

Abstract
Alterations in mitochondrial structure and functions have long been observed in cancer cells. Targeting mitochondria as a cancer therapeutic strategy has gained momentum in the recent years. The signaling pathways that govern mitochondrial function, apoptosis and molecules that affect mitochondrial integrity and cell viability have been important topics of the recent review in the literature. In this article, we first briefly summarize the rationale and biological basis for developing mitochondrial-targeted compounds as potential anticancer agents, and then provide key examples of small molecules that either directly impact mitochondria or functionally affect the metabolic alterations in cancer cells with mitochondrial dysfunction. The main focus is on the small molecular weight compounds with potential applications in cancer treatment. We also summarize information on the drug developmental stages of the key mitochondria-targeted compounds and their clinical trial status. The advantages and potential shortcomings of targeting the mitochondria for cancer treatment are also discussed.
Keywords: Mitochondria, Warburg effect, Glycolysis, Reactive oxygen species, Apoptosis, Anticancer agents, Therapeutic selectivity
1. Introduction
As an important organelle in the cells, mitochondria not only play a central role in energy metabolism and calcium homeostasis (Clapham, 2007), but also are essential components of the apoptotic machinery and sites of reactive oxygen species (ROS) generation (Green and Reed, 1998). Under physiological conditions, mitochondria are considered as the main powerhouse of the cells, since this organelle can use glucose, fatty acids, and certain amino acids as the fuel sources to generate ATP through oxidative phosphorylation. The Krebs cycle within the mitochondrial provides an essential metabolic platform for effective conversion of various metabolic intermediates and the production of NADH as the substrate (electron donor) for the mitochondrial respiratory chain, through which electrons are transported in an orderly manner and transmembrane potential is established. The energy stored in the form of transmembrane potential across the inner mitochondrial membrane is then used for ATP generation in complex V. In addition to energy production, the important roles of mitochondria in calcium uptake/homeostasis and in lipid metabolism are essential for normal cellular function and survival. Importantly, mitochondria also contain potentially lethal molecules, which when released from this organelle, may cause the death of the host cells. For instance, cytochrome c is a normal component of the mitochondrial respiratory chain and plays an essential function in electron transport. However, when cytochrome c is released from the mitochondria to the cytosol, it binds to the cytosolic protein Apaf-1 in the presence of dATP to form a protein complex known as apoptosome, which activates the caspase protease cascade leading to cell destruction. Another mitochondrial protein known as apoptosis-inducing factor (AIF) can cause caspase-independent apoptosis when it is translocated to the nucleus. Many factors, including exogenous and endogenous stimuli, can induce the change of mitochondrial membrane permeability and cause the release of apoptotic factors.
Because of their important functions in energy production and in regulation of cell death, mitochondria have been considered to be a potentially important target for anticancer drug development, and this strategy has recently gained momentum (Fantin et al., 2002; Toogood, 2008). Interestingly, numerous notable differences in the structure and function of mitochondria between cancer cells and normal cells have been reported (Modica-Napolitano and Singh, 2004). For instance, there are various changes in the size, shape and number of the mitochondria in liver cancer cells when compared to the corresponding normal cells. It has also been observed that certain fast growing tumor cells seemed to have fewer and smaller mitochondria than slowly growing tumors, while certain relatively benign tumors (such as oncocytic adenomas) exhibited large numbers of mitochondria and high levels of oxidative enzymes (Maximo and Sobrinho-Simoes, 2000; Modica-Napolitano and Singh, 2002). Alterations of the inner mitochondrial membrane proteins have also been observed in hepatoma cells (Chang et al., 1971; Irwin et al., 1978). Consistent with these structural changes, mitochondrial dysfunction have been observed in various types of cancer cells, evident by a compromised capacity in mitochondrial ATP generation and an abnormal increase in ROS generation (Chen, 2007; Pelicao, 2004). It should be pointed out, however, that the mitochondrial morphological changes and functional alterations observed in one cancer type or cell line should not be generalized as common mitochondrial abnormalities in all cancer cells. Although mitochondrial dysfunction is often observed in cancer, it is likely that the specific alterations may vary depending in the cancer types, tissue origins, disease stages, proliferation and differentiation states, and microenvironment such as hypoxia.
A prominent metabolic alteration in cancer cells is that they exhibit a substantial increase in aerobic glycolysis and seem to rely more on this non-oxidative glucose metabolism for generation of ATP and for production of other molecules for cell growth and proliferation. This phenomenon, known as the Warburg effect, has been observed in a variety of cancer types including solid tumors and leukemia (Warburg, 1956). The successful use of positron emission tomography (PET) imaging in clinical diagnosis of cancer, based on the increased uptake of glucose in tumor tissues, is an excellent example of the clinical relevance of the Warburg theory. Although the underlying mechanisms responsible for the Warburg effect remain to be defined, it has been postulated that mitochondrial dysfunction (respiration injury) may be a key event that compromises the cancer cell’s ability to generate ATP through oxidative phosphorylation, and thus forces them to increase glucose fermentation to compensate the energy supply (Warburg, 1956). Mitochondrial mutations, oncogenic signals, and metabolic stress are possible factors that can lead to mitochondrial dysfunction (Chen et al., 2007). In fact, electron transport chain complex activities in mitochondria have been found to be decreased in certain cancer cells such as hepatoma (Chan and Barbour, 1983; Sun et al., 1981), and tumor cells (Morris hepatomas and ascites tumor cells) showed markedly reduced ATPase activity when compared with that of the normal cells(Pedersen and Morris, 1974). Mitochondrial DNA (mtDNA, which encodes for 13 key electron transport components) mutations were detected in various cancers, and certain mutations seemed to be associated with specific cancers (Modica-Napolitano and Singh, 2004). Moreover, some studies showed that cancer cells have increased mitochondrial transmembrane potential (Bernal et al., 1982; Johnson et al., 1981). In addition, mitochondria are major sites of ROS generation in the cells, and increased ROS generation in cancer cells may reflect an increased leakage of electrons from the transport complexes (complex I and complex III). The capture of an electron by molecular oxygen may lead to a formation of superoxide, which can then be further converted to hydrogen peroxide. Thus, elevated ROS generation in cancer cells may be considered as an indication of mitochondrial dysfunction, although it should be noted that ROS generation can also occur by other mechanisms outside the mitochondria, such as reactions catalyzed by NAD(P)H oxidase (NOX) complex and xanthine oxidase.
These differences between normal cells and cancer cells in their mitochondrial structure and functions may provide a biological basis to preferentially target cancer cells using agents that either directly interact with mitochondria or functionally impact the metabolic alterations as the consequences of mitochondrial dysfunction in cancer cells. As illustrated in Fig 1, the increase of mitochondrial transmembrane potential in cancer cells may provide a possibility of utilizing compounds such as rhodamine123 with certain biochemical properties that can be selectively taken up by the cancer mitochondria and preferentially disturb cancer cell metabolism or activate the apoptotic cell death process. Similarly, the structural/functional mitochondrial alterations in cancer cells may render them more vulnerable to damage by certain pharmacological agents leading to release of apoptotic factors. For instance, targeting Bcl-2 family proteins may cause the opening of mitochondrial permeability pore and release of apoptotic factors. Furthermore, the increased dependence on glycolysis as a consequence of mitochondrial dysfunction in cancer cells may serve as a biochemical basis to preferentially kill malignant cells using proper glycolytic inhibitors such as 3-bromopyruvate (3-BrPA) and lonidamine. The phenomenon of mitochondrial dysfunction, the signaling pathways that mitochondrial affect function and regulate apoptosis, and agents that affect mitochondrial integrity and cell viability have been reviewed previously (Armstrong, 2006; Dias and Bailly, 2005; Galluzzi et al., 2006; Morrison et al., 2009; Ralph and Neuzil, 2009). In this article, we summarize the recent advances in this research area and review key examples of small molecules that preferentially target mitochondria in cancer cells directly or preferentially interfere with cancer metabolism, with a focus on those compounds that may have potential utility in cancer treatment. The chemical structure these compounds and their status in drug development will also be indicated when such information is available.
Figure 1.

Overview of possible sites and functions of mitochondria as potential targets for anticancer therapy. (1) Certain compound, often with positive charge, may preferentially accumulate in the mitochondria of cancer cells due to the elevated transmembrane potential (Δψm), with the inner surface of the inner mitochondrial membrane being highly negatively-charged. (2) Mitochondrial DNA, which encodes 13 key components of the respiratory chain complexes, are vulnerable to damage by certain DNA-interacting compounds due to the unique structure of mitochondrial DNA, lack of histone protection, and relatively weak DNA repair capacity in the mitochondria. (3) Inhibition of mitochondrial respiration through targeting the electron transport complexes has been shown to elevate ROS production, deplete ATP and induce apoptosis. (4) Targeting the mitochondriapermeability transition pore (MPTP) may alter Δψm, induce change in membrane permeability, and result in a release of apoptotic factors. (5) Enzymes of the glycolytic pathways are often found elevated in cancer cells with mitochondrial dysfunction, likely being a mechanism to compensate energy supply and provide metabolic intermediates for cell growth and proliferation. Inhibition of glycolytic enzymes has been shown to preferentially kill cancer cells, especially those with significant mitochondrial dysfunction or under hypoxic conditions. 3BrPA, 3-bromopyruvate; HXK, hexokinase; LDH, lactate dehydrogenase; PDH, pyruvade dehydrogenase; PDK, pyruvate dehydrogenase kinase; Δψm, mitochondria transmembrane potential; mtDNA, mitochondrial DNA (Δψm). MPTP, mitochondrial permeability transition pore.
2. Small molecules that directly target mitochondria
2.1 Compounds targeting mitochondria based on altered mitochondrial transmembrane potential (Δψm)
It has been known for sometime that mitochondria of cancer cells and transformed cells have significantly higher transmembrane potential than normal cells (Dairkee and Hackett, 1991; Davis et al., 1985; Johnson et al., 1980; Modica-Napolitano and Aprille, 1987; Summerhayes et al., 1982). This biological property has been used as a basis to develop compounds that may preferentially accumulate within the mitochondria of cancer cells. A group of molecules known as delocalized lipophilic cations (DLCs), with highly hydrophobic structures and positive charge, seem to be able to accumulate in the tumor mitochondria of tumor due to the highly negatively-charged microenvironment within the mitochondrial matrix (Modica-Napolitano and Aprille, 2001). Several members of DLCs have been found to exhibit some degree of efficacy in killing cancer cells or inhibiting their growth. Although increased transmembrane potential in cancer cells is necessary for DLCs to achieve selective cytotoxictiy, the overall effectiveness of these compounds depends on other factors. For example, cytoplasmic characteristics are involved in the kinetics of uptake and retention of these compounds (Modica-Napolitano and Singh, 2002) as well as the ability of these agents to disrupt mitochondrial function once they have accumulated within the organelle. Although elevation of mitochondrial transmembrane potential has been observed in various types of cancer cells, it is important to compare cancer cells with the normal cells of same tissue origins and at similar developmental stages for their transmembrane potential and cytotoxic responses to these compounds. This would be particularly important for the evaluation of the anticancer selectivity of compounds whose action is membrane potential-dependent, including DLCs, rhodamine-123, and MKT-077 (see below).
