

Abstract
Lipoapoptosis of pancreatic β cells caused by elevated circulating free fatty acids (FFAs) has now been recognized to be a pivotal factor contributing to β cellular dysfunction and β-mass lose in type 2 diabetes. Although recent studies suggested an important role for the ceramide pathway in the late destructive phase of lipid overload in the pancreatic β cells, the overall underlying mechanisms leading to lipoapoptosis, however, remained poorly understood. mir-375 was recently characterized to be a pancreatic islet-specific miRNA implicated in the regulation of insulin secretion and β-mass turnover. In the present study we further examined its effect on palmitate-induced lipoapoptosis in NIT-1 cells, a NOD-derived β-cell line. It was found that NIT-1 cells with ectopic mir-375 expression were much more susceptible to palmitate-induced lipoapoptosis. In contrast, knockdown of endogenous pri-mir-375 expression by a modified antisense oligo, 2'-O-me-375, almost completely protected NIT-1 cells from palmitate-induced lipoapoptosis. We further demonstrated that mir-375 could target V1 mRNA and repress its translation. Consistent with this assumption, NIT-1 cells transfected with 2'-O-me-375 showed significant higher levels of V1 protein after palmitate induction. Together, our data suggest that mir-375 could be a potential therapeutic target for prevention and intervention of β-cell dysfunction and β-mass lose in type 2 diabetes.
Keywords: Lipoapoptosis, mir-375, NIT-1 cells, β-cell dysfunction, β-mass, type 2 diabetes
Introduction
Type 2 diabetes is tightly linked to obesity characterized by hyperlipidemia and elevated circulating free fatty acids (FFAs) [1;2]. Excessive accumulation of unoxidized long-chain fatty acids can lead to the overflow of lipid to non-adipose tissues such as pancreatic islets [3-5]. Intracellular accumulation of lipid in the pancreatic islets is associated with β cellular dysfunction and death and ultimately contributes to the pathogenesis of type 2 diabetes [6-8]. Therefore, a progressive deterioration of β-mass and reduced β-cell function were observed in patients with type 2 diabetes [9]. Studies have shown that islet function was about 50% of normal upon the onset of diabetes, while β- mass remained only about 40% of normal [10]. There is compelling evidence that the reduction of β-mass is attributable to the accelerated apoptosis [11-13]. Factors responsible for the progressive loss of β-cell function and β-mass include glucotoxicity [14], lipotoxicity [15-17], pro-inflammatory cytokines [18;19], leptin [20;21], and islet amyloidosis [22]. Conventionally, type 2 diabetes has been defined in a glucocentric perspective (insulin resistance), while elevated systemic levels of fatty acids has now been considered a significant contributor towards the pathophysiological aspects, as dysregulated lipid homeostasis not only contributes to the development of insulin resistance but also plays a primary role in the progressive loss of pancreatic β cells [16;23]. As a result, chronic exposure to long-chain FFAs is associated with reduced insulin content, defective insulin secretion and β-cell apoptosis (lipoapoptosis). Despite recent extensive studies, the underlying molecular mechanisms leading to lipoapoptosis, however, remained poorly understood.
MicroRNAs (miRNAs) are short, noncoding RNAs that have recently been found to be pivotal for the regulation of gene expressions [24;25]. miRNA genes are transcribed primarily by RNA polymerase II into long precursor molecules which are then processed via RNase III enzymes Drosha and Dicer into the matured 21 - 23 nu-cleotide miRNA. Matured miRNAs subsequently bind to the specific sequences in the 3'UTR of mRNAs to regulate protein translation or mRNA stability. miRNAs are now known to be virtually ubiquitous among vertebrates, and are involved in a remarkable array of key cellular activities including differentiation, proliferation and apoptosis. Studies in animal model suggested that mir-375 is a pancreatic specific miRNA implicated in the maintenance of normal pancreatic α- and β-mass [26]. More interestingly, recent studies further demonstrated that mir-375 is also important in the regulation of β-cell apoptosis and insulin secretion [27-30]. Based on these observations, we sought to test the hypothesis that mir-375 is also important in the regulation of β-cell lipoapoptosis. Previously, we used NIT-1 cells, a non-obese diabetic (NOD) mouse derived β-cell line, to demonstrate the impact of G protein-coupled receptor 40 (GPR40) on lipoapotosis in β cells [31]. We now also used NIT-1 cells to address the above hypothesis. By transfection of NIT-1 cells with mir-375 duplexes or an antisense oligo specific mir-375 we have clearly demonstrated that mir-375 is a pivotal regulator for β-cell lipoapoptosis and, as a result, it could be a potential therapeutic target for prevention of β-cell lipoapoptosis during type 2 diabetes.
