

Abstract
Telomerase activity is readily detected in most cancer biopsies, but not in premalignant lesions or in normal tissue samples with a few exceptions that include germ cells and hemopoietic stem cells. Telomerase activity may, therefore, be a useful biomarker for diagnosis of malignancies and a target for inactivation in chemotherapy or gene therapy. These observations have led to the hypothesis that activation of telomerase may be an important step in tumorigenesis. To test this hypothesis, we studied telomerase activity in isogeneic samples of uncultured and cultured specimens of normal human uroepithelial cells (HUCs) and in uncultured and cultured biopsies of superficial and myoinvasive transitional cell carcinoma (TCC) of the bladder. Our results demonstrated that four of four TCC biopsies, representing both superficial and myoinvasive TCCs, were positive for telomerase activity, but all samples of uncultured HUC were telomerase negative. However, when the same normal HUC samples were established as proliferating cultures in vitro, telomerase activity was readily detected but usually at lower levels than in TCCs. Consistent with the above observation of the telomerase activity in HUCs, telomeres did not shorten during the HUC in vitro lifespan. Demonstration of telomerase in proliferating human epithelial cells in vitro was not restricted to HUCs, because it was also present in prostate and mammary cell cultures. Notably, telomerase activity was relatively low or undetectable in nonproliferating HUC cultures. These data do not support a model in which telomerase is inactive in normal cells and activated during tumorigenic transformation. Rather, these data support a model in which the detection of telomerase in TCC biopsies, but not uncultured HUC samples, reflects differences in proliferation between tumor and normal cells in vivo.
Keywords: tumor biomarkers, human uroepithelial cells, mammary epithelial cells, prostate epithelial cells
Telomeres are repetitive sequences at the ends of chromosomes that protect chromosomes from incomplete replication, nuclease degradation, and end-to-end fusion during replication (for review, see ref. 1). Telomeres are required for chromosome segregation during meiosis and mitosis (2). Telomerase is a ribonucleoprotein complex responsible for de novo telomere synthesis and addition of telomeric repeats to existing telomeres (3). Telomerase activity can be measured in vitro by using the telomeric repeat amplification protocol (TRAP) (4). This assay has been used extensively to study telomerase activity in uncultured and cultured samples of normal and tumor tissue from many cell types.
Telomerase activity has been demonstrated in a high percent of extracts from most tumor types (4). For example, telomerase has been demonstrated in 75% of oral carcinomas (5), 80% of lung cancers (6), 84% of prostate cancers (7), 85% of liver cancers (8), 93% of breast cancers (9), 94% of neuroblastomas (10), 95% of colorectal cancers (11), and 98% of bladder cancers (12, 13). Recently, telomerase activity was detected in exfoliated cells found in the urine of bladder cancer patients (14). Cell lines, immortalized either spontaneously or after transformation by oncogenic viruses, such as simian virus 40 or human papillomvirus types 16 or 18, are usually telomerase-positive (4, 15, 16). However, telomerase activity is not always detectable in immortal cell lines (17). Such observations led to the current hypothesis that telomerase is activated during immortalization in vitro and tumorigenesis in vivo (18, 19).
Telomerase activity has also been assessed in many normal tissue types. Most results showed that normal somatic cells were telomerase-negative, whereas stem cells such as in the germ-line and hemopoietic tissues were telomerase-positive (20, 21). Several studies using normal fibroblasts demonstrated age-associated telomere shortening in vivo and in vitro, consistent with the above model. In addition, it has been suggested that normal cells contain an inhibitor of telomerase, possibly on chromosome 3 (22–24), whose deletion or inactivation is required for immortalization and tumorigenic transformation. In contrast to the above findings, telomerase activity has been detected in the basal layer of epidermis, where most proliferation occurs (25, 26). In addition, telomerase activity was demonstrated in endometrial tissue biopsies, but only during the proliferative phase of the menstrual cycle (27). Finally, telomerase activity has been demonstrated in normal oral mucosa, a highly proliferative tissue (5). These latter studies are not consistent with a model in which activation of telomerase occurs during tumorigenic transformation. Rather, they suggest that telomerase activity may be associated with cell proliferation. There are still many unanswered questions concerning telomerase activity in normal vs. cancer cells (28).
We tested the hypothesis that telomerase activity is associated with cell proliferation rather than with tumorigenic transformation. To test this hypothesis, we examined telomerase activity in uncultured ureteral tissues, cultured normal human uroepithelial cells (HUCs) and uncultured and cultured biopsies of superficial and muscle invasive bladder transitional cell carcinomas (TCCs). We further addressed the hypothesis that telomerase activity is associated with proliferation by examining telomerase activity in nonproliferating, senescent and quiescent cultures from the same tissue samples. In this report, we demonstrate by using both normal and tumorous human uroepithelial tissues that telomerase activity is a marker of cell proliferation, not malignant transformation.