Rhodamine-123
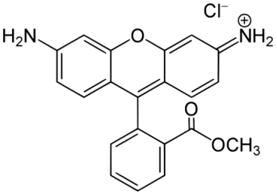
Studies in the early 1980s showed that Rhodamine-123 could function as a specific chemical probe for the localization of mitochondria in living cells. After cells were incubated with Rhodamine-123, mitochondria were preferentially stained and revealed as clusters of organelle in the perinuclear region of src-transformed cells (Chen et al., 1982; Johnson et al., 1980). The accumulation of Rhodamine-123 in mitochondria depends on its lipophilic and cationic properties, which helps it cross the double mitochondrial membranes and stay within the mitochondrial matrix where the microenvironment is negatively charged (Johnson et al., 1981; Lampidis et al., 1984). By depolarizing the plasma membrane, it was revealed that the uptake of Rhodamine-123, driven mainly by the mitochondrial membrane potential, was greater in cancer cells than in normal cells (Davis et al., 1985; Lampidis et al., 1985). High retention of Rhodamine-123 in mitochondria was observed in transitional cell carcinoma, adenocarcinoma, chemical carcinogen-transformed epithelial cell lines, and squamous cell carcinoma lines. Interestingly, the mitochondria of these cancer cells retained Rhodamine-123 for 2 to 5 days, whereas normal cells released Rhodamine-123 within a few hours (Summerhayes et al., 1982).
Kidney and breast cancer cells that retained Rhodamine-123 longer than non-tumorigenic epithelial cells were highly sensitive to this compound, as evidenced by significant inhibition of colony formation (Bernal et al., 1982). Rhodamine-123 also showed selective anticancer activity in animal tumor models, and this selective toxicity could be further potentiated by addition of 2-deoxyglucose, an inhibitor of glycolysis (Arcadi, 1986; Bernal et al., 1983; Herr et al., 1988). A phase I clinical trial of Rhodamine-123 was carried in hormone refractory prostate cancer patients to determine the maximum tolerated dose (MTD) and its safety/toxicity profile (Jones et al., 2005). The MTD of Rhodamine-123 was estimated at 96 mg/m2 and that this compound could be safely administered at monthly intervals. This drug administration schedule resulted in preferential drug retention in prostatic tumor tissue without detectable drug accumulation in serum. Therapeutic efficacy, as assessed by the lengthening of PSA doubling time, was observed in this clinical trial but the data did not reach statistical significance. Thus, monthly administration of Rhodamine-123 at a dose of 96 mg/m2 seems not an effective therapeutic schedule. Dose and schedule optimization and combination of Rhodamine-123 with other therapeutic modalities may improve therapeutic efficacy. Since a decrease in mitochondrial production of ATP may be compensated by an increase in glycolysis in the cytosol to generate ATP, a combination of rhodamine-123 and glycolytic inhibitors such as 2-deoxyglucose seems to be a logical strategy. It should be noted that some cancer cells may use fatty acids and amino acids such as glutamine as alternative energy sources for ATP generation (Vander Heiden et al, 2009), inhibition glycolysis alone may not be sufficient to cause ATP deprivation. A combination of a glycolytic inhibitor and compounds that disrupt mitochondria or suppress the alternative energy metabolic pathways in the mitochondria would be more effective in killing these cancer cells.
MKT-077
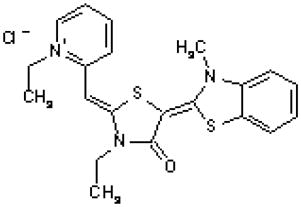
The rhodacyanine analogue MKT-077 (also known as FJ-776) is localized in the mitochondria after the compound enters the cells. Like other lipophilic cations, MKT-077 accumulates preferentially in the mitochondria of tumor cells due to a higher mitochondrial membrane potential. A previous study showed that MKT-077 selectively damages mitochondria of carcinoma cells, as evidenced by a 4-fold higher drug concentration required to obtain a similar inhibition of respiration in normal cells (Modica-Napolitano et al., 1996). It was also observed that carcinoma cells exposed to MKT-077 led to a preferential damage to mitochondrial DNA without a significant loss of nuclear DNA. Interestingly, MKT-077 exhibited a growth inhibitory effect in human breast, ovary, endometrial, colon and non-small cell lung cancer cell lines, but not in normal epithelial cells (Petit et al., 1999). In vivo, MKT-077 was able to inhibit the growth of implanted human renal carcinoma and prostate carcinoma in mice, and prolonged the animal survival (Koya et al., 1996). A phase I study has been conducted to evaluate the safety and pharmacokinetics of MKT-077 in advanced solid tumors. This study showed that MKT-077 was well tolerated in this group of patients with a recommended dose of 126 mg/m2/week. The main toxicity was hypomagnesemia due to renal loss of magnesium, which was manageable by administration of magnesium (Britten et al., 2000).
F16
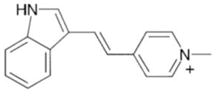
Through a cell-based high throughput screening of a chemical library, Fantin et al identified a novel molecule designated as F16 that specifically inhibited the proliferation of Her2-overexpressing mammary epithelial cells. This molecule exhibited low binding to the mitochondrial membranes and high accumulation in the mitochondrial matrix, resulting in mitochondrial damage, opening of the permeability transition pore, cytochrome c release, cell cycle arrest, and cell death. Besides the apoptosis effect triggered by F16, this compounds also induced necrosis in tumor cells (Fantin et al., 2002).
Dequalinium
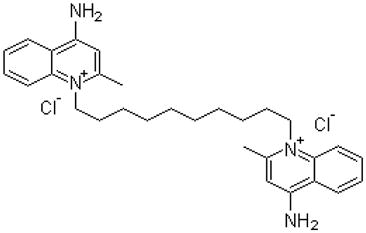
Dequalinium is a lipophilic compound with two positive charges and a C-10 aliphatic side chain. Clinically, this compound has been used as a topical antimicrobial agent for half a century (Babbs et al., 1956). Like other lipophilic cationic compounds, dequalinium mainly localized to the mitochondria of tumor cells. Prolonged exposure to dequalinium drastically altered the morphology of the mitochondria, causing globular change and perinuclear distribution (Weiss et al., 1987). This compound also inhibits the ability of calmodulin, a calcium-binding protein, to activate phosphodiesterase and thereby suppresses cell proliferation (Bodden et al., 1986). Furthermore, dequalinium selectively targeted cancer cells, impaired carcinoma cell proliferation, migration and invasion and prolonged survival of mice with implanted bladder and colon cancer (Bleday et al., 1986; Helige et al., 1992; Weiss et al., 1987).
2.2 Compounds that target cancer mitochondrial respiration
Similar to that observed in embryos or neonates, mitochondria in tumor cells seem to maintain a low electron transfer activity at approximately 20–30% of that seen in normal quiescent organelles (Galli et al., 2003). In colorectal cancer, NADH-cytochrome c reductase complex I and cytochrome oxidase, part of complex IV in the mitochondrial electron transport chain were observed to be decreased in tumor and peritumor tissues. The enzyme activity of superoxide dismutase 1 (SOD1), an antioxidant which converts superoxide to hydrogen peroxide, decreased while the mitochondrial nitric oxide synthase (mtNOS) activity increased in tumor tissue in advanced stage as compared with the initial stage (Sanchez-Pino et al., 2007). The relatively lower mitochondrial electron transport chain activity in cancer cells seems to reflect dysfunction of oxidative phosphorylation of cancer mitochondria. Because the electron transport chain is a major site of reactive oxygen species (ROS) generation due to capture of electrons by molecular oxygen, compounds that interfere with the respiratory chain may promote leakage of electrons and thus, increase the production of ROS, leading to mitochondrial damage and activation of apoptosis in cancer cells. The killing of cancer cells by ROS-mediated mechanisms is considered to have therapeutic selectivity against malignant cells due to their intrinsic high ROS generation (Trachootham et al., 2009).
Arsenic trioxide
![]()
Arsenic trioxide (As2O3) has long been used in traditional Chinese medicine to treat a variety of diseases. In the 1970s, As2O3 was introduced for the treatment of acute promyelocytic leukemia (APL) and showed a striking therapeutic effectiveness, with complete remission rates ranging from 65.6% to 84% in clinical studies conducted in the northeastern region of China (Shen et al., 1997). Interestingly, As2O3 exerted a dose dependent dual effect on APL cell lines in vitro, triggering apoptosis at high concentrations (0.5–2.0umol/L) while inducing differentiation at lower concentrations (0.1–0.5umol/L). Several studies showed As2O3 caused generation of superoxide and hydrogen peroxide, leading to a decrease of mitochondria membrane potential and apoptosis (Chen et al., 1998; Iwama et al., 2001; Wang et al., 1996). A recent study showed that interfering with the mitochondrial electron transport chain may be an important mechanism by which As2O3 promoted ROS generation (Pelicano et al., 2003). In leukemia cells, As2O3 was capable of inhibiting mitochondrial respiration, resulting in a substantial decrease of oxygen consumption as early as 3 hours, concurrent with the increase of ROS generation. Since cell death was detected after 24 h of drug treatment, inhibition of respiration seemed to be a primary event rather than the consequence of cell death. Moreover, in the presence of the mitochondrial complex I inhibitor rotenone, As2O3 caused further inhibition which suggested they may act in series. It appears that by interfering with electron transport in the mitochondria, As2O3 may cause an increase in electron leakage and therefore promotes ROS generation. Furthermore, a study using respiration-deficient cells revealed that the rho-0 cells were resistant to As2O3, confirming that the respiratory chain activity is important for the cytotoxic action of this drug (Pelicano et al., 2003).