Materials and methods
Cell culture and treatment
NIT-1 cells originated from NOD mice were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS, Hyclone, USA) and 1% penicillin-streptomycin. The cells were incubated at 37°C in a humidified atmosphere of 5% CO2. Culture medium was refreshed every three days and NIT-1 cells with passages 20 - 40 in actively growing condition were used for the experiment. Long chain saturated palmitic fatty acid (palmitate, Sigma, USA) was employed to induce lipoapoptosis in NIT-1 cells. After 2 h of preincubation in serum-free condition, NIT-1 cells were cultured in the presence of 500 μM BSA-bound palmitate or control medium for 48 h as previously reported [31].
mir-375 preparation and transfection
mir-375 duplexes (5'- UUU GUU CGU UCG GCU CGC GUG A-3’ and 5'-ACG CGA GCC GAA CGA ACA A AU U-3') were commercially synthesized from GenePharma (Shanghai, China). An unrelated known miRNA (mir-375-NC, 5'-UUC UCC GAA CGU GUC ACG UTT-3’ and 5'-ACG UGA CAC GUU CGG AGA ATT-3', Invitrogen, China) was used as a negative control. Knockdown of mir-375 was carried out by using 2'-O-me-375 (5'-CAG UAC UUU UGU GUA GUA CAA-3'), and a control oligo named inhibitor-NC (5'-CAG UAC UUU UGU GUA GUA CAA-3') was used as a negative control. NIT-1 cells were prepared at 30 - 50% confluence at the time of transfection. Lipofectamine 2000 transfection reagent (Invitrogen, China) was used to transfeet NIT-1 cells. A total of 80 pmol mir-375 duplex, 2'-O-me-375, mir-375-NC, and inhibitor-NC were used along with 2 μl Lipofectamine 2000 per well with each containing 5 × 105 cells (six-well plate), respectively. Cells with Lipofectamine 2000 only (lipo2000) were used as a control.
MiroRNA qRT-PCR
Relative mir-375 expression levels were determined using a TaqMan MicroRNA Assay kit (ABI, USA) according to the manufacturer's instruction. Total RNAs from transfected NIT-1 cells were extracted with an RNeasy mini kit (Qiagen, USA), and 10 ng of total RNAs were then used for reverse transcription (RT) using the TaqMan MicroRNA Reverse Transcription kit (ABI, USA) in the presence of TaqMan MicroRNA Assay RT primer (1 μl). The reactions were carried out at 16°C for 30 min followed by 42°C for 30 min and 85°C for 5 min. The resulting cDNA products were diluted at 20×, and 1.33 μl of the diluted cDNA were used for PCR reaction (total volume 20 μl) with 1 μl of TaqMan MicroRNA Assay mix and 10 μl of TaqMan 2× Universal PCR Master Mix. The PCR reaction was conducted at 95°C 10 min followed by 40 cycles of 95°C 15s and 60°C 60s in an ABI 7900HT fast real-time PCR system. The real-time PCR results were analyzed as relative mir-375 expression of CT (threshold cycle) value to U6 internal control, which were then converted to fold changes.
Apoptosis assay
Palmitate-induced NIT-1 cell apoptosis was evaluated by terminal deoxynucleotidyl trans-ferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) and flow cytometry. For TUNEL assay, the cells were harvested after 48 h of palmitate treatment. After washing with PBS, the cells were fixed and permeabilized, followed by TUNEL labeling using a One Step TUNEL Apoptosis Assay Kit (Beyotime, China) as instructed. The percentage of apoptotic cells was estimated by the percentage of cells with positive TUNEL staining of five randomly selected fields in each slide under a fluorescent microscope. At least 100 cells were assessed in each selected field. Flow cytometry analysis of apoptotic NIT-1 cells was carried out using an Annexin V-FITC/PI staining kit (BD Biosciences, USA). After washes with cold PBS, the cells were resuspended in 1× binding buffer (0.1 M HEPES/ NaOH, pH 7.4, 1.4 M NaCI, and 25 mM CaCI2) followed by staining with AnnexinV-FITC/PI at RT in darkness for 15 min. Apoptotic cells were then evaluated by gating both PI and Annexin V positive cells on a FACSCalibur (BD Bio-science, USA). All experiments were performed in triplicates
Western blotting
Total proteins were prepared from NIT-1 cells using RIPA lysis buffer supplemented with protease inhibitors. Protein concentrations for all preparations were determined using the Bradford method (Bio-Rad, USA). Equal amount (25 - 50 μg) of cellular lysates was loaded onto a 12% SDS-polyacrylamide gel and run for 2 h at constant voltage (200V). Resolved proteins were then electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes, which were blocked for 1 h in blocking buffer containing 5% Non-Fat Dry Milk in TBST (10 mM Tris pH 7.6, 150 mM NaCI, 0.05% Tween-20) at room temperature. The membranes were first incubated with a primary antibody (sc-28416, Santa Cruz, USA, 1:500) at 4°C overnight. After washes with TBST buffer, the membranes were then incubated with a secondary antibody conjugated to peroxidase (BA1050, Boster, China, 1:10000) for 1 h. After extensive washes, the immunoreactive bands were visualized using a chemiluminescent substrate (M502, Jingmei Biotech, China), β-actin was used for normalization. The relative intensity for the target bands was analyzed by the Kodak Digital Science 1D analysis software (version 2.0). The results are present as a ratio with β-actin.
