.Kristensen GB, Vergote I, Stuart G, Del Campo JM, Kaern J, Lopez AB, et al. First-line treatment of ovarian cancer FIGO stages IIb-IV with paclitaxel/epirubicin/carboplatin versus paclitaxel/carboplatin. Int J Gynecol Cancer. 2003;13(Suppl 2):172–7. doi: 10.1111/j.1525-1438.2003.13363.x. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14656276. [DOI] [PubMed]
- 24.Muggia FM, Braly PS, Brady MF, Sutton G, Niemann TH, Lentz SL, et al. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: a gynecologic oncology group study. J Clin Oncol. 2000;18(1):106–15. doi: 10.1200/JCO.2000.18.1.106. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10623700. [DOI] [PubMed]