Vitamin E analogues
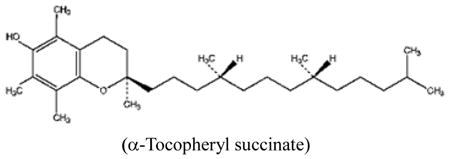
Alpha-tocopheryl succinate (α-TOS), a redox-inactive vitamin E analogue, exhibited strong pro-apoptotic and anticancer activity by causing rapid production of ROS (Stapelberg et al., 2005; Wang et al., 2005; Weber et al., 2003).α-TOS also seems to be able to inhibit the anti-apoptotic function of Bcl-xl and Bcl-2 by disrupting their binding by the Bak BH3 peptide (Shiau et al., 2006). The molecular target of α-TOS was recently identified (Dong et al., 2008). Dong et al revealed that this compound is a competitive inhibitor of the UbQ sites (Qp and QD) in Complex II thus inhibiting succinate dehydrogenase (SDH) activity in the mitochondrial electron transport chain. Knockdown of CybL made cancer cells resistant to α-TOS mediated killing, and addition of MitoQ overcame the α-TOS-mediated inhibition of MTT reduction driven by succinate. Reconstitution of a functional complex II in the CybL mutant cells led to normalization of SDH activity and reestablished sensitivity to α-TOS (Dong et al., 2008). In addition, Vitamin E analogues selectively inhibited a diverse range of malignant cells including melanomas, colon cancer and breast cancer in experimental animals (Dong et al., 2009; Neuzil et al., 2007). Higher esterase activities in normal cells can hydrolyze α-TOS into α-TOC (alpha-tocopheryl) and attenuate their activity. The selectivity of Vitamin E analogues against cancer cells seems to depend on their ability to target and down-regulate certain abnormally activated pathways such as PI3K/AKT and NF-kB (Constantinou et al., 2008).
Resveratrol
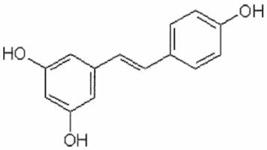
Resveratrol (3,5,4′-trihydroxystilbene) is a natural compound found in certain fruits, particularly in some grapes and blueberries, and has been suggested to have cancer chemopreventive properties. This compound seems to inhibit cellular events associated with tumor initiation, promotion, and progression (Jang et al., 1997). It was found that the mitochondrial respiratory chain (complexes I–III) was inhibited by resveratrol in mice. By competing with DUQH2, resveratrol decreased complex III activity and lowered ROS levels (Zini et al., 1999). Though considered as an antioxidant, resveratrol was shown to induce apoptosis through the mitochondrial pathway (Juan et al., 2008; Vetvicka et al., 2007). Interestingly, resveratrol seems to interfere with important signaling pathways such as PI3K/AKT, JAK/STAT, and the MAPK cascade (Filomeni et al., 2007; Madan et al., 2008; Roy et al., 2009). Moreover, this compound attenuated HIF-1α and VEGF expression induced by lysophosphatidic acid (Park et al., 2007).
Rotenone
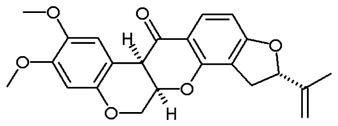
Rotenone, a strong inhibitor of mitochondria complex I (Chance et al., 1963), is often used as a pesticide and insecticide. Interestingly, this compound at the concentration which blocks electron flow through complex I, did not significantly affect cell viability or growth in B lymphoma cells (Armstrong et al., 2001), suggesting ATP generated outside the mitochondria might be sufficient to support cell survival in these lymphoma cells. It has been observed that chronic rotenone treatment could result in increased superoxide production, leading to mitochondrial alteration (Koopman et al., 2005). However, reports on the effect of rotenone on cellular ROS levels have been conflicting, with both increase and decrease of ROS observed in cells treated with rotenone (Deshpande et al., 2000; Torres-Roca et al., 2000; Vrablic et al., 2001 ; Armstrong et al., 2001; Kitamura et al., 2002; Koopman et al., 2005; Pelicano et al., 2003). The reasons for these opposite results are unclear. Differences in the cellular genetic backgrounds, mitochondrial functional states and different rotenone concentrations used in these studies might possibly contribute to the variations in ROS observed. While rotenone is a potent and specific inhibitor of complex I and has been commonly used as a biochemical tool, its utility as an anticancer agent is unclear, although its cancer preventive potential has been suggested (Yoshitani et al., 2001).
2.3 Compounds affecting cancer mitochondrial membrane permeability
A traditional model for mitochondrial membrane permeability transition pore (MPTP) was a multi-protein structure between the inner and outer mitochondrial membranes comprised of several proteins such as adenine nucleotide transporter (ANT), voltage dependent anion channel (VDAC) and cyclophilin D (Chipuk et al., 2006). As a voltage-dependent, cyclosporine-A-sensitive and high-conductance inner membrane channel, MPTP can depolarize the mitochondrial potential by its opening. Abnormal opening of MPTP may result in the collapse of mitochondrial membrane potential and the release of apoptotic factors from the mitochondria to cytosol, leading to cell death. Some studies also found certain differences in MPT between normal and malignant cells. For instance, in mitochondria of certain hepatoma cells, the adenine nucleotide exchange function of ANT was decreased and the sensitivity to bongkrekic acid (an inhibitor of adenine nucleotide exchange and formation of MPTP) was also diminished. The expression of ANT2, a gene that is usually repressed in quiescent cells was up-regulated in several tumor cells (Modica-Napolitano and Singh, 2002). These differences may provide an opportunity to preferentially target MPTP in cancer cells for therapeutic purpose. However, it should be noted that the absence of one or more ANT or VDAC isoforms in animal models by gene knockout did not inactivate the mitochondrial pore, suggesting that what would constitute the necessary components for the standard model of MPTP complexes might need to be reconsidered and further characterized (Baines et al., 2007; Berridge et al., 2009; Kokoszka et al., 2004; Krauskopf et al., 2006). Regardless of the putative roles of ANT or VDAC as the components of MPTP, several compounds have been shown to effectively induce changes in mitochondrial membrane permeability and exhibit anticancer activity.
Honokiol
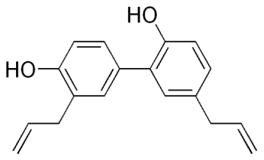
Extracted from Magnolia officinals, honokiol has been used for the treatment of thrombotic stroke, anxiety and gastrointestinal diseases for a long time. Honokiol can induce not only cancer cell death through caspase-dependent and independent apoptosis but also necrosis through mitochondrial permeability transition pore (Filomeni et al., 2007; Ishitsuka et al., 2005). Honokiol also exhibited an antioxidant effect and inhibited lipid peroxidation in the mitochondria (Haraguchi et al., 1997; Lo et al., 1994). Recently, it was found that honokiol could selectively kill esophageal adenocarcinoma cells which expressed higher levels of antioxidant molecules and seemed well adapted to tolerate ROS stress (Chen et al., 2009a). Mechanistically, honokiol was able to cause a decrease in mitochondrial transmembrane potential, which could be inhibited by pretreatment with cyclosporine A, suggesting that the cyclosporine-sensitive MPTP is a possible target of Honokiol. Although treatment of esophageal cells with honokiol led to increased ROS generation, this seemed not the critical event in honokiol cytotoxicity, since antioxidants didn’t affect honokiol induced cell death. Cyclophilin D seems to play a key role in mediating the cytotoxicity of honokiol, since a knockdown of cyclophilin D by siRNA partially suppressed honokiol-induced cell death (Chen et al., 2009a).
Betulinic acid
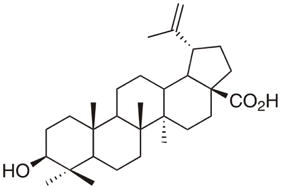
3β-Hydroxy-lup-20(29)-en-28-oic acid (Betulinic acid), a plant-derived pentacyclic triterpenoid, possesses some intriguing pharmacological effects including anti-inflammation, anti-HIV-1, and anticancer effects (Cichewicz and Kouzi, 2004). Betulinic acid was reported to exhibit selective cytotoxicity against several types of tumors including neuroectodermal tumor, colon cancer, and melanoma (Fulda and Debatin, 2000; Fulda et al., 1999; Jung et al., 2007; Pisha et al., 1995). A study found that betulinic acid triggered an apoptotic pathway independent of CD95 ligand/receptor interaction. The cytotoxic action of this compound appeared to be p53-independent. Moreover, betulinic acid showed high efficacy in neuroblastoma cells resistant to CD95 and doxorubicin (Fulda et al., 1997). Using isolated mitochondria in vitro, Fulda et al demonstrated that betulinic acid could directly trigger mitochondrial permeability transition without the involvement of a Z-VAD-fmk-inhibitable caspase (Fulda et al., 1998). Another study also found that betulinic acid induced a significant mitochondrial depolarization independent of caspase activation, while cyclosporine A (CsA) completely prevented mitochondrial depolarization caused by cyclophilin D (Mullauer et al., 2009). Upon mitochondrial permeability transition, apoptogenic factors such as cytochrome c and AIF were released into the cytosol and caspase 8 was cleaved (Fulda et al., 1998).
Bcl-2 inhibitors
Bcl-2 is an integral inner mitochondrial membrane protein, the overexpression of which blocks apoptosis (Hockenbery et al., 1990). In some tumors like non-Hodgkin’s lymphoma, Bcl-2 is overexpressed or deregulated, which is correlated with drug resistance and poor prognosis. Antisense oligonucleotides specific for Bcl-2 RNA sequences have been shown to reduce Bcl-2 protein level and specifically suppressed proliferation of cancer cells or enhanced their sensitivity to chemotherapeutic drugs (Campos et al., 1994; Kitada et al., 1994). Antisense oligonucleotides form specific DNA:RNA duplexes leading to RNA degradation and causing cell death (Manion and Hockenbery, 2003). The apoptotic effect of antisense Bcl-2 was mediated by caspase 9, caspase 3 and cytochrome c release from mitochondria. Pretreatment of cancer cells with antisense Bcl-2 potentiated apoptosis induced by dexamethasone, adenovirus-mediated delivery of p53, or paclitaxel in drug resistant myeloma cells (Dong et al., 2008). Mechanistically, a decrease of Bcl-2 protein would release Bad from the Bcl-2/Bad complex, leading to association of Bad with the MPTP component and opening of the pore. G3139, also known as oblimersen sodium, is an 18-mer synthetic phosphorothioate oligodeoxynucleotide with a sequence complementary to a portion of human Bcl-2 mRNA (O’Brien et al., 2005). Combination of G3139 with doxorubicin exhibited a synergistic effect in a breast cancer xenograft model (Lopes de Menezes et al., 2000). G3139 has now entered clinical trails in combination with chemotherapeutic agents in a variety of tumors such as leukemia, small cell lung cancer, multiple myeloma and prostate cancer (Badros et al., 2005; Iwama et al., 2001; O’Brien et al., 2005; Pepper et al., 1999; Rudin et al., 2004).