Statistical analysis
All of our data are present as mean ± SD unless otherwise indicated. Comparisons between groups for NIT-1 cell apoptosis were accomplished by one-way ANOVA using SPS 11.5 for windows. Mean values for microRNA expression and target protein expression were compared by unpaired Student's t-test. P < 0.05 was considered statistically significant.
Results
Palmitate is a strong inducer for lipoapoptosis in NIT-1 cells
Although previous studies suggested the role of palmitate in the induction of β-cell lipoapoptosis, its effect on NIT-1 cells in our current system is unknown. Therefore, we first sought to examine the effect of palmitate on NIT-1 cell lipoapoptosis in our culture system. For this purpose, NIT-1 cells were cultured in the presence of palmitate (500μM) or control medium for 48 h and then harvested for analysis of lipoapoptosis by TUNEL and flow cytometry assays. Consistent with previous report, palmitate is also a strong lipoapoptosis inducer for NIT-1 cells. Both in situ TUNEL staining and Annexin V/PI staining indicated significant higher levels of NIT-1 cells undergoing apoptosis after palmitate treatment. As shown in Figure 1, in average around 15.3% of palmitate treated NIT-1 cells became apoptotic. In sharp contrast, only 3.5% of cells cultured with control medium showing apoptosis (p < 0.0001).
Figure 1.

Palmitate is potent to induce NIT-1 cell lipoapoptosis. A. A representative results for TUNEL staining of apoptotic NIT-1 cells. B. A representative results for flow cytometry analysis of apoptotic NIT-1 cells. C. A bar graph showing the average apoptosis of NIT-1 cells cultured in the presence of palmitate and control medium. The data are present as mean ± SD of three independent experiments performed.
2'-O-me-375 is potent for knockdown of the endogenous pri-mir-375 expression in NIT-1 cells
Next, we sought to screen antisense oligonu-cleotides with high potency for specific knockdown of endogenous pri-mir-375 expression in NIT-1 cells. To this end, we have commercially synthesized three 2'-OMe modified antisense oligos, which were then transfected into NIT-1 cells as described earlier. As 2'-O-methyl oli-gonucleotides are refractory to nucleolytic cleavage by cellular ribonucleases, they are more stable than that of unmodified counterparts. Transfection of an unrelated oligo (inhibitor-NC) was served as a control. Real-time PCR was then employed to assess the endogenous pri-mir-375 expressions in the transfected NIT-1 cells. As expected, the unrelated control oligo did not show perceptible effect on pri-mir-375 expressions. Of important note, one particular antisense oligo named 2'-O-me-375 showed high potency for knockdown of pri-mir-375 expressions. We then performed similar assays with different doses of 2'-O-me-375 and found that the highest reduction for pri-miRNA was achieved when cells transfected with 60 - 80 pmol of 2'-O-me-375 (data not shown). As can be seen in Figure 2, pri-mir-375 expressions in NIT-1 cells had been reduced by 70% when cells transfected with 80 pmol of 2'-O-me-375. In contrast, the other two 2'-OMe modified oligos, oligo-2 and olig-3, only showed minor effect for knockdown of endogenous pri-mir-375 expressions (Figure 2). Together, our data indicate that 2'-O-me-375 possesses high potency for knockdown of pri-mir-375 expressions in NIT- 1 cells.
Figure 2.
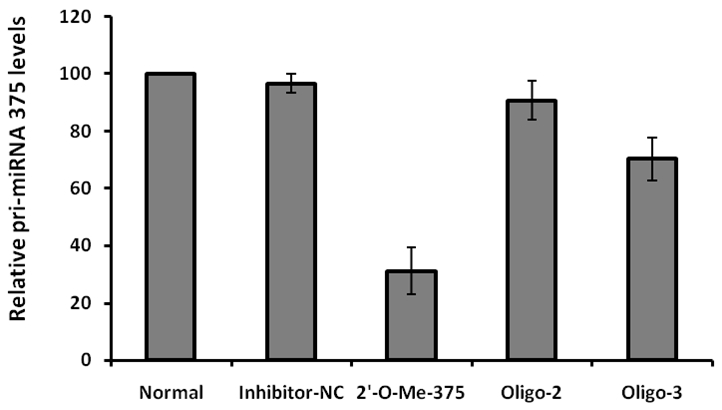
2'-O-Me-375 is potent to repress endogenous pri-mir-375 expressions in NIT-1 cells. The relative expression levels for pri-mir-375 were determined by real-time PCR as described. The expression levels of pri-mir-375 in NIT-1 cells cultured with control medium (Normal) were considered as 100%, its relative expression levels in cells transfected with inhibitor-NC, 2'-O-Me-375, oligo-2 and oligo-3 are present as a ratio with that of control cells. The data were derived from three replicates.