MATERIALS AND METHODS
Tissues and Cell Culture.
HUCs were obtained from residual ureteral segments after kidney transplantation (29). The normal bladder sample was obtained from a cadaver, and the bladder epithelial cultures (BECs) were derived from explant cultures of the same bladder mucosa. TCC biopsies were provided by the urologic oncology surgeons in the Department of Surgery at the University of Wisconsin Hospital and Clinics. Three of the TCCs were superficial noninvasive grade I tumors (TCCs 1, 3, and 4), and two TCCs were muscle invading grade II and III (TCCs 5 and 2, respectively). David Jarrard (Department of Surgery, University of Wisconsin Medical School) provided the normal human prostate epithelial cell (PEC) cultures. The human mammary epithelial cell (MEC) cultures were grown from cryopreserved samples obtained from reduction mammoplasty and provided by Michael Gould (Department of Human Oncology, University of Wisconsin Medical School). All uroepithelial cells (HUCs, TCCs, and BECs) were cultured on collagen-coated plastic tissue culture dishes in a supplemented Ham’s F-12 medium with 1% fetal bovine serum added (1%FBS/F12+) (29). MECs and PECs were cultured on collagen-coated dishes in 1%FBS/F12+ with bovine pituitary extract added at 35 μg/ml.
Telomerase activity was tested in primary ureter tissues and cultures of proliferating, senescent, confluent, or growth arrested HUCs. Various histologic sections of ureter tissue were collected for telomerase analysis as follows. The first sample was a cross-section of the ureter including both the mucosal and submucosal layers. The mucosa was then stripped from the submucosa and a sample of each piece was taken for extraction of telomerase. Explant cultures were then established from the remaining mucosal tissue to generate isogeneic proliferating HUCs. Cell extracts were collected from these HUCs during logarithmic phase to analyze telomerase activity in proliferating cultures. Postconfluent nonproliferating HUC cultures were established exactly as described (29, 30). Briefly HUCs were grown to confluence and maintained for 7–10 days after becoming confluent with frequent medium changes. Senescent cultures were obtained by serially passing HUCs until they reached senescence as characterized by morphological changes and senescence-associated (SA)-β-galactosidase activity (31). TCC biopsies were divided into three portions: one sample was analyzed to determine the pathology of the tumor, one sample was prepared for telomerase extraction, and the final sample was used to establish explant cultures. Two spontaneously immortalized TCC cell lines, TCC 94–10 and TCC 96–2, were used as a positive and a negative control for the TRAP assay.
Measurement of Telomerase Activity by TRAP.
Telomerase extracts were prepared and analyzed as described (4, 32). Briefly tissues were washed in PBS and then lysed in ice-cold lysis buffer (10 mM Tris⋅HCl, pH 7.4/1 mM MgCl2/1 mM EGTA/0.1 mM phenylmethylsulfonyl fluoride/5 mM 2-mercaptoethanol/0.5% CHAPS/10% glycerol). All cell extracts were immediately frozen and stored at −70°C. Usually the whole-cell extracts were thawed immediately prior to the TRAP assay. First, extracts were centrifuged at 16,000 × g for 30 min at 4°C, then supernatants were transferred to a clean tube, and protein concentrations were measured by using the Bio-Rad protein assay kit. Finally, TRAP, which is a one-tube PCR-based assay, was performed (32), as described below.