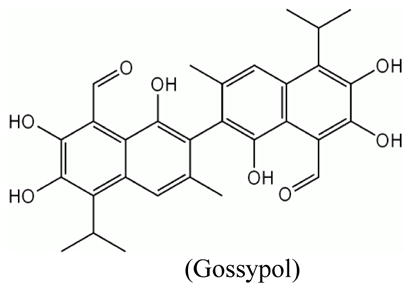
Small molecular weight compounds have been developed as potent inhibitors of Bcl-2 family proteins. Gossypol, a natural compound initially identified as an antifertility agent for male in China, is the prototype of BH3 interacting small molecule that showed inhibition of Bcl-2, Bcl-XL and Mcl-1 (Kang and Reynolds, 2009). Gossypol seems to be able to directly act upon Bcl-2 molecules present on the mitochondrial outer membrane. It can block Bcl-XL heterodimerization with Bax or Bad, and promote caspase-3 activation and cytochrome c release in Bcl-2 and Bcl-XL overexpressing cells (Meng et al., 2008; Oliver et al., 2005; Shiau et al., 2006). Since gossypol exhibited potent killing in multiple cancer cells (Meng et al., 2007; Vetvicka et al., 2007), it has been introduced into clinical trials in CLL, hormone refractory prostate cancer, and advanced breast cancer (Politzer, 2008; Stein et al., 1992; Van Poznak et al., 2001). Several gossypol derivatives are currently under development as potential anticancer agents. Another small molecule, ABT-737 was identified by using nuclear magnetic resonance (NMR)-based screening. ABT-737 binds with high affinity to Bcl-XL, Bcl-2, and Bcl-w, and antagonizes their anti-apoptotic effect, leading to sensitization of cancer cells to chemotherapy and radiation (Oltersdorf et al., 2005). Besides inducing the classic features of apoptosis such as caspase activation and cytochrome c release, ABT-737 also induced mitochondrial inner membrane permeabilization (MIMP) leading to outer mitochondrial membrane rupture and matrix swelling in leukemia and lymphoma cells (Vogler et al., 2008). The apoptotic function of ABT-737 was also found to be associated with diminishing intracellular GSH and increased ROS (Howard et al., 2009). Moreover, the combination of ABT-737 and CPT-11 or bortezomib can upregulate pro-apoptotic molecule Noxa expression in colorectal cancer cells and melanoma cells (Miller et al., 2009; Okumura et al., 2008).
2.4 Compounds targeting mtDNA
The human mitochondrial genome contains 16.5 kb DNA which encodes 13 respiratory chain subunits. The lack of protection by histones and the relatively weak DNA repair capacity in mitochondria seems to make mitochondrial DNA more vulnerable to damage. By comparing mtDNA from primary bladder, head and neck, and lung cancers with the mtDNA from the blood samples in all cases, and the corresponding normal tissues where available, Fliss et al revealed a high frequency of mtDNA mutations in tumor cells (Fliss et al., 2000). Most of the mutations were T-to-C and G-to-A base transitions, which might be related to ROS derived mutagens. A majority of the mutations were homoplasmic which implies the mtDNA mutation gain substantial replicative advantage. Seventy percent of colorectal cancer cell lines were found with mtDNA mutations, most of which were transitions at purines, again consistent with a ROS related derivation (Polyak et al., 1998). MtDNA also plays a role in affecting cellular response to chemotherapy drugs (Singh et al., 1999). Due to the important role of respiratory chain in mitochondrial ATP generation, compounds that target mtDNA are likely to significantly affect cellular energy metabolism and cell viability.
Cisplatin
Cisplatin (CDDP) was found to preferentially bind to mtDNA about 50 times more than to nuclear DNA, resulting in NADH-ubiquinone reductase inhibition and decreased ATP generation (Murata et al., 1990). After exposure to one dose of cisplatin during gestation, mice showed higher cisplatin-mtDNA adduction levels than corresponding genomic DNA adduct levels in maternal and fetal brain and liver tissues. The high levels of cisplatin adduction in mtDNA may impair its function and lead to cell death (Giurgiovich et al., 1997). Since CDDP binds to nuclear DNA and mtDNA, both mechanisms of action likely contribute to the anticancer activity of this compound.
Topoisomerase inhibitors
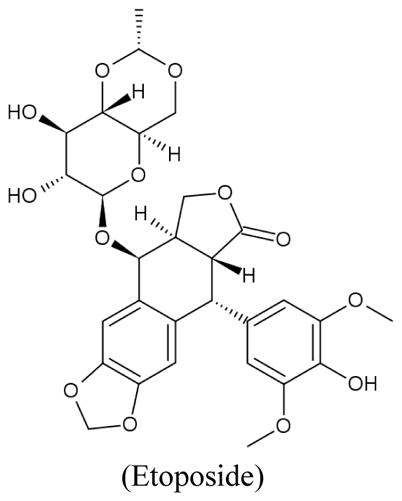
The 4-quinolone antibiotics nalidixic acid and ciprofloxacin, inhibitors of the bacterial type II topoisomerase, caused a time-dependent decrease in mtDNA content associated with a reduction in mitochondrial respiration and an increase of lactate production due to upregulation of glycolysis (Petit et al., 1999). DNA topoisomerase II isolated from calf thymus mitochondria was inhibited in vitro by amsacrine (m-AMSA), etoposide (VP-16) and teniposide (VM-26) (Lin and Castora, 1991). Etoposide, a topoisomerase II poison widely used in cancer treatment, can induce cytochrome c release in a caspase-independent manner. Low concentrations of etoposide predominantly caused nuclear DNA damage, while higher concentrations result in a direct effect on the mitochondria through mitochondrial pore transition (Robertson et al., 2000). The contribution of mtDNA damage induced by topoisomerase inhibitors to their anticancer activity is unclear. Interference of nuclear DNA processing is considered the main mechanism by which this class of compounds exerts their anticancer activity. It should also be pointed out that mitochondrial damage has been associated with cardiotoxicity of doxorubicin.
Ditercalinium
Ditercalinium (NSC 335153) is a bis-intercalating agent which accumulates mainly in the mitochondria (Fellous et al., 1988). Treating mouse and human cells with ditercalinium caused a specific elimination of mtDNA and inhibited its replication (Okamaoto et al., 2003). Ultrastructural studies showed a complete loss of mitochondrial cristae and depletion of mtDNA after ditercalinium treatment (Segal-Bendirdjian et al., 1988). This compound exhibits in vivo antitumor activity in animal models.
Vitamin K3
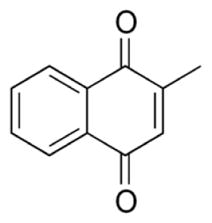
Although vitamin K3 (menadione) does not target mitochondrial DNA directly, this compound exhibited specific inhibitory affect on DNA polymerase γ (pol γ), the mitochondrial enzyme responsible for mtDNA replication. A recent study showed that at the concentration of 30 μM, Vitamin K3 inhibited pol γ activity by 80% without affecting other DNA polymerases, and led to mitochondrial dysfunction, ROS generation, and apoptosis (Sasaki et al., 2008). Vitamin K3 also induced mitochondria-related apoptosis in breast cancer cells (Akiyoshi et al., 2009; Marchionatti et al., 2009). Some recent studies have found its inhibitory effect on pancreatic cancer cells (Osada and Yoshida, 2009). The effect of Vitamin K3 on blood coagulation and its clinical usefulness have long been recognized, but its use as a potential anticancer agent still remains to be evaluated.
3. Compounds that target altered metabolisms associated with mitochondrial dysfunction
In cancer cells with mitochondrial dysfunction, one important metabolic alteration is the increase of glycolytic activity, which provides ATP as well as other metabolic intermediates for cancer cells to survive and proliferate. Altered expression and activities of enzymes in glycolysis and the tricarboxylic acid (TCA) cycle have been observed in various cancers (Kroemer and Pouyssegur, 2008). These enzyme changes contribute to or promote the preference of cancer cells to aerobic glycolysis and thus have prompted researchers to test whether targeting these enzymes will have any therapeutic benefit. Some of these compounds described below have indeed been shown to be useful in targeting cancer cells with high ‘glycolytic phenotype’ and also enhance their sensitivity towards other chemotherapeutic agents.
3-Bromoyruvate
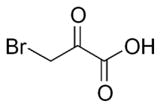
Hexokinase (HXK) (D-hexose 6-phosphotransferase) is the first enzyme in the glycolytic pathway and converts glucose to glucose-6-phosphate via transfer of a phosphate group from ATP to the 6-carbon of glucose. Four isoforms of HXK exist (I–IV) and are expressed in various tissues ((Katzen and Schimke, 1965)). HXKII has been found to bind onto the mitochondria (Robey and Hay, 2006) and has been shown to have elevated activity in H-91 hepatoma cells compared to the control liver samples (Bustamante and Pedersen, 1977). Known for high glucose catabolism ((Shatton et al., 1969; Weinhouse et al., 1972) a hepatocellular carcinoma model, generated via liver implantation of the rabbit VX2 tumor, was tested with 3-bromopyruvate (3BrPA) (Ko et al., 2004). Isolated tissue samples exhibited greater lactate production and higher hexokinase activity in implanted VX2 tumors compared to normal liver samples isolated from the same animal. In addition, the VX2 tumors showed greater distribution of HXK activity to the mitochondria compared to the normal liver tissue. Total mitochondrial HXK activity and mitochondrial respiration was inhibited with 3BrPA (Ko et al., 2004). Recent work by Chen et al found 3BrPA to cause a covalent modification of mitochondria-bound HXKII, leading to disruption of interaction between AIF (Apoptosis Inducing Factor) and HXKII (Chen et al., 2009b). HXKII and AIF released from the mitochondria leads to induction of cell death, revealing another mechanism of action of 3BrPA (Chen et al., 2009b). In addition, 3BrPA exhibited greater potency against lymphoma and colon cancer cells cultured under hypoxic conditions, and was very effective in killing cancer cells with defective mitochondria (Xu et al., 2005).
Dichloroacetate
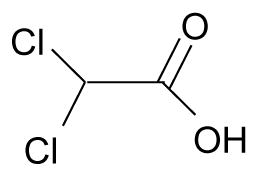
A key mechanism of action of dichloroacetate (DCA) is via inhibition of pyruvate dehydrogenase kinase (PDK), leading to activation of pyruvate dehydrogenase (PDH). PDH is responsible for the conversion of pyruvate into acetyl-CoA thereby allowing it to enter the TCA cycle. Phosphorylation of PDH by PDK results in inhibition of the pyruvate dehydrogenase activity. Bonnet et al showed DCA was able to reduce hyperpolarized mitochondrial membrane potential in glioblastoma, non-small-cell lung and breast cancer cells but not in noncancerous cell lines (Bonnet et al., 2007). Analysis of metabolic parameters upon DCA treatment in A549 lung cancer cells showed it was able to reduce both glycolysis and fatty acid oxidation while increasing glucose oxidation. In addition, DCA was able to induce apoptosis, elevate H2O2 production and activate the Kv1.5 potassium channel in A549 cells (Bonnet et al., 2007). Treatment of head and neck squamous cell carcinomas by DCA showed a dose dependent reduction in phosphorylated PDHα in primary UM-22A cells (McFate et al., 2008). However, such reduction was not seen in the metastatic UM-22B cell line which exhibited a higher ratio of phosphorylated PDHα to total PDHα, greater basal PDK1 levels and lactate. Interestingly, the UM-22B cells were more sensitive to DCA than the UM-22A cells.