mir-375 enhances the susceptibility of NIT-1 cells to palmitate-induced lipoapoptosis
To demonstrate the effect of mir-375 on palmitate-induced lipoapoptosis in NIT-1 cells, we next transfected NIT-1 cells with 80 pmol of mir-375 duplex (mir-375) and 2'-O-me-375, respectively. Transfection of NIT-1 cells with an unrelated known miRNA (mir-375-NC) was severed as a control. NIT-1 cells cultured with lipofec-tamine 2000 only (lipo2000) or normal medium (normal) were used as negative controls. Following 72 h of transfection, the cells were then replaced with medium containing 500 μM of palmitate. After culturing the cells with another 48 h, the cells were harvested and subjected to analysis of apoptosis by in situ TUNEL staining and flow cytometry analysis as above. It was interestingly found that transfection of NIT-1 cells with mir-375 duplexes significantly enhanced palmitate-induced lipoapoptosis as compared with that of control cells and cells transfected with control oligos (Figure 3, 31.2 ± 5.3% vs. 15.4 ± 6.5% for Normal; 14.7 ± 4.7% for mir-375-NC; and 16.5 ± 4.1% for lipo2000, p < 0.001).
Figure 3.

Ectopic mir-375 duplexes enhance palmitate-induced NIT-1 cell lipoapoptosis. A. A representative results for TUNEL staining of apoptotic NIT-1 cells. B. A representative results for flow cytometry analysis of apoptotic NIT-1 cells. C. A bar graph showing the average apoptosis of NIT-1 cells transfected with mir-375-NC, mir-375 duplexes and 2'-O-Me-375. NIT-1 cells cultured with normal medium (Normal) or in the presence of same amount Lipofactamine 2000 (lipo2000) were served as controls. The data are present as mean ± SD of three independent experiments performed.
In line with above observations, knockdown of endogenous mir-375 expressions by transfection of 2'-O-me-375 almost completely protected NIT-1 cells from palmitate-induced lipoapoptosis. As shown in Figure 3, only about 4.8% of 2'-O-me-375 transfected cells were undergoing apoptosis, which is comparable to those cells without palmitate treatment (3.5%, Figure 1). Combining all of these data together, our results suggest that mir-375 plays a pivotal role in palmitate-induced lipoapoptosis in NIT-1 cells, and as a result, it could be potential therapeutic target for prevention of β-cell dysfunction and β-mass loss during type 2 diabetes.
mir-375 negatively regulates myotrophin (V1) expression in NIT-1 cells after palmitate treatment
To demonstrate the underlying mechanism through which mir-375 promotes palmitate-induced lipoapoptosis in NIT-1 cells, we examined the expression levels for myotrophin (V1), a pivotal factor implicated in the regulation of a variety of genes responsible for cellular apoptosis and proliferation. High levels of V1 protein were detected in NIT-1 cells in the normal culture condition. To our surprise, a significant reduction of V1 protein levels was observed in NIT-1 cells upon palmitate treatment. As shown in Figure 4, the protein levels for V1 in NIT-1 cells were decreased by 2-fold as compared with those NIT-1 cells cultured with control medium. A recent study suggested that V1 could be a specific target for mir-375 [27], the above results prompted us to check the effect of mir-375 on V1 expression in NIT-1 cells. For this purpose, we performed similar studies as above by transfection of NIT-1 cells with mir-375 duplexes and 2'-O-Me-375. Transfection of NIT-1 cells with mir-375-NC was used as a negative control. The cells were harvested after 72 h of transfection and then subjected to semi-quantitative Western blot analysis of V1 protein levels. As expected, transfection of mir-375-NC did not show discernible effect on V1 expression. However, Ectopic mir-375 duplexes reduced V1 protein levels in NIT-1 cells by 3-fold, while inhibition of the endogenous mir-375 expression by transfection of 2'-O-Me-375 increased V1 protein levels by 1-fold (Figure 5).
Figure 4.
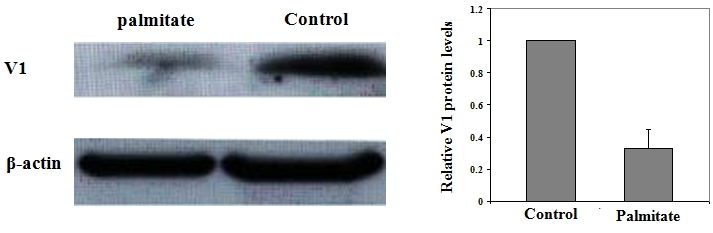
Palmitate treatment is associated with a significant reduction of V1 protein in NIT-1 cells. Left: A representative of Western blot results for V1 protein levels in NIT-1 cells treated with palmitate or control medium. Right: A bar graph showing the relative V1 expression levels in NIT-1 cells treated with palmitate. The relative intensity of each band was determined as a ratio with its corresponding β-actin band. V1 relative expression levels are present as fold changes compared with control cells. The dada are present as mean ± SD of three independent experiments performed.
Figure 5.