Briefly, the TS oligonucleotide (5′-AATCCGTCGAGCAGAGTT-3′) served as both the telomerase template and the forward primer for the PCR, and the CX oligonucleotide (3′-AATCCC(ATTCCC)3-5′) was the reverse PCR primer. Telomerase activity in 5 μg of protein was measured by using a 1-hr incubation at 22°C for telomere extension followed by 34 cycles of PCR amplification (90°C, 90 sec; 50°C, 30 sec; 72°C, 45 sec; 94°C, 30 sec). Each TRAP assay included the following controls. Because telomerase contains a critical RNA template, a sample of each extract was treated with 1 unit of RNase A as a control for telomerase specificity. Every assay included a telomerase-positive (TCC 94–10) and a telomerase-negative (TCC 96–2) control extract. Each assay also contained an extract-free lane that contained only the reaction mixture to detect PCR amplification of primer dimers. Only assays in which all control lanes showed the expected results were included in this study. To test for inhibitors of Taq DNA polymerase that might be present in negative samples, mixtures of negative and positive extracts were tested for telomerase activity, by using a 1:1 ratio. Controls were done in which telomerase-positive extracts were mixed with lysis buffer also at a 1:1 ratio. As another approach to test for the presence of inhibitors, serial dilutions of the positive samples were done. Lysis buffer was used for dilution and the final volume was the same for all samples. There was no evidence of Taq DNA polymerase inhibitors. All telomerase reactions were done in a total volume of 50 μl, and 40 μl of this was loaded on a 10% nondenaturing polyacrylamide gel, which was then electrophoresed for 45 min at 175 V followed by 85 min at 280 V. Gels were stained for 45 min in SYBRGreen I (Molecular Probes) and analyzed by a Fluorimager SI (Molecular Dynamics). Usually telomerase signals were visible after 45 min. However, gels that were negative were stained longer and tested at different Fluorimager exposure ranges to increase sensitivity. Telomerase activity was manifested in this assay by the presence of a characteristic 6-bp ladder (33).
Southern Blot Analysis for Telomeres.
HUCs from four independent ureters were grown to senescence, and DNA was collected at different passages and at senescence. Senescence was determined by cessation of proliferation, morphological alterations, and the SA-β-galactosidase method (31). The ages of the donors were 52, 49, 23, and 17 years old. DNA was prepared from the two immortal TCC cell lines described above. DNA extracts were prepared by SDS lysis and proteinase K digestion essentially as described (34). Concentrations of DNA were determined by the absorbence at 260/280 nm. Fifteen micrograms of DNA were digested with RsaI/HinfIII, then electrophoresed on a 0.8% agarose gel. One lane always contained 1-kb molecular markers. Separated DNAs were transferred to a Hybond N membrane and then probed with the Sty11 telomere probe (courtesy of Titia de Lange at The Rockefeller University) by using the Vistra fluorescent random prime DNA labeling and signal amplification modules (Amersham). The terminal restriction fragments detected by the probe appeared as a smear on the gel due to variation on each chromosome and between cells within a population, in the subtelomeric region size and the number of TTAGGG repeats (17). The median of the length of the smear for each cell culture was compared.
RESULTS
Telomerase Activity Was Not Detected in Uncultured Normal Human Ureteral Tissues.
We tested the current model that uncultured samples of normal somatic tissues do not have detectable telomerase activity by examining different tissue layers in independent uncultured ureters. Telomerase was undetectable in all samples including a cross-section of the entire ureter, the stripped mucosal layer (which is primarily epithelial cells), and the submucosal layer (which is mostly fibroblasts and other mesenchymal cell types) (Fig. 1 and Table 1). The same results were obtained with three independent ureters in reproducible experiments in which the control samples from the telomerase-positive (TCC 94–10) cell line was positive. Longer exposure did not result in visible telomerase ladders. These results demonstrate that telomerase activity was not detectable in uncultured samples of normal ureteral tissues under these conditions.
Figure 1.
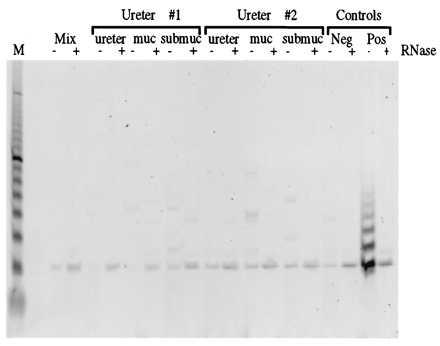
Telomerase activity is not detected in ureteral tissues. Results of the TRAP assay to detect telomerase activity in ureter, ureteral mucosa, and ureteral submucosa from two tissue samples (Ureters 1 and 2) are shown. Controls included a sample of every extract treated with RNase (−/+ RNase), the reaction mixture with no cell extract added (Mix), and samples containing extracts from both a telomerase-negative (TCC 96–2) and a telomerase-positive (TCC 94–10) TCC line. A 10-bp DNA ladder is included in lane M for size reference.
Table 1.
Telomerase activity in human epithelial cells
| Cell type | Total samples, n | Telomerase positive fraction |
|---|---|---|
| Uncultured | ||
| Ureter | 3 | 0/3 |
| Mucosa | 3 | 0/3 |
| Submucosa | 3 | 0/3 |
| TCC | 5 | 5/5 |
| Proliferating | ||
| HUC | 10 | 10/10 |
| BEC | 1 | 1/1 |
| MEC | 3 | 3/3 |
| PEC | 3 | 3/3 |
| TCC | 5 | 5/5 |
| Senescent | ||
| HUC | 2 | 0/2 |
| TCC | 2 | 0/2 |
| Growth arrested HUC | 3 | 0/3 |
Telomerase Activity Was Detected in Cultured Proliferating Normal Human HUCs, BECs, MECs, and PECs.