Lonidamine
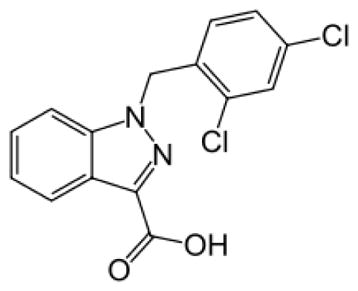
Lonidamine (LND) derived from indazole-3-carboxylic acid, exhibits both antispermatogenic and antineoplastic properties. LND exhibits a plethora of effects on cells, its primary effect being an energy modulator in cancer cells. LND has been shown to inhibit glycolysis due to inhibition of hexokinase II (Floridi et al., 1981a) which is often found elevated in cancers (Bustamante and Pedersen, 1977). Use of LND was found to deplete ATP, inhibit oxygen consumption in Ehrlich’s ascites tumor cells and lactate production under aerobic and anaerobic conditions (Floridi et al., 1998; Floridi et al., 1981a; Floridi et al., 1981b). Minimal apoptosis was observed with individual LND or radiation treatment but was significantly increased upon their combination in radio-resistant malignant melanoma cells (Miyato and Ando, 2004). LND was also found to enhance the cytotoxicity of several chemotherapeutic agents including doxorubicin ((Floridi et al., 1998; Zupi et al., 1986), cisplatin (Raaphorst (Pratesi et al., 1996; Raaphorst et al., 1991) and others such as carmustine (BCNU) and 4-hydroperoxycyclophosphamide (4-HC) (Rosbe et al., 1989). Multiple clinical trials of lonidamine with or without other chemotherapy drugs have been carried on in non small cell lung cancer, breast cancer, ovary cancer and glioblastoma (Berruti et al., 2002; De Lena et al., 2001; De Marinis et al., 1999; Oudard et al., 2003; Portalone et al., 1999).
Oxamate
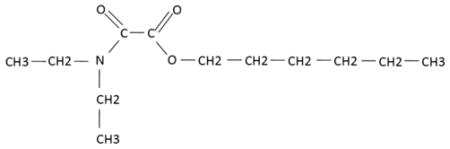
Oxamate inhibits lactate dehydrogenase (LDH), an NADH-dependent enzyme responsible for the conversion of pyruvate to lactate. LHD-A activity was shown to be elevated in several mouse tumor mammary epithelial cell lines in comparison to immortalized, non-transformed mammary epithelial cells (Fantin et al., 2002). Goldberg et al showed that the glycolytic rate was reduced, via measurement of lactate production, in HeLa S3 cells treated with oxamate (Goldberg and Colowick, 1965). Furthermore, cell growth and glucose uptake were also inhibited by this compound. This study also supported LDH as a target of oxamate by using α-ketobutyrate as a substitute for pyruvate. Addition of α-ketobutyrate reversed the inhibition of growth and glucose uptake in the presence of oxamate in HeLa cells. However, α-ketobutyrate by itself inhibited cell growth. Generation of recombinant human LDH-A showed oxamate competitively inhibited the enzyme, and was also confirmed in vitro (Thornburg et al., 2008). Both in vitro and in vivo growth inhibition was seen with oxamate treatment in MDA-MB-231 breast adenocarcinoma cells. Depletion of ATP in HeLa cells by oxamate also appears to contribute to its growth inhibition and enhance doxorubicin-mediated cytotoxicity (Hamilton et al., 1995). Additionally, oxamate was shown to inhibit the activity of human recombinant aspartate aminotransferase (AAT) ((Rej, 1979; Thornburg et al., 2008). AAT functions with malate dehydrogenase, a TCA enzyme which oxidizes malate to oxaloacetate, to help shuttle the two metabolites between the mitochondria and cytosol.
4. Summary
Conventional chemotherapy using cytotoxic agents tends to damage both tumor and normal cells and cause significant toxic side effects, which compromise the therapeutic outcomes. The emergence of targeted therapy which is based on the concept of specifically targeting critical molecules unique to cancer cells may provide promising means of selectively killing malignant cells. However, cancer may be caused by multiple genetic alterations and environmental factors. As such, there are many potential targets in cancer cells but no single critical target can be readily identified in most cancer types, perhaps with an exception of Bcr-Abl in CML. This situation makes the development of targeted therapy a challenging task (Green, 2004). Mitochondria structural and functional alterations associated with malignant transformation seem to be a common phenomenon observed in many types of cancers. Utilizing these differences to preferentially target mitochondria of cancer cells may be a logical strategy to achieve therapeutic selectivity. Several classes of small molecules that directly target mitochondria at specific sites or indirectly impact the metabolic alterations in cancer cells with dysfunctional mitochondria seem to exhibit promising anticancer activity. Since many of these small molecules have mainly been tested in cancer cell lines and in animal tumor models, clinical trials are needed to further test their potential for use in clinical treatment of cancer patients. Table 1 provides a list of compounds that target mitochondria or impact the metabolic alterations associated with mitochondrial dysfunction in cancer cells. The modes of action of these compounds and their clinical trial status are also indicated. It is important to recognize that cancer cells may be able to tolerate inhibition of mitochondrial function by upregulation of glycolysis and other survival mechanisms. As such, a combination of mitochondria-targeted agents with glycolytic inhibitors and other chemotherapeutic drugs may be required to achieve maximum efficacy. However, caution must be exercised to prevent potential increase in toxic side effects. A comprehensive understanding of mitochondrial biology in cancer cells and the interaction between cellular metabolism and drug action is essential in developing mitochondrial-targeted agents for cancer treatment.
Table 1.
Compounds that target mitochondria or impact the metabolic alterations associated with mitochondrial dysfunction in cancer cells.
| Class | Compounds name | Status | Cancer type | Reference |
|---|---|---|---|---|
| Targeting mitochondria transmembrane potential | ||||
| Rhodamine123 | Phase I clinical trial | Prostate cancer | Jones et al., 2005 | |
| MKT-077 | Phase I clinical trial | Solid tumors |
Britten et al., 2000 Propper et al.,1999 |
|
| F16 | Pre-clinical testing | Breast cancer, fibrosarcoma, intestinal cancer | Fantin et al., 2002 | |
| Dequalinium | Pre-clinical testing | Colon cancer, meloma, bladder cancer | Bleday et al., 1986; Helige et al., 1992; Weiss et al., 1987 | |
| Targeting cancer mitochondrial respiration | ||||
| Arsenic trioxide | In clinical use and clinical trials in various cancer types. | APL, Liver cancer, melanoma, AML, glioma, CLL, lymphoma, breast cancer, non-small cell lung cancer, pancreatic cancer | Bael et al.,2008, Chang et al., 2009, Fox et al.,2008, Shen et al., 1997, Kindler et al.,2008, Niu et al.,1999, Tarhini et al.,2008 | |
| Vitamin E analogue | Phase II study | Melanoma, prostate cancer, colorectal cancer, mesothelioma, breast cancer | Tsavachidou et al.,2009 Neuzil et al., 2007 |
|
| Resveratrol | Phase II clinical trials | Colorectal cancer, myeloma, follicular lymphoma | Jang et al., 1997; Goldberg et al.,2003; Walle et al.,2004; Meng et al., 2004; Boocock et al.,2007; Vitaglione et al.,2005; Levi et al., 2005 | |
| Rotenone | Experimental tool | Chance et al., 1963 | ||
| Targeting mitochondrial membrane permeability | ||||
| Honokiol | Pre-clinical testing | Esophagus cancer | Chen et al., 2009a | |
| Betulinic acid | Pre-clinical testing | Brain tumor, colon cancer, melanoma | Fulda & Debatin, 2000; Fulda et al., 1999; Jung et al., 2007; Pisha et al., 1995 | |
| Bcl-2 inhibitors (Gossypol) | Phase III clinical trial | Small cell lung cancer, prostate cancer, breast cancer, lymphoma, leukemia |
Van et al., 2001 O’Brien et al., 2005 Glenn et al., 2009 |
|
| Targeting mtDNA | ||||
| Cisplatin | In clinical use | Lung cancer, breast cancer, ovarian cancer, germ cell tumor, sarcoma | Sculier et al., 2009, Besse et al., 2009, Decatris et al., 2004, Murata et al., 1990, De et al.,1989 | |
| Topoisomerase inhibitors (Etoposide) | In clinical use | Lymphoma, lung cancer, brain tumor, germ cell tumor | Fischer et al., 2008, Kondaqunta et al, 2006, Mascaux et al., 2000 Lawrence et al., 1993 | |
| Ditercalinium | Pre-clinical testing | Fibrosarcoma, cervical cancer | Rodríguez et al., 2009, Okamaoto et al., 2003 |
|
| Vitamin K3 | Pre-clinical testing | Pancreatic cancer |
Akiyoshi et al., 2009 Marchionatti et al., 2009 Osada & Yoshida, 2009 |
|
| Targeting metabolic alterations | ||||
| 3-Bromopyruvate | Pre-clinical testing | Hepatoma, colon cancer, lymphoma | Bustamante and Pedersen, 1977; | |
| Dichloroacetate | Phase II clinical trial | Brain tumor, some solid tumor, Non-small cell | Xu et al., 2005 Bonnet et al., 2007; McFate et al., 2008 | |
| Lonidamine | Phase III clinical trial | lung cancer, breast cancer | De Marinis et al., 1999; Berruti et al., 2002 | |
| Oxamate | Pre-clinical testing | Cervical cancer, | Goldberg et al., 1965; Hamilton et al., 1995 | |
Abbreviations
- PET
positron emission tomography
- ATP
adenosine triphosphate
- mtDNA
mitochondrial DNA
- DLCs
delocalized lipophilic cations
- SOD1
superoxide dismutase1
- mtNOS
mitochondrial nitric oxide synthase
- ROS
reactive oxygen species
- As2O3
arsenic trioxide
- APL
acute promyelocytic leukemia
- NAC
n-acetyl cysteine
- α-TOS
alpha-tocopheryl succinate
- UbQ
ubiquinone
- Cyb
cytochrome B
- SDH
succinate dehydrogenase
- α-TOC
alpha tocopherol
- dUQH2
decyl-ubiquinol
- Mn-SOD
mitochondria SOD
- MPTP
mitochondrial membrane permeability transition pore
- ANT
adenine nucleotide transporter
- VDAC
voltage dependent anion channel
- MMP
mitochondrial inner membrane permeabilization
- mt
mitochondrial
- m-AMSA
amsacrine
- etoposide
VP-16
- teniposide
VM-26
- TCA
tri-carboxylic acid
- HXK
hexokinase
- 3BrPa
3-bromopyruvate
- DCA
dichloroacetate
- PDK
pyruvate dehydrogenase kinase
- PDH
pyruvate dehydrogenase
- H2O2
hydrogen peroxide
- LND
lonidamine
- BCNU
carmustine
- 4-HC
4-hydroperoxycyclophosphamide
- LDH
lactate dehydrogenase
- AAT
aspartate aminotransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyoshi T, Matzno S, Sakai M, Okamura N, Matsuyama K. The potential of vitamin K(3) as an anticancer agent against breast cancer that acts via the mitochondria-related apoptotic pathway. Cancer Chemother Pharmacol. 2009 doi: 10.1007/s00280-009-1016-7. [DOI] [PubMed] [Google Scholar]