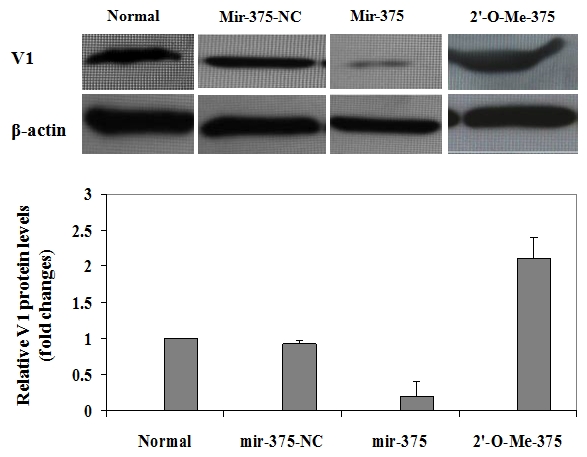
Ectopic mir-375 duplexes significantly reduce V1 protein levels in NIT-1 cells. NIT-1 cells were harvested after 72 h of transfection, and cell lysates were prepared and subjected to Western blot analysis of V1 protein expressions. Upper panel: A representative of Western blot results for V1 protein levels in NIT-1 cells cultured with control medium or transfected with mir-375-NC, mir-375 duplexes and 2'-O-Me-375, respectively. Lower panel: A bar graph showing the relative V1 protein levels in each above indicated experimental group. Similar as Figure 4, V1 relative protein levels are present as fold changes compared with that of control cells (Normal). The dada are present as mean ± SD of three independent experiments performed.
To further confirm the above observations, we next checked the effect of mir-375 on V1 protein expression after palmitate induction. To this end, the cells were treated with 500 μM of palmitate for 48 h after 72 h of transfection. The corresponding cell lysates were then prepared for Western blot analysis as above. Consistently, only very low levels of V1 protein can be detected in normal control cells and cells transfected with mir-375-NC. Of important note, V1 protein was almost undetectable in cells with ectopic mir-375 duplexes, while more than 3-fold higher V1 protein was observed in cells transfected with 2'-O-Me-375 (Figure 6). All together, our data strongly suggest that mir-375 could target V1 mRNA and affect its translation, which then promotes pamitate-induced lipoapoptosis in NIT-1 cells.
Figure 6.
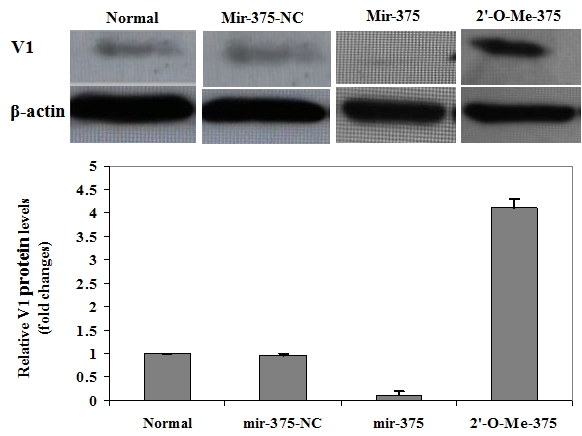
Repression of endogenous pri-mir-375 expression prevents palmitate-induced V1 reduction in NIT-1 cells. NIT-1 cells after 72 h of transfection were treated with 500μM of palmitate for 48 h, followed by Western blot analysis of cell lysates for V1 protein expression. Upper panel: A representative of Western blot results for V1 protein levels in each indicated transfected NIT-1 cells after palmitate treatment. Lower panel: A bar graph showing the relative V1 protein levels in each transfected NIT-1 cells after palmitate induction. Similarly, V1 relative protein levels are present as fold changes compared with that of control cells (Normal). The dada are present as mean ± SD of three independent experiments performed. It was found that repression of endogenous pri-mir-375 expressions by 2'-O-Me-375 prevented NIT-1 cells from palmitate-induced V1 reduction.
Discussion
Previous studies have consistently demonstrated that there is a progressive deterioration in β-cell function and β-mass in type 2 diabetes [10], and the reduction of β-mass is attributable to the accelerated apoptosis, in which elevated circulating free fatty acids (FFAs) are one of the pivotal predisposing factors responsible for β-cell apoptosis [16;17]. Recent studies highlighted the importance for one particular islet specific miRNA, mir-375, in the regulation of β-cell function and the maintenance of normal pancreatic α- and β-mass [26;27]. In the present study, we further examined the role of mir-375 in the regulation of palmitate-induced β-cell lipoapoptosis. We found that NIT-1 cells, a NOD-derived β-cell line, with ectopic mir-375 duplexes are much more susceptible to palmitate-induced lipoapoptosis. In contrast, NIT-1 cells are remarkably resistant to palmitate-induced lipoapoptosis once the endogenous mir-375 was suppressed by a specific antisense oligo, 2'-O-Me-375. Our study also demonstrates that the expression levels of V1 protein are inversely associated with β-cell lipoapoptosis. Since V1 could be a target for mir-375 [27], our data suggest that mir-375 probably enhances β-cell lipoapoptosis by downregulation of V1 expressions in β cells.