Next, to determine whether the failure to detect telomerase activity in ureter tissue was a result of the low proliferative capacity of the tissue in vivo, we cultured HUCs from the mucosal layer and tested telomerase activity during HUC proliferation and at senescence in two independent HUCs (Fig. 2 and Table 1). Results showed telomerase activity, but only in the proliferating cultures. To further test the correlation between telomerase activity and HUC proliferation, telomerase activity was tested in 10 independent proliferating HUC samples. In all cases telomerase was detected (Table 1).
Figure 2.
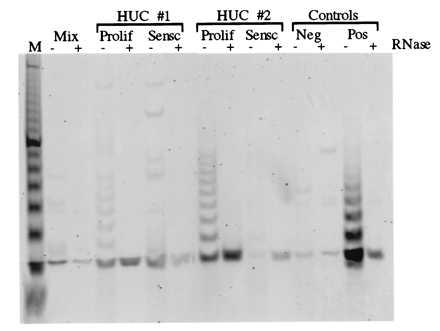
Telomerase activity is detected in proliferating HUCs. Results of the TRAP assay to test telomerase activity in two independent HUC cell cultures (HUCs 1 and 2) established from explant cultures of Ureters 1 and 2, respectively, as shown in Fig. 1, are shown. Telomerase activity was analyzed in both proliferating (Prolif) and senescent (Sensc) cultures. The control lanes are as described in Fig. 1.
We next tested whether cells growth-arrested by confluence and by senescence were telomerase negative. For this purpose, we tested telomerase activity in extracts from four samples from one ureter including: uncultured mucosal tissue, proliferating HUCs, senescent HUCs, and postconfluent HUC cultures. The results reproducibly showed telomerase activity only in the proliferating HUC culture (Fig. 3 and Table 1). The failure to detect telomerase in postconfluent HUCs was confirmed by using two independent ureter samples. To determine whether the negative result was due to the presence of a Taq DNA polymerase inhibitor, extracts from cells at senescence were mixed with positive cells. No evidence was obtained that an inhibitor was present. In addition, serial dilutions to titrate out a possible Taq polymerase inhibitor failed to uncover any telomerase activity.
Figure 3.
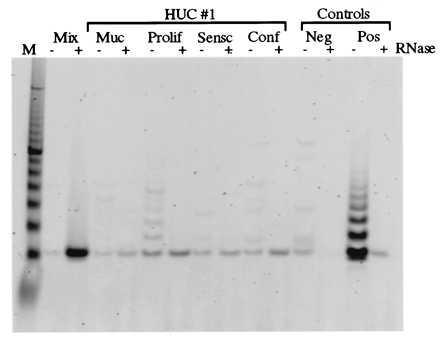
Association of telomerase activity with HUC proliferation. The TRAP assay results shown compare telomerase activity in isogeneic samples of uroepithelial cells. Samples included uncultured mucosa (muc), proliferating (Prolif) HUC, senescent (Sensc) HUC, and confluent (Conf) HUC cultures. The mucosa and all cultures were derived from ureter 1. The control lanes are as described in Fig. 1.
Finally, to test whether telomerase activity could be detected in other normal human epithelial cell types, we tested proliferating normal epithelial cell cultures from three other tissues. These included normal MECs from three donors, normal PECs from three donors, and BECs from one donor. Results reproducibly showed telomerase activity in all these proliferating epithelial cultures (Fig. 4 and Table 1).
Figure 4.
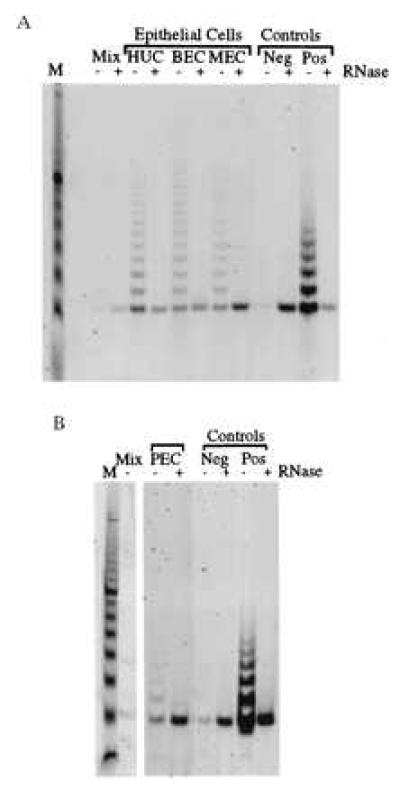
Telomerase activity is detected in proliferating epithelial cells. Results of the TRAP assay comparing proliferating cultures of ureter (HUC), bladder (BEC), and mammary (MEC) epithelial cells (A) and prostate (PEC) epithelial cells (B). The control lanes are as described in the legend to Fig. 1.