- Arcadi JA. Rhodamine-123 as effective agent in rat prostate tumor R3327-H. Preliminary report. Urology. 1986;28:501–503. doi: 10.1016/0090-4295(86)90152-4. [DOI] [PubMed] [Google Scholar]
- Armstrong JS. Mitochondria: a target for cancer therapy. Br J Pharmacol. 2006;147:239–248. doi: 10.1038/sj.bjp.0706556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS, Hornung B, Lecane P, Jones DP, Knox SJ. Rotenone-induced G2/M cell cycle arrest and apoptosis in a human B lymphoma cell line PW. Biochem Biophys Res Commun. 2001;289:973–978. doi: 10.1006/bbrc.2001.6054. [DOI] [PubMed] [Google Scholar]
- Babbs M, Collier HO, Austin WC, Potter MD, Taylor EP. Salts of decamethylene-bis-4-aminoquinaldinium (dequadin); a new antimicrobial agent. J Pharm Pharmacol. 1956;8:110–119. doi: 10.1111/j.2042-7158.1956.tb12138.x. [DOI] [PubMed] [Google Scholar]
- Badros AZ, Goloubeva O, Rapoport AP, Ratterree B, Gahres N, Meisenberg B, Takebe N, Heyman M, Zwiebel J, Streicher H, et al. Phase II study of G3139, a Bcl-2 antisense oligonucleotide, in combination with dexamethasone and thalidomide in relapsed multiple myeloma patients. J Clin Oncol. 2005;23:4089–4099. doi: 10.1200/JCO.2005.14.381. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal SD, Lampidis TJ, McIsaac RM, Chen LB. Anticarcinoma activity in vivo of rhodamine 123, a mitochondrial-specific dye. Science. 1983;222:169–172. doi: 10.1126/science.6623064. [DOI] [PubMed] [Google Scholar]
- Bernal SD, Lampidis TJ, Summerhayes IC, Chen LB. Rhodamine-123 selectively reduces clonal growth of carcinoma cells in vitro. Science. 1982;218:1117–1119. doi: 10.1126/science.7146897. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Herst PM, Lawen A. Targeting mitochondrial permeability in cancer drug development. Mol Nutr Food Res. 2009;53:76–86. doi: 10.1002/mnfr.200700493. [DOI] [PubMed] [Google Scholar]
- Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, De Matteis A, Nuzzo F, Giardina G, Danese S, De Lena M, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. J Clin Oncol. 2002;20:4150–4159. doi: 10.1200/JCO.2002.08.012. [DOI] [PubMed] [Google Scholar]
- Bleday R, Weiss MJ, Salem RR, Wilson RE, Chen LB, Steele G., Jr Inhibition of rat colon tumor isograft growth with dequalinium chloride. Arch Surg. 1986;121:1272–1275. doi: 10.1001/archsurg.121.11.1272. [DOI] [PubMed] [Google Scholar]
- Bodden WL, Palayoor ST, Hait WN. Selective antimitochondrial agents inhibit calmodulin. Biochemical and biophysical research communications. 1986;135:574–582. doi: 10.1016/0006-291x(86)90032-x. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Britten CD, Rowinsky EK, Baker SD, Weiss GR, Smith L, Stephenson J, Rothenberg M, Smetzer L, Cramer J, Collins W, et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin Cancer Res. 2000;6:42–49. [PubMed] [Google Scholar]
- Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:3735–3739. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos L, Sabido O, Rouault JP, Guyotat D. Effects of BCL-2 antisense oligodeoxynucleotides on in vitro proliferation and survival of normal marrow progenitors and leukemic cells. Blood. 1994;84:595–600. [PubMed] [Google Scholar]
- Chan SH, Barbour RL. Adenine nucleotide transport in hepatoma mitochondria. Characterization of factors influencing the kinetics of ADP and ATP uptake. Biochim Biophys Acta. 1983;723:104–113. doi: 10.1016/0005-2728(83)90014-2. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR, Hollunger G. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. The Journal of biological chemistry. 1963;238:418–431. [PubMed] [Google Scholar]
- Chang LO, Schnaitman CA, Morris HP. Comparison of the mitochondrial membrane proteins in rat liver and hepatomas. Cancer Res. 1971;31:108–113. [PubMed] [Google Scholar]
- Chen G, Izzo J, Demizu Y, Wang F, Guha S, Wu X, Hung MC, Ajani J, Huang P. Different redox states in malignant and non-malignant esophageal epithelial cells and differential cytotoxic responses to bile acid and honokiol. Antioxid Redox Signal. 2009a doi: 10.1089/ars.2008.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB, Summerhayes IC, Johnson LV, Walsh ML, Bernal SD, Lampidis TJ. Probing mitochondria in living cells with rhodamine 123. Cold Spring Harb Symp Quant Biol . 1982;46(Pt 1):141–155. doi: 10.1101/sqb.1982.046.01.018. [DOI] [PubMed] [Google Scholar]
- Chen YC, Lin-Shiau SY, Lin JK. Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol. 1998;177:324–333. doi: 10.1002/(SICI)1097-4652(199811)177:2<324::AID-JCP14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007;39:267–274. doi: 10.1007/s10863-007-9086-x. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang H, Lu W, Huang P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochimica et biophysica acta. 2009b;1787:553–560. doi: 10.1016/j.bbabio.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- Cichewicz RH, Kouzi SA. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med Res Rev. 2004;24:90–114. doi: 10.1002/med.10053. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123:739–752. doi: 10.1002/ijc.23689. [DOI] [PubMed] [Google Scholar]
- Dairkee SH, Hackett AJ. Differential retention of rhodamine 123 by breast carcinoma and normal human mammary tissue. Breast Cancer Res Treat. 1991;18:57–61. doi: 10.1007/BF01975444. [DOI] [PubMed] [Google Scholar]
- Davis S, Weiss MJ, Wong JR, Lampidis TJ, Chen LB. Mitochondrial and plasma membrane potentials cause unusual accumulation and retention of rhodamine 123 by human breast adenocarcinoma-derived MCF-7 cells. The Journal of biological chemistry. 1985;260:13844–13850. [PubMed] [Google Scholar]
- De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, Guida M, Catino A, Crucitta E, Sambiasi D, Mazzei A. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. 2001;37:364–368. doi: 10.1016/s0959-8049(00)00400-7. [DOI] [PubMed] [Google Scholar]
- De Marinis F, Rinaldi M, Ardizzoni A, Bruzzi P, Pennucci MC, Portalone L, D’Aprile M, Ripanti P, Romano F, Belli M, et al. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: a phase III randomized FONICAP trial. Italian Lung Cancer Task Force. Tumori. 1999;85:177–182. doi: 10.1177/030089169908500306. [DOI] [PubMed] [Google Scholar]
- Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- Dias N, Bailly C. Drugs targeting mitochondrial functions to control tumor cell growth. Biochemical pharmacology. 2005;70:1–12. doi: 10.1016/j.bcp.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Dong LF, Freeman R, Liu J, Zobalova R, Marin-Hernandez A, Stantic M, Rohlena J, Valis K, Rodriguez-Enriquez S, Butcher B, et al. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex II. Clin Cancer Res. 2009;15:1593–1600. doi: 10.1158/1078-0432.CCR-08-2439. [DOI] [PubMed] [Google Scholar]
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008;27:4324–4335. doi: 10.1038/onc.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2:29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]
- Fellous R, Coulaud D, el Abed I, Roques BP, Le Pecq JB, Delain E, Gouyette A. Cytoplasmic accumulation of ditercalinium in rat hepatocytes and induction of mitochondrial damage. Cancer Res. 1988;48:6542–6549. [PubMed] [Google Scholar]
- Filomeni G, Graziani I, Rotilio G, Ciriolo MR. trans-Resveratrol induces apoptosis in human breast cancer cells MCF-7 by the activation of MAP kinases pathways. Genes Nutr. 2007;2:295–305. doi: 10.1007/s12263-007-0059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- Floridi A, Bruno T, Miccadei S, Fanciulli M, Federico A, Paggi MG. Enhancement of doxorubicin content by the antitumor drug lonidamine in resistant Ehrlich ascites tumor cells through modulation of energy metabolism. Biochemical pharmacology. 1998;56:841–849. doi: 10.1016/s0006-2952(98)00054-9. [DOI] [PubMed] [Google Scholar]
- Floridi A, Paggi MG, D’Atri S, De Martino C, Marcante ML, Silvestrini B, Caputo A. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer research. 1981a;41:4661–4666. [PubMed] [Google Scholar]
- Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, De Martino C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. Journal of the National Cancer Institute. 1981b;66:497–499. [PubMed] [Google Scholar]
- Fulda S, Debatin KM. Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med Pediatr Oncol. 2000;35:616–618. doi: 10.1002/1096-911x(20001201)35:6<616::aid-mpo27>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Fulda S, Friesen C, Los M, Scaffidi C, Mier W, Benedict M, Nunez G, Krammer PH, Peter ME, Debatin KM. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997;57:4956–4964. [PubMed] [Google Scholar]
- Fulda S, Jeremias I, Steiner HH, Pietsch T, Debatin KM. Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. Int J Cancer. 1999;82:435–441. doi: 10.1002/(sici)1097-0215(19990730)82:3<435::aid-ijc18>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Fulda S, Scaffidi C, Susin SA, Krammer PH, Kroemer G, Peter ME, Debatin KM. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem. 1998;273:33942–33948. doi: 10.1074/jbc.273.51.33942. [DOI] [PubMed] [Google Scholar]
- Galli S, Labato MI, Bal de Kier Joffe E, Carreras MC, Poderoso JJ. Decreased mitochondrial nitric oxide synthase activity and hydrogen peroxide relate persistent tumoral proliferation to embryonic behavior. Cancer Res. 2003;63:6370–6377. [PubMed] [Google Scholar]
- Galluzzi L, Larochette N, Zamzami N, Kroemer G. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene. 2006;25:4812–4830. doi: 10.1038/sj.onc.1209598. [DOI] [PubMed] [Google Scholar]
- Giurgiovich AJ, Diwan BA, Olivero OA, Anderson LM, Rice JM, Poirier MC. Elevated mitochondrial cisplatin-DNA adduct levels in rat tissues after transplacental cisplatin exposure. Carcinogenesis. 1997;18:93–96. doi: 10.1093/carcin/18.1.93. [DOI] [PubMed] [Google Scholar]
- Goldberg EB, Colowick SP. The Role of Glycolysis in the Growth of Tumor Cells. 3. Lactic Dehydrogenase as the Site of Action of Oxamate on the Growth of Cultured Cells. The Journal of biological chemistry. 1965;240:2786–2790. [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Green MR. Targeting targeted therapy. N Engl J Med. 2004;350:2191–2193. doi: 10.1056/NEJMe048101. [DOI] [PubMed] [Google Scholar]