The cellular population for a particular organ is determined by the balance between the rates of cell division and programmed cell death named apoptosis. Once the apoptosis rate for a particular cell type becomes higher than that of cell replication, a reduction for cell population would occur, which subsequently leads to organ decompensation. Loss of β-mass late in the course of obesity is a typical example that β-cell apoptosis exceeds cell division, which then predisposes to the development of type 2 diabetes. Lipoapoptosis, a type of apoptosis caused by dysregulated lipid metabolism, has recently been recognized to occur in obesity and aging. Excessive accumulation of long-chain fatty acids is associated with altered leptin liporegulation and, as a result, when nonadipose tissues such as pancreatic islets are exposed to an excess of long-chain fatty acids, the cell would undergo lipoapoptosis. The effect of thermal, hypoxic or cytotoxic factors on apoptosis has been well demonstrated [32], while metabolic causes of programmed cell death, particularly lipoapoptosis, have received less attention. Although recent studies in ZDF (fa/fa) rats suggested an important role for the ceramide pathway in the late destructive phase of lipid overload in the pancreatic β cells [16], the overall underlying mechanisms leading to lipoapoptosis remained poorly understood. Our studies in the current report provided strong evidence indicating a pivotal role for miRNA in the regulation of β-cell lipoapoptosis. Although we are only beginning to appreciate the immense potential of miRNAs as controllers of gene networks, there is already substantial evidence that these small noncoding RNA molecules play a central role in a variety of physiological processes, including tissue differentiation, cell proliferation, and apoptosis. miRNAs are a growing class of non-coding RNAs involved in the regulation of gene expression by translational repression. Islet-specific mir-375 is originally characterized by Poy and colleagues from analysis of miRNAs in endocrine cell types of the pancreas [27]. Recent functional studies indicated that mir-375 plays an indispensable role in normal glucose homeostasis, β-mass turnover in newborns and adaptive β-cell expansion in response to increasing insulin demand in insulin resistance [26]. Our data now provide evidence indicating that mir-375 could also act as a proapoptotic factor implicated in the pathogenesis of β-cell lipoapoptosis.
Studies have shown that some evolutionarily conserved proteins characterized in neuronal cells are also present in the pancreatic β cells. V1 is such a protein that is highly expressed in both neuronal cells and pancreatic β cells. V1 (also known as myotrophin) is a 12-kD protein, which was originally identified from the hypertrophied ventricles of spontaneously hypertensive rats and rat cerebellum [33;34]. By 3-dimensional alignment, Knuefermann and colleagues demonstrated that mammalian V1 resembles a truncated IkBα without the signal response domain and PEST sequence [35]. Therefore, V1 was later validated as an antiapoptotic factor in cardiac myocytes, neuronal cells and fibroblasts [36-38]. In the current study, we further noticed that the expression levels for V1 in NIT-1 cells are inversely associated with palmitate-induced lipoapoptosis, indicating that V1 could also act as an antiapoptotic factor in maintaining β-cell viability.
Of important note, we limited our study in the current report to one of the pivotal targets for mir-375, the pancreatic islet-specific V1 protein. It is possible that mir-375 also targets additional factors implicated in the regulation of β-cell apoptosis. For example, a recent study also suggests that mir-375 could target PDK1 mRNA, a key molecule for PI 3-kinase signaling in the pancreatic β cells [29], which has been demonstrated to be involved in the regulation of cell survival and proliferation [39;40]. Therefore, multiple signaling pathways could be regulated by mir-375, by which mir-375 enhances β-cell lipoapoptosis.
The modified synthetic anti-miRNA oligonucleotides (AMOs) were found to be useful tools for specific repression of targeted miRNA expressions, thereby helping to unravel the function of miRNAs and their targets. The 2'-O-methyl oligonucleotides have been shown to irreversibly inhibit small RNA function in vitro and in intact cells in a sequence-specific fashion, presumably by stoichiometric binding to RISCs containing the cognate miRNA, and thus preventing interaction with its mRNA targets [41]. Similar to antisense-based oligonucleotides (ASOs), AMOs may contribute to the prioritization of pharmaceutical targets and have the potential to eventually progress into a new class of therapeutic agents. Here we have designed a 2'-OMe modified anti-mir-375 oligonucleotide, 2'-O-me-375, and demonstrated its high potency for repression of endogenous mir-375 expressions in NIT-1 cells (Figure 2). As 2'-O-me-375 transfected NIT-1 cells were almost completely protected from palmitate-induced lipoapoptosis (Figure 3), 2'-O-me-375 could be a useful “off-target” reagent for mir-375 with significant implications in both experimental and clinical studies.
Impaired β-cell function and possibly β-mass deficit appear to be reversible, particularly at the early stage of disease when the limiting threshold for reversibility of the decreased β-mass has probably not been passed. Our data in the current report indicate that mir-375, a pancreatic islet-specific miRNA, could be a pivotal regulator of lipoapoptosis in the pancreatic β cells. Taking into account that mir-375 also regulates insulin secretion, mir-375 could emerge as a potential therapeutic target for prevention and intervention of β-cell dysfunction and β-mass loss in type 2 diabetes.
Acknowledgments
This work was partly supported by the Natural Science Foundation of China (30671974) and by the Guangdong Provincial Natural Science Foundation of China (6021329). All authors declare that they have no competing financial interest.