Telomerase Activity Was Detectable in Both Uncultured and Proliferating TCCs.
Next, we tested whether differences in telomerase activity could be detected between uncultured and cultured TCCs. We also tested whether differences in telomerase activity could be detected between superficial TCCs and myoinvasive TCCs. Results showed that telomerase activity was readily detectable in biopsies of superficial (TCCs 1, 3, and 4) and myoinvasive (TCCs 2 and 5) uncultured TCC biopsies. Telomerase activity was consistently higher in cultured TCC samples compared with uncultured samples (Fig. 5 and Table 1). Telomerase activity was tested in two senescent TCC cultures to test the hypothesis that nonproliferating TCCs would have decreased telomerase activity. As with normal HUCs, telomerase activity was reduced at senescence in TCCs (Table 1).
Figure 5.
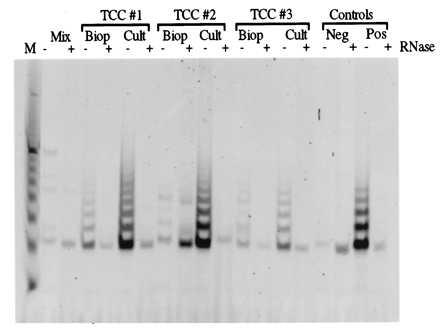
Telomerase activity in uncultured and cultured TCCs. Representative results of the TRAP assay comparing uncultured biopsies (Biop) of TCCs and proliferating TCC cultures (Cult) from three TCCs (TCCs 1, 2, and 3) are shown. TCCs 1 and 3 are superficial grade I, and TCC 2 is myoinvasive grade III. The controls are as described in Fig. 1.
Telomeres Were Stable in Cultured HUC.
We tested the hypothesis that telomeres of normal diploid cells shorten during their limited lifespan in vitro by using HUCs. For this purpose, we examined telomere length of HUC cultures from four ureters at different passages and at senescence. The range of the HUC telomere lengths was 6–14 kb. Results showed no decrease in average telomere length at senescence in any of the samples (Fig. 6). Furthermore, although the ages of the four donors differed by 39 years, the average lengths of telomeres of the derived HUC cultures were essentially the same. Thus results also showed that the length of uroepithelial telomeres do not decrease with age in vivo. Telomeres of the positive (TCC 94–10 P57) and negative (TCC 96–1 P24) cell lines were analyzed to compare the effect of telomerase status on telomere length. Results showed that the presence of telomerase did not necessarily correspond to longer telomeres (Fig. 7).
Figure 6.

Lack of telomere shortening in HUC in vitro and in vivo. Results of Southern blot analysis to determine whether telomere length changes after serial passage of HUC in vitro by using cultures (HUCs 3 and 4) established from two different ureters. The donor of ureter 3 was 53 years old, and the donor of ureter 4 was 23.
Figure 7.

Lack of correlation between telomerase activity and telomere length in two TCC lines. A Southern blot analysis is shown demonstrating shorter telomeres (3–5 kb) in a TCC line that shows telomerase activity (P) compared with a TCC line that is negative (N) for telomerase activity (5–10 kb) (Figs. 1, 2, 3, 4, 5 above). Telomere length of early-passage (P1) and senescent (P4) cultures of HUC 5 are shown for comparison. The donor of ureter 5 was 17 years old.
DISCUSSION
In the present study, we demonstrated, by using isogeneic samples of cultured and uncultured HUCs and TCCs, that telomerase activity was associated with cellular proliferation and not cellular transformation in human uroepithelial cells. We also demonstrated telomerase activity in normal proliferating mammary and prostate epithelial cultures.