- Hamilton E, Fennell M, Stafford DM. Modification of tumour glucose metabolism for therapeutic benefit. Acta oncologica (Stockholm, Sweden) 1995;34:429–433. doi: 10.3109/02841869509094003. [DOI] [PubMed] [Google Scholar]
- Haraguchi H, Ishikawa H, Shirataki N, Fukuda A. Antiperoxidative activity of neolignans from Magnolia obovata. J Pharm Pharmacol. 1997;49:209–212. doi: 10.1111/j.2042-7158.1997.tb06781.x. [DOI] [PubMed] [Google Scholar]
- Helige C, Smolle J, Zellnig G, Fink-Puches R, Kerl H, Tritthart HA. Effect of dequalinium on K1735-M2 melanoma cell growth, directional migration and invasion in vitro. Eur J Cancer. 1992;29A:124–128. doi: 10.1016/0959-8049(93)90589-8. [DOI] [PubMed] [Google Scholar]
- Herr HW, Huffman JL, Huryk R, Heston WD, Melamed MR, Whitmore WF., Jr Anticarcinoma activity of rhodamine 123 against a murine renal adenocarcinoma. Cancer Res. 1988;48:2061–2063. [PubMed] [Google Scholar]
- Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- Howard AN, Bridges KA, Meyn RE, Chandra J. ABT-737, a BH3 mimetic, induces glutathione depletion and oxidative stress. Cancer Chemother Pharmacol. 2009 doi: 10.1007/s00280-009-1001-1. [DOI] [PubMed] [Google Scholar]
- Irwin CC, Malkin LI, Morris HP. Differences in total mitochondrial proteins and proteins synthesized by mitochondria from rat liver and Morris hepatomas 9618A, 5123C, and 5123tc. Cancer Res. 1978;38:1584–1588. [PubMed] [Google Scholar]
- Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106:1794–1800. doi: 10.1182/blood-2005-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama K, Nakajo S, Aiuchi T, Nakaya K. Apoptosis induced by arsenic trioxide in leukemia U937 cells is dependent on activation of p38, inactivation of ERK and the Ca2+-dependent production of superoxide. Int J Cancer. 2001;92:518–526. doi: 10.1002/ijc.1220. [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Walsh ML, Bockus BJ, Chen LB. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J Cell Biol. 1981;88:526–535. doi: 10.1083/jcb.88.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV, Walsh ML, Chen LB. Localization of mitochondria in living cells with rhodamine 123. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:990–994. doi: 10.1073/pnas.77.2.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LW, Narayan KS, Shapiro CE, Sweatman TW. Rhodamine-123: therapy for hormone refractory prostate cancer, a phase I clinical trial. J Chemother. 2005;17:435–440. doi: 10.1179/joc.2005.17.4.435. [DOI] [PubMed] [Google Scholar]
- Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem. 2008;56:4813–4818. doi: 10.1021/jf800175a. [DOI] [PubMed] [Google Scholar]
- Jung GR, Kim KJ, Choi CH, Lee TB, Han SI, Han HK, Lim SC. Effect of betulinic acid on anticancer drug-resistant colon cancer cells. Basic Clin Pharmacol Toxicol. 2007;101:277–285. doi: 10.1111/j.1742-7843.2007.00115.x. [DOI] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen HM, Schimke RT. Multiple forms of hexokinase in the rat: tissue distribution, age dependency, and properties. Proceedings of the National Academy of Sciences of the United States of America. 1965;54:1218–1225. doi: 10.1073/pnas.54.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada S, Takayama S, De Riel K, Tanaka S, Reed JC. Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2 gene expression. Antisense Res Dev. 1994;4:71–79. doi: 10.1089/ard.1994.4.71. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Inden M, Miyamura A, Kakimura J, Taniguchi T, Shimohama S. Possible involvement of both mitochondria- and endoplasmic reticulum-dependent caspase pathways in rotenone-induced apoptosis in human neuroblastoma SH-SY5Y cells. Neurosci Lett. 2002;333:25–28. doi: 10.1016/s0304-3940(02)00964-3. [DOI] [PubMed] [Google Scholar]
- Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, Hullihen J, Pedersen PL. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochemical and biophysical research communications. 2004;324:269–275. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJ, Verkaart S, Visch HJ, van der Westhuizen FH, Murphy MP, van den Heuvel LW, Smeitink JA, Willems PH. Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am J Physiol Cell Physiol. 2005;288:C1440–1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido, Chen LB. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–543. [PubMed] [Google Scholar]
- Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(−/−) mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lampidis TJ, Hasin Y, Weiss MJ, Chen LB. Selective killing of carcinoma cells “in vitro” by lipophilic-cationic compounds: a cellular basis. Biomed Pharmacother. 1985;39:220–226. [PubMed] [Google Scholar]
- Lampidis TJ, Salet C, Moreno G, Chen LB. Effects of the mitochondrial probe rhodamine 123 and related analogs on the function and viability of pulsating myocardial cells in culture. Agents Actions. 1984;14:751–757. doi: 10.1007/BF01978920. [DOI] [PubMed] [Google Scholar]
- Lin JH, Castora FJ. DNA topoisomerase II from mammalian mitochondria is inhibited by the antitumor drugs, m-AMSA and VM-26. Biochem Biophys Res Commun. 1991;176:690–697. doi: 10.1016/s0006-291x(05)80239-6. [DOI] [PubMed] [Google Scholar]
- Lo YC, Teng CM, Chen CF, Chen CC, Hong CY. Magnolol and honokiol isolated from Magnolia officinalis protect rat heart mitochondria against lipid peroxidation. Biochem Pharmacol. 1994;47:549–553. doi: 10.1016/0006-2952(94)90187-2. [DOI] [PubMed] [Google Scholar]
- Lopes de Menezes DE, Hudon N, McIntosh N, Mayer LD. Molecular and pharmacokinetic properties associated with the therapeutics of bcl-2 antisense oligonucleotide G3139 combined with free and liposomal doxorubicin. Clin Cancer Res. 2000;6:2891–2902. [PubMed] [Google Scholar]
- Madan E, Prasad S, Roy P, George J, Shukla Y. Regulation of apoptosis by resveratrol through JAK/STAT and mitochondria mediated pathway in human epidermoid carcinoma A431 cells. Biochemical and biophysical research communications. 2008;377:1232–1237. doi: 10.1016/j.bbrc.2008.10.158. [DOI] [PubMed] [Google Scholar]
- Manion MK, Hockenbery DM. Targeting BCL-2-related proteins in cancer therapy. Cancer Biol Ther. 2003;2:S105–114. [PubMed] [Google Scholar]
- Marchionatti AM, Picotto G, Narvaez CJ, Welsh J, Tolosa de Talamoni NG. Antiproliferative action of menadione and 1,25(OH)2D3 on breast cancer cells. J Steroid Biochem Mol Biol. 2009;113:227–232. doi: 10.1016/j.jsbmb.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Maximo V, Sobrinho-Simoes M. Hurthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Arch. 2000;437:107–115. doi: 10.1007/s004280000219. [DOI] [PubMed] [Google Scholar]
- McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. The Journal of biological chemistry. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Li Y, Li J, Li H, Fu J, Liu Y, Liu H, Chen X. (−)Gossypol and its combination with imatinib induce apoptosis in human chronic myeloid leukemic cells. Leuk Lymphoma. 2007;48:2204–2212. doi: 10.1080/10428190701583991. [DOI] [PubMed] [Google Scholar]
- Meng Y, Tang W, Dai Y, Wu X, Liu M, Ji Q, Ji M, Pienta K, Lawrence T, Xu L. Natural BH3 mimetic (−)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol Cancer Ther. 2008;7:2192–2202. doi: 10.1158/1535-7163.MCT-08-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LA, Goldstein NB, Johannes WU, Walton CH, Fujita M, Norris DA, Shellman YG. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J Invest Dermatol. 2009;129:964–971. doi: 10.1038/jid.2008.327. [DOI] [PubMed] [Google Scholar]
- Miyato Y, Ando K. Apoptosis of human melanoma cells by a combination of lonidamine and radiation. Journal of radiation research. 2004;45:189–194. doi: 10.1269/jrr.45.189. [DOI] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Aprille JR. Basis for the selective cytotoxicity of rhodamine 123. Cancer Res. 1987;47:4361–4365. [PubMed] [Google Scholar]
- Modica-Napolitano JS, Aprille JR. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv Drug Deliv Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, Chen LB. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996;56:544–550. [PubMed] [Google Scholar]
- Modica-Napolitano JS, Singh KK. Mitochondria as targets for detection and treatment of cancer. Expert Rev Mol Med. 2002;4:1–19. doi: 10.1017/S1462399402004453. [DOI] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–762. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Morrison BJ, Andera L, Reynolds BA, Ralph SJ, Neuzil J. Future use of mitocans against tumour-initiating cells? Mol Nutr Food Res. 2009;53:147–153. doi: 10.1002/mnfr.200800254. [DOI] [PubMed] [Google Scholar]
- Mullauer FB, Kessler JH, Medema JP. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent, permeability transition pore dependent fashion. Apoptosis. 2009;14:191–202. doi: 10.1007/s10495-008-0290-x. [DOI] [PubMed] [Google Scholar]
- Murata T, Hibasami H, Maekawa S, Tagawa T, Nakashima K. Preferential binding of cisplatin to mitochondrial DNA and suppression of ATP generation in human malignant melanoma cells. Biochem Int. 1990;20:949–955. [PubMed] [Google Scholar]
- Neuzil J, Tomasetti M, Zhao Y, Dong LF, Birringer M, Wang XF, Low P, Wu K, Salvatore BA, Ralph SJ. Vitamin E analogs, a novel group of “mitocans,” as anticancer agents: the importance of being redox-silent. Mol Pharmacol. 2007;71:1185–1199. doi: 10.1124/mol.106.030122. [DOI] [PubMed] [Google Scholar]
- O’Brien SM, Cunningham CC, Golenkov AK, Turkina AG, Novick SC, Rai KR. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23:7697–7702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- Okamaoto M, Ohsato T, Nakada K, Isobe K, Spelbrink JN, Hayashi J, Hamasaki N, Kang D. Ditercalinium chloride, a pro-anticancer drug, intimately associates with mammalian mitochondrial DNA and inhibits its replication. Curr Genet. 2003;43:364–370. doi: 10.1007/s00294-003-0393-4. [DOI] [PubMed] [Google Scholar]