References
- 1.Charles MA, Eschwege E, Thibult N, Claude JR, Warnet JM, Rosselin GE, Girard J, Balkau B. The role of non-esterified fatty acids in the deteriora tion of glucose tolerance in Caucasian subjects: results of the Paris Prospective Study. Diabetologia. 1997;40(9):1101–6. doi: 10.1007/s001250050793. Sep; [DOI] [PubMed] [Google Scholar]
- 2.Schulz LO, Bennett PH, Ravussin E, Kidd JR, Kidd KK, Esparza J, Valencia ME. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care. 2006;29(8):1866–71. doi: 10.2337/dc06-0138. Aug; [DOI] [PubMed] [Google Scholar]
- 3.Bluher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117(6):241–50. doi: 10.1055/s-0029-1192044. Jun; [DOI] [PubMed] [Google Scholar]
- 4.Surampudi PN, John-Kalarickal J, Fonseca VA. Emerging concepts in the pathophysiology of type 2 diabetes mellitus. Mt Sinai J Med. 2009;76(3):216–26. doi: 10.1002/msj.20113. Jun; [DOI] [PubMed] [Google Scholar]
- 5.Davis N, Forges B, Wylie-Rosett J. Role of obesity and lifestyle interventions in the prevention and management of type 2 diabetes. Minerva Med. 2009;100(3):221–8. Jun. [PubMed] [Google Scholar]
- 6.Johnson JD. Proteomic identification of carboxypeptidase E connects lipid-induced beta-cell apoptosis and dysfunction in type 2 diabetes. Cell Cycle. 2009;8(1):38–42. doi: 10.4161/cc.8.1.7343. Jan 1. [DOI] [PubMed] [Google Scholar]
- 7.Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296(4):E690–E701. doi: 10.1152/ajpendo.90525.2008. Apr; [DOI] [PubMed] [Google Scholar]
- 8.El-Assaad W, Buteau J, Peyot ML, Nolan C, Ro-duit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144(9):4154–63. doi: 10.1210/en.2003-0410. Sep; [DOI] [PubMed] [Google Scholar]
- 9.Funakoshi S, Fujimoto S, Hamasaki A, Fujiwara H, Fujita Y, Ikeda K, Hamamoto Y, Hosokawa M, Seino Y, Inagaki N. Analysis of factors influencing pancreatic beta-cell function in Japanese patients with type 2 diabetes: association with body mass index and duration of diabetic exposure. Diabetes Res Clin Pract. 2008;82(3):353–8. doi: 10.1016/j.diabres.2008.09.010. Dec; [DOI] [PubMed] [Google Scholar]
- 10.Wajchenberg BL. beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28(2):187–218. doi: 10.1210/10.1210/er.2006-0038. Apr; [DOI] [PubMed] [Google Scholar]
- 11.Campbell RK. Fate of the beta-cell in the patho physiology of type 2 diabetes. J Am Pharm Assoc (2003) 2009;49(Suppl 1):S10–S15. doi: 10.1331/JAPhA.2009.09076. Sep; [DOI] [PubMed] [Google Scholar]
- 12.Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009 doi: 10.1007/s10495-009-0339-5. Mar 26. [DOI] [PubMed] [Google Scholar]
- 13.Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord. 2008;9(4):329–43. doi: 10.1007/s11154-008-9101-5. Dec; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poitout V, Amyot J, Semache M, Zarrouki B, Hag-man D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbalip.2009.08.006. Aug 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, von EA. Low- and high-density lipo-proteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology. 2009;150(10):4521–30. doi: 10.1210/en.2009-0252. Oct; [DOI] [PubMed] [Google Scholar]
- 16.Unger RH, Zhou YT. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes. 2001;50(Suppl 1):S118–>S121. doi: 10.2337/diabetes.50.2007.s118. Feb; [DOI] [PubMed] [Google Scholar]
- 17.Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273(49):32487–90. doi: 10.1074/jbc.273.49.32487. Dec 4. [DOI] [PubMed] [Google Scholar]
- 18.Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Sterling J, Rosenberg L, Billestrup N, Maysinger D, Mandrup-Poulsen T. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009;58(8):1807–15. doi: 10.2337/db08-0178. Aug; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology. 2008;149(5):2208–18. doi: 10.1210/en.2007-1059. May; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okuya S, Tanabe K, Tanizawa Y, Oka Y. Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology. 2001;142(11):4827–3O. doi: 10.1210/endo.142.11.8494. Nov. [DOI] [PubMed] [Google Scholar]
- 21.Brown JE, Dunmore SJ. Leptin decreases apoptosis and alters BCL-2 : Bax ratio in clonal rodent pancreatic beta-cells. Diabetes Metab Res Rev. 2007;23(6):497–502. doi: 10.1002/dmrr.726. Sep. [DOI] [PubMed] [Google Scholar]