These findings have several important implications for understanding the mechanisms involved in tumorigenic transformation. First, our findings alter the current model in which immortalization in vitro and tumorigenesis in vivo require telomerase activation, putatively by a genetic event, as a critical early step (18, 19, 35–38). The current model was based on the observation that few normal tissues, but most tumors and immortalized cell lines, had telomerase activity. In this paper we demonstrate that normal cells do have the capability to express telomerase activity given proliferative conditions in vitro. Our findings are consistent with other recent reports demonstrating that telomerase activity is present in highly proliferative normal tissues in vivo including hemopoietic cells (21), the oral mucosa (5), and endometrial tissue from the proliferative phase of the menstrual cycle (27). Thus we propose a new model in which telomerase activity is associated with proliferation and not malignant transformation. We would therefore attribute the relatively high telomerase activity in tumor biopsies at least in part to their high proliferative ability. Notably, our data do not preclude the possibility that genetic or epigenetic alterations leading to increased telomerase activity also may contribute to the higher telomerase levels seen in tumors, including TCCs.
Second, our demonstration that biopsies of bladder tumors, but not normal tissue specimens, are telomerase-positive supports the use of telomerase as a biomarker for detection of tumors. An advantage of using telomerase activity as a biomarker arises when cells may be obtained noninvasively, such as from urine, cervical smears, or sputum. Our observation that normal bladder epithelium is negative for telomerase activity is consistent with the observation that normal bladder epithelium does not proliferate in vivo, except in cases where healing may occur such as during infection. Examination of exfoliated bladder cells extracted from the urine of healthy sources demonstrated that normal bladder cells were telomerase-negative in vivo (14). However, our data suggest that using telomerase as a biomarker or a target for cancer therapy may have some limitations. The use of telomerase activity as a biomarker may generate false positives depending upon the proliferative state of the tissue in question. It has been suggested, in reports using liver, colorectal, and prostate cells, for example, that telomerase activity can differentiate between premalignant and malignant cells (7, 8, 10, 11, 39). Our results with bladder tissues do not support this hypothesis. Many reports suggest that telomerase could be targeted in cancer therapy (19, 24, 35, 38, 40, 41). Our demonstration of telomerase activity in normal cells from three independent tissue types suggests that targeting telomerase activity during cancer therapy may have widespread consequences for normal tissue renewal and healing due to lack of specificity toward tumor cells. One of the disadvantages of conventional cancer treatments that target the highly proliferative tumor cells is the elimination of healthy proliferating cells of the immune system and other high turnover organs such as gastrointestinal tract, which causes serious side effects.
Third, our results showing that telomeres of HUCs do not shorten with age in vitro are also significant and interesting for several reasons. The majority of studies associating shortening of telomeres with age in vivo and with lifespan in vitro were done by using fibroblasts (42–45). Thus there may be cell type differences in the ability to express telomerase and maintain telomere length. Telomerase activity is not detected in extracts of fibroblast cultures (26). This cannot be explained by lack of proliferation, because fibroblasts in general proliferate more easily in vitro than epithelial cells and have a longer lifespan. Thus, our data suggest that telomere shortening is not responsible for triggering senescence.
Finally, an interesting observation is that the telomerase-negative TCC cell line had longer telomeres than the telomerase-positive TCC cell line. There are several other reports of immortalized human cells that are telomerase-negative (17, 37). This suggests that there may be methods other than telomerase to actively maintain telomere length (17). This is consistent with reports suggesting alternate methods to maintain telomeres. For example, studies of yeast with mutant telomerase showed that telomere length can be maintained by recombination (46, 47). Because telomeres are protective sequences at the ends of chromosomes and telomere shortening is correlated with chromosome instability (15, 17, 48, 49), it makes evolutionary sense that multiple pathways were devised to maintain such a critical component of the cell.
In conclusion, the results of this study provide further insight into the possible regulation of telomerase activity. Our results suggest that targeting telomerase in cancer treatments may produce unwanted side effects if the loss of telomerase activity leads to death of stem cells and chromosome instability. Telomerase may be useful as a diagnostic tool for cancer detection in tissues with low proliferative capacity in vivo; however, conditions that would cause proliferation might lead to false positives. Further in depth study of the regulation of telomerase during the cell cycle should help to further elucidate this complicated story.
Acknowledgments
We thank the pathologists of the University of Wisconsin Hospital and Clinics for their analysis of the TCC biopsies. Ms. Patricia Lopez, who participated in this project, was supported by the Short Term Research Training Program for Minority Students sponsored by the University of Wisconsin Environmental Toxicology Center through a grant from the National Institutes of Health. This work was supported by National Institutes of Health Grants RO1 CA29525-17 and RO1 CA67158-03 to C.A.R. and a gift from the Haertle Family to the University of Wisconsin Comprehensive Cancer Center. C.D.B. was partially supported by Training Grant T32 ES07015 from the National Institute of Environmental Health Sciences to the University of Wisconsin Environmental Toxicology Center.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: HUC, human uroepithelial cell; TRAP, telomeric repeat amplification protocol; TCC, transitional cell carcinoma; BEC, bladder epithelial culture; PEC, prostate epithelial cell; MEC, mammary epithelial cell.