- Okumura K, Huang S, Sinicrope FA. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res. 2008;14:8132–8142. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE. (−)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Osada S, Yoshida K. A Novel Strategy for Advanced Pancreatic Cancer - Progression of Molecular Targeting Therapy. Anticancer Agents Med Chem. 2009 doi: 10.2174/187152009789124691. [DOI] [PubMed] [Google Scholar]
- Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF, Dutrillaux B, Andrieu JM, Delattre JY. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2003;63:81–86. doi: 10.1023/a:1023756707900. [DOI] [PubMed] [Google Scholar]
- Park SY, Jeong KJ, Lee J, Yoon DS, Choi WS, Kim YK, Han JW, Kim YM, Kim BK, Lee HY. Hypoxia enhances LPA-induced HIF-1alpha and VEGF expression: their inhibition by resveratrol. Cancer Lett. 2007;258:63–69. doi: 10.1016/j.canlet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Pedersen PL, Morris HP. Uncoupler-stimulated adenosine triphosphatase activity. Deficiency in intact mitochondria from Morris hepatomas and ascites tumor cells. The Journal of biological chemistry. 1974;249:3327–3334. [PubMed] [Google Scholar]
- Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. The Journal of biological chemistry. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Pepper C, Thomas A, Hoy T, Cotter F, Bentley P. Antisense-mediated suppression of Bcl-2 highlights its pivotal role in failed apoptosis in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;107:611–615. doi: 10.1046/j.1365-2141.1999.01726.x. [DOI] [PubMed] [Google Scholar]
- Petit T, Izbicka E, Lawrence RA, Nalin C, Weitman SD, Von Hoff DD. Activity of MKT 077, a rhodacyanine dye, against human tumor colony-forming units. Anticancer Drugs. 1999;10:309–315. doi: 10.1097/00001813-199903000-00010. [DOI] [PubMed] [Google Scholar]
- Pisha E, Chai H, Lee IS, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CW, Fong HH, Kinghorn AD, Brown DM, et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat Med. 1995;1:1046–1051. doi: 10.1038/nm1095-1046. [DOI] [PubMed] [Google Scholar]
- Politzer WM. Long-term clinical remission of a patient with chronic lymphocytic leukemia using alternative treatment option: cottonseed oil (gossypol) Phytomedicine. 2008;15:563–565. doi: 10.1016/j.phymed.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- Portalone L, Lombardi A, Antilli A, Cruciani AR, Magliacani V, Mugnaini L, Nunziati F, Perrone N, Signora M, Salvati F. Treatment of inoperable non-small cell lung carcinoma stage IIIb and IV with cisplatin, epidoxorubicin, vindesine and lonidamine: a phase II study. Tumori. 1999;85:239–242. doi: 10.1177/030089169908500405. [DOI] [PubMed] [Google Scholar]
- Pratesi G, De Cesare M, Zunino F. Efficacy of lonidamine combined with different DNA-damaging agents in the treatment of the MX-1 tumor xenograft. Cancer chemotherapy and pharmacology. 1996;38:123–128. doi: 10.1007/s002800050459. [DOI] [PubMed] [Google Scholar]
- Raaphorst GP, Ko D, Feeley MM, Danjoux CE, Maroun J, Evans WK. The effect of lonidamine alone and in combination with cisplatin on in vitro growth and viability of lung squamous cell carcinoma cell lines. Anticancer research. 1991;11:41–47. [PubMed] [Google Scholar]
- Ralph SJ, Neuzil J. Mitochondria as targets for cancer therapy. Mol Nutr Food Res. 2009;53:9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- Rej R. Measurement of aspartate aminotransferase activity: effects of oxamate. Clinical chemistry. 1979;25:555–559. [PubMed] [Google Scholar]
- Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Distinct pathways for stimulation of cytochrome c release by etoposide. J Biol Chem. 2000;275:32438–32443. doi: 10.1074/jbc.C000518200. [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- Rosbe KW, Brann TW, Holden SA, Teicher BA, Frei E., 3rd Effect of lonidamine on the cytotoxicity of four alkylating agents in vitro. Cancer chemotherapy and pharmacology. 1989;25:32–36. doi: 10.1007/BF00694335. [DOI] [PubMed] [Google Scholar]
- Roy P, Kalra N, Prasad S, George J, Shukla Y. Chemopreventive potential of resveratrol in mouse skin tumors through regulation of mitochondrial and PI3K/AKT signaling pathways. Pharm Res. 2009;26:211–217. doi: 10.1007/s11095-008-9723-z. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Kozloff M, Hoffman PC, Edelman MJ, Karnauskas R, Tomek R, Szeto L, Vokes EE. Phase I study of G3139, a bcl-2 antisense oligonucleotide, combined with carboplatin and etoposide in patients with small-cell lung cancer. J Clin Oncol. 2004;22:1110–1117. doi: 10.1200/JCO.2004.10.148. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pino MJ, Moreno P, Navarro A. Mitochondrial dysfunction in human colorectal cancer progression. Front Biosci. 2007;12:1190–1199. doi: 10.2741/2137. [DOI] [PubMed] [Google Scholar]
- Sasaki R, Suzuki Y, Yonezawa Y, Ota Y, Okamoto Y, Demizu Y, Huang P, Yoshida H, Sugimura K, Mizushina Y. DNA polymerase gamma inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci. 2008;99:1040–1048. doi: 10.1111/j.1349-7006.2008.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal-Bendirdjian E, Coulaud D, Roques BP, Le Pecq JB. Selective loss of mitochondrial DNA after treatment of cells with ditercalinium (NSC 335153), an antitumor bis-intercalating agent. Cancer Res. 1988;48:4982–4992. [PubMed] [Google Scholar]
- Shatton JB, Morris HP, Weinhouse S. Kinetic, electrophoretic, and chromatographic studies on glucose-ATP phosphotransferases in rat hepatomas. Cancer research. 1969;29:1161–1172. [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- Shiau CW, Huang JW, Wang DS, Weng JR, Yang CC, Lin CH, Li C, Chen CS. alpha-Tocopheryl succinate induces apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 function. The Journal of biological chemistry. 2006;281:11819–11825. doi: 10.1074/jbc.M511015200. [DOI] [PubMed] [Google Scholar]
- Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999;18:6641–6646. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- Stapelberg M, Gellert N, Swettenham E, Tomasetti M, Witting PK, Procopio A, Neuzil J. Alpha-tocopheryl succinate inhibits malignant mesothelioma by disrupting the fibroblast growth factor autocrine loop: mechanism and the role of oxidative stress. The Journal of biological chemistry. 2005;280:25369–25376. doi: 10.1074/jbc.M414498200. [DOI] [PubMed] [Google Scholar]
- Stein RC, Joseph AE, Matlin SA, Cunningham DC, Ford HT, Coombes RC. A preliminary clinical study of gossypol in advanced human cancer. Cancer Chemother Pharmacol. 1992;30:480–482. doi: 10.1007/BF00685601. [DOI] [PubMed] [Google Scholar]
- Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, Shepherd EL, Chen LB. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:5292–5296. doi: 10.1073/pnas.79.17.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AS, Sepkowitz K, Geller SA. A study of some mitochondrial and peroxisomal enzymes in human colonic adenocarcinoma. Lab Invest. 1981;44:13–17. [PubMed] [Google Scholar]
- Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood PL. Mitochondrial drugs. Curr Opin Chem Biol. 2008;12:457–463. doi: 10.1016/j.cbpa.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Torres-Roca JF, Tung JW, Greenwald DR, Brown JM, Herzenberg LA, Katsikis PD. An early oxygen-dependent step is required for dexamethasone-induced apoptosis of immature mouse thymocytes. J Immunol. 2000;165:4822–4830. doi: 10.4049/jimmunol.165.9.4822. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, Borgen P, Gollub M, Bacotti D, Yao TJ, et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat. 2001;66:239–248. doi: 10.1023/a:1010686204736. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetvicka V, Volny T, Saraswat-Ohri S, Vashishta A, Vancikova Z, Vetvickova J. Glucan and resveratrol complex--possible synergistic effects on immune system. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2007;151:41–46. doi: 10.5507/bp.2007.007. [DOI] [PubMed] [Google Scholar]
- Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, Dyer MJ, Cohen GM. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. 2008;15:820–830. doi: 10.1038/cdd.2008.25. [DOI] [PubMed] [Google Scholar]
- Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15:1739–1744. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
- Wang TS, Kuo CF, Jan KY, Huang H. Arsenite induces apoptosis in Chinese hamster ovary cells by generation of reactive oxygen species. J Cell Physiol. 1996;169:256–268. doi: 10.1002/(SICI)1097-4652(199611)169:2<256::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Wang XF, Witting PK, Salvatore BA, Neuzil J. Vitamin E analogs trigger apoptosis in HER2/erbB2-overexpressing breast cancer cells by signaling via the mitochondrial pathway. Biochemical and biophysical research communications. 2005;326:282–289. doi: 10.1016/j.bbrc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weber T, Dalen H, Andera L, Negre-Salvayre A, Auge N, Sticha M, Lloret A, Terman A, Witting PK, Higuchi M, et al. Mitochondria play a central role in apoptosis induced by alpha-tocopheryl succinate, an agent with antineoplastic activity: comparison with receptor-mediated pro-apoptotic signaling. Biochemistry. 2003;42:4277–4291. doi: 10.1021/bi020527j. [DOI] [PubMed] [Google Scholar]
- Weinhouse S, Shatton JB, Criss WE, Morris HP. Molecular forms of enzymes in cancer. Biochimie. 1972;54:685–693. doi: 10.1016/s0300-9084(72)80167-6. [DOI] [PubMed] [Google Scholar]
- Weiss MJ, Wong JR, Ha CS, Bleday R, Salem RR, Steele GD, Jr, Chen LB. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5444–5448. doi: 10.1073/pnas.84.15.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer research. 2005;65:613–621. [PubMed] [Google Scholar]
- Yoshitani SI, Tanaka T, Kohno H, Takashima S. Chemoprevention of azoxymethane-induced rat colon carcinogenesis by dietary capsaicin and rotenone. Int J Oncol. 2001;19:929–939. doi: 10.3892/ijo.19.5.929. [DOI] [PubMed] [Google Scholar]
- Zini R, Morin C, Bertelli A, Bertelli AA, Tillement JP. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp Clin Res. 1999;25:87–97. [PubMed] [Google Scholar]
- Zupi G, Greco C, Laudonio N, Benassi M, Silvestrini B, Caputo A. In vitro and in vivo potentiation by lonidamine of the antitumor effect of adriamycin. Anticancer research. 1986;6:1245–1249. [PubMed] [Google Scholar]