- 22.Guardado-Mendoza R, Davalli AM, Chavez AO, Hubbard GB, Dick EJ, Majluf-Cruz A, Tene-Perez CE, Goldschmidt L, Hart J, Perego C, Comuzzie AG, Tejero ME, et al. Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci U S A. 2009;106(33):13992–7. doi: 10.1073/pnas.0906471106. Aug 18; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusminski CM, Shetty S, Orci L, Unger RH, Scherer PE. Diabetes and apoptosis: lipotoxicity. Apoptosis. 2009 doi: 10.1007/s10495-009-0352-8. May 8. [DOI] [PubMed] [Google Scholar]
- 24.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11(3):228–34. doi: 10.1038/ncb0309-228. Mar; [DOI] [PubMed] [Google Scholar]
- 25.Rogaev EI, Borinskaia SA, Islamgulov DV, Grigorenko AP. [Human microRNA in norm and pathology] Mol Biol (Mosk) 2008;42(5):751–64. Sep; [PubMed] [Google Scholar]
- 26.Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci U S A. 2009;106(14):5813–8. doi: 10.1073/pnas.0810550106. Apr7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–30. doi: 10.1038/nature03076. Nov 11; [DOI] [PubMed] [Google Scholar]
- 28.Lynn FC. Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol Metab. 2009;20(9):452–9. doi: 10.1016/j.tem.2009.05.007. Nov; [DOI] [PubMed] [Google Scholar]
- 29.El OA, Baroukh N, Martens GA, Lebrun P, Pipeleers D, van OE. miR-375 targets 3'-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes. 2008;57(10):2708–17. doi: 10.2337/db07-1614. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker MD. Role of MicroRNA in pancreatic beta-cells: where more is less. Diabetes. 2008;57(10):2567–8. doi: 10.2337/db08-0934. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Xu M, Zhang S, Yan L, Yang C, Lu W, Li Y, Cheng H. The role of G protein-coupled receptor 40 in lipoapoptosis in mouse beta-cell line NIT-1. J Mol Endocrinol. 2007;38(6):651–61. doi: 10.1677/JME-06-0048. Jun; [DOI] [PubMed] [Google Scholar]
- 32.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–6. doi: 10.1038/35037710. Oct 12; [DOI] [PubMed] [Google Scholar]
- 33.Sen S, Kundu G, Mekhail N, Castel J, Misono K, Healy B. Myotrophpurification of a novel peptide from spontaneously hypertensive rat heart that influences myocardial growth. J Biol Chem. 1990;265(27):16635–43. Sep 25; [PubMed] [Google Scholar]
- 34.Taoka M, Yamakuni T, Song SY, Yamakawa Y, Seta K, Okuyama T, Isobe T. A rat cerebellar protein containing the cdc10/SWI6 motif. Eur J Biochem. 1992;207(2):615–20. doi: 10.1111/j.1432-1033.1992.tb17088.x. Jul 15; [DOI] [PubMed] [Google Scholar]
- 35.Knuefermann P, Chen P, Misra A, Shi SP, Abdel-latif M, Sivasubramanian N. Myotrophin/V-1, a protein up-regulated in the failing human heart and in postnatal cerebellum, converts NFkappa B p50-p65 heterodimers to p50-p50 and p65-p65 homodimers. J Biol Chem. 2002;277(26):23888–97. doi: 10.1074/jbc.M202937200. Jun 28. [DOI] [PubMed] [Google Scholar]
- 36.Young D, Popovic ZB, Jones WK, Gupta S. Blockade of NF-kappaB using IkappaB alpha dominant-negative mice ameliorates cardiac hypertrophy in myotrophin-overexpressed transgenic mice. J Mol Biol. 2008;381(3):559–68. doi: 10.1016/j.jmb.2008.05.076. Sep 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Wang H, Qiu P, Luo H. Proteomic profiling of proteins associated with methamphetamine-induced neurotoxicity in different regions of rat brain. Neurochem Int. 2008;52(1-2):256–64. doi: 10.1016/j.neuint.2007.06.014. Jan. [DOI] [PubMed] [Google Scholar]
- 38.Gupta S, Purcell NH, Lin A, Sen S. Activation of nuclear factor-kappaB is necessary for myotrophin-induced cardiac hypertrophy. J Cell Biol. 2002;159(6):1019–28. doi: 10.1083/jcb.200207149. Dec 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito K, Akazawa H, Tamagawa M, Furukawa K, Ogawa W, Yasuda N, Kudo Y, Liao CH, Yama-moto R, Sato T, Molkentin JD, Kasuga M, et al. PDK1 coordinates survival pathways and beta-adrenergic response in the heart. Proc Natl Acad Sci U S A. 2009;106(21):8689–94. doi: 10.1073/pnas.0900064106. May 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi JH, Yang YR, Lee SK, Kim SH, Kim YH, Cha JY, Oh SW, Ha JR, Ryu SH, Suh PG. Potential inhibition of PDK1/Akt signaling by phenothiazines suppresses cancer cell proliferation and survival. Ann N Y Acad Sci. 2008;1138:393–403. doi: 10.1196/annals.1414.041. Sep. [DOI] [PubMed] [Google Scholar]
- 41.Leaman D, Chen PY, Fak J, Yalcin A, Pearce M, Unnerstall U, Marks DS, Sander C, Tuschl T, Gaul U. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell. 2005;121(7):1097–108. doi: 10.1016/j.cell.2005.04.016. Jul 1. [DOI] [PubMed] [Google Scholar]