References
- 1.Gall J G. In: Telomeres. Blackburn E H, Greider C W, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 1–10. [Google Scholar]
- 2.Kirk K E, Harmon B P, Reichardt I K, Sedat J W, Blackburn E H. Science. 1997;275:1478–1481. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- 3.Greider C W. In: Telomeres. Blackburn E H, Greider C W, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 35–68. [Google Scholar]
- 4.Kim N W, Piatyszek M A, Prowse K R, Harley C B, West M D, Ho P L C, Coviello G M, Wright W E, Weinrich S L, Shay J W. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 5.Kannan S, Tahara H, Yokozaki H, Mathew B, Nalinakumari K R, Nair M K, Tahara E. Cancer Epidem Biomarkers Prevent. 1997;6:413–420. [PubMed] [Google Scholar]
- 6.Hiyama K, Ishioka S, Shirotani Y, Inai K, Hiyama E, Murakami I, Isobe T, Inamizu T, Yamakido M. Oncogene. 1995;10:937–944. [PubMed] [Google Scholar]
- 7.Sommerfield H-J, Meeker A K, Piatyszek M A, Bova G S, Shay J W, Coffey D S. Cancer Res. 1996;56:218–222. [PubMed] [Google Scholar]
- 8.Tahara H, Nakanishi T, Kitamoto M, Nakashio R, Shay J W, Tahara E, Kajiyama G, Ide T. Cancer Res. 1995;55:2734–2736. [PubMed] [Google Scholar]
- 9.Hiyama E, Gollahon L, Kataoka T, Kuroi K, Yokoyama T, Gasdar A F, Hiyama K, Piatyszek M A, Shay J W. J Natl Cancer Inst. 1996;88:116–122. doi: 10.1093/jnci/88.2.116. [DOI] [PubMed] [Google Scholar]
- 10.Hiyama E, Hiyama K, Yokoyama T, Matsuura Y, Piatyszek M A, Shay J W. Nat Med. 1995;1:249–255. doi: 10.1038/nm0395-249. [DOI] [PubMed] [Google Scholar]
- 11.Tahara H, Kumiyasu H, Yokozaki H, Yasui W, Shay J W, Ide T, Tahara E. Clin Cancer Res. 1995;1:1245–1251. [PubMed] [Google Scholar]
- 12.Kyo S, Kunimio K, Uchibayashi T, Namiki M, Inoue M. Am J Clin Pathol. 1997;107:555–560. doi: 10.1093/ajcp/107.5.555. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Miyamoto H, Fujinami K, Uemura H, Hosaka M, Iwasaki Y, Kubota Y. Clin Cancer Res. 1996;2:929–932. [PubMed] [Google Scholar]
- 14.Kinoshita H, Ogawa O, Kakehi Y, Mishina M, Mitsumori K, Itoh N, Yamada H, Terachi T, Yoshida O. J Natl Cancer Inst. 1997;89:724–730. doi: 10.1093/jnci/89.10.724. [DOI] [PubMed] [Google Scholar]
- 15.Counter C M, Avilion A A, LeFeuvre C E, Stewart N G, Greider C W, Harley C B, Bacchetti S. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klingelhutz A J, Barber S A, Smith P P, Dyer K, McDougall J K. Mol Cell Biol. 1994;14:961–969. doi: 10.1128/mcb.14.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryan T M, Englezou A, Gupta J, Bacchetti S, Reddel R R. EMBO J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Lange T. Proc Natl Acad Sci USA. 1994;91:2882–2885. doi: 10.1073/pnas.91.8.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greider C W, Blackburn E H. Sci Am. 1996;274:92–97. doi: 10.1038/scientificamerican0296-92. [DOI] [PubMed] [Google Scholar]
- 20.Hiyama, K., Hirai, Y., Kyoizumi, S., Aklyama, M., Hiyama, E., Piatyszek, M. A., Shay, J. W., Ishioka, S. & Yamakido, M. (1995) J. Immunol. 3711–3715. [PubMed]
- 21.Buchkovich K J, Greider C W. Mol Biol Cell. 1996;7:1443–14454. doi: 10.1091/mbc.7.9.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steenbergen R D M, Walboomers J M M, Meijer C J L M, van der Raaj-Helmer E M H, Parker J N, Chow l T, Broker T R, Snijders P J F. Oncogene. 1996;13:1249–1257. [PubMed] [Google Scholar]
- 23.Ohmura H, Tahara H, Suzuki M, Ide T, Shimizu M, Yoshida M A, Tahara E, Shay J W, Barrett J C, Oshimura M. Jpn J Cancer Res. 1995;86:899–904. doi: 10.1111/j.1349-7006.1995.tb02998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seachrist L. Science. 1995;268:29–30. doi: 10.1126/science.7701337. [DOI] [PubMed] [Google Scholar]
- 25.Yasumoto S, Kumimura C, Kikuchi K, Tahara H, Ohji H, Yamamoto H, Ide T, Utakoji T. Oncogene. 1996;13:433–439. [PubMed] [Google Scholar]
- 26.Harle-Bachor C, Boukamp P. Proc Natl Acad Sci USA. 1996;93:6576–6481. doi: 10.1073/pnas.93.13.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyo S, Takakura M, Kohama T, Inoue M. Cancer Res. 1997;57:610–614. [PubMed] [Google Scholar]
- 28.Lundblad V, Wright W E. Cell. 1996;87:369–375. doi: 10.1016/s0092-8674(00)81358-6. [DOI] [PubMed] [Google Scholar]
- 29.Reznikoff C A, Loretz L J, Presciotta D M, Oberley T D, Ignjatovic M M. J Cell Physiol. 1987;131:285–301. doi: 10.1002/jcp.1041310302. [DOI] [PubMed] [Google Scholar]
- 30.Reznikoff C A, Belair C D, Savelieva E, Zhai Y, Pfeifer K, Yeager T, Thompson K J, DeVries S, Bindley C, Newton M A, Sekhon G, Waldman F. Genes Dev. 1994;8:2227–2240. doi: 10.1101/gad.8.18.2227. [DOI] [PubMed] [Google Scholar]
- 31.Dimri G P, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E E, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piatyszek M A, Kim N W, Weinrich S L, Hiyama K, Hiyama E, Wright W E, Shay J W. Methods Cell Sci. 1995;17:1–15. [Google Scholar]
- 33.Morin G B. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 34.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. In: Preparation of Genomic DNA from Mammalian Tissues. Chanda V B, editor. Vol. 1. New York: Wiley; 1997. [Google Scholar]
- 35.Shay J W. Mol Med Today. 1995;1:376–382. doi: 10.1016/s1357-4310(95)93872-9. [DOI] [PubMed] [Google Scholar]
- 36.Holt S E, Wright W E, Shay J W. Mol Cell Biol. 1996;16:2932–2939. doi: 10.1128/mcb.16.6.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitaker N J, Bryan T M, Bonnefin P, Chang A C-M, Musgrove E A, Braithwaite A W, Reddel R R. Oncogene. 1995;11:971–976. [PubMed] [Google Scholar]
- 38.Harley C B. J NIH Res. 1995;7:64–68. [Google Scholar]
- 39.Hiyama E, Kodama T, Shinbara K, Iwao T, Itoh M, Hiyama K, Shay J W, Matsuura Y, Yokoyama T. Cancer Res. 1997;57:326–331. [PubMed] [Google Scholar]
- 40.Marx J. Science. 1994;265:1656–1658. doi: 10.1126/science.7521969. [DOI] [PubMed] [Google Scholar]
- 41.Haber D A. N Engl J Med. 1995;332:955–956. doi: 10.1056/NEJM199504063321412. [DOI] [PubMed] [Google Scholar]
- 42.Allsopp R C, Vasiri H, Patterson C, Goldstein S, Younglai E V, Futcher A B, Greider C W, Harley C B. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harley C B, Futcher A B, Greider C W. Nature (London) 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 44.Murnane J P, Sabatier L, Marder B A, Morgan W F. EMBO J. 1994;13:4953–4962. doi: 10.1002/j.1460-2075.1994.tb06822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slagboom P E, Droog S, Boomsma D I. Am J Hum Genet. 1994;55:876–882. [PMC free article] [PubMed] [Google Scholar]
- 46.McEachern M J, Blackburn E H. Nature (London) 1995;376:403–409. doi: 10.1038/376403a0. [DOI] [PubMed] [Google Scholar]
- 47.Zakian V A. In: Telomeres. Blackburn E H, Greider C W, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 107–137. [Google Scholar]
- 48.Holzmann K, Blin N, Welter C, Zang K D, Seitz G, Henn W. Genes Chromosomes Cancer. 1993;6:178–181. doi: 10.1002/gcc.2870060308. [DOI] [PubMed] [Google Scholar]
- 49.de Lange T. In: Telomeres. Blackburn E H, Greider C W, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. pp. 265–293. [Google Scholar]