

Abstract
Background
Sudden infant death syndrome (SIDS) is one of the leading causes of death during the first year of life. Long QT syndrome (LQTS)-associated mutations may be responsible for 5-10% of SIDS. Recently, we established CAV3-encoded caveolin-3 as a novel LQTS-associated gene with mutations producing a gain-of-function, LQT3-like molecular/cellular phenotype.
Objective
To determine the prevalence and functional properties of CAV3 mutations in SIDS.
Methods
Using PCR, DHPLC, and DNA sequencing, postmortem genetic testing of CAV3 was performed on genomic DNA isolated from frozen necropsy tissue on a population-based cohort of unrelated cases of SIDS (N = 134, 57 females, average age = 2.7 months). CAV3 mutations were engineered using site-directed mutagenesis and heterologously expressed in HEK293 cell lines stably expressing the SCN5A-encoded cardiac sodium channel.
Results
Overall, 3 distinct CAV3 mutations (V14L, T78M, and L79R) were identified in 3/50 black infants (6 month-old male, 2 month-old female, and 8 month-old female), while no mutations were detected in 83 white infants (p value < 0.05). CAV3 mutations were more likely in decedents > 6 months of age (2/12) then infants who died before 6 months (1/124, p value = 0.02). Voltage clamp studies showed that all 3 CAV3 mutations caused a significant five-fold increase in late sodium current compared to controls.
Conclusions
This report provides the first molecular and functional evidence to implicate CAV3 as a pathogenic basis of SIDS. The LQT3-like phenotype of increased late sodium current supports an arrhythmogenic mechanism for some cases of SIDS.
INTRODUCTION
Sudden infant death syndrome (SIDS) is the sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of clinical history.(1) These inexplicable tragedies remain the leading cause of infant death beyond the neonatal period and the third leading cause of infant death overall in the United States.(2-5) In fact, in 2003, there were 2,162 SIDS deaths in the United States according to the National Center for Heath Statistics.(6)
While many pathophysiologic mechanisms for SIDS have been proposed, including respiratory dysfunction, cardiac dysrhythmias, cardiorespiratory instability, and inborn errors of metabolism, definitive pathogenic mechanisms precipitating an infant’s sudden death remain elusive.(7-10) However, Ackerman and colleagues and Crotti and colleagues have provided molecular and functional evidence establishing long QT syndrome (LQTS)-conferring cardiac channel mutations as the pathogenic basis for an estimated 5-10% of SIDS involving Caucasian infants.(11-13)
LQTS is one of the most common cardiac channelopathies affecting an estimated 1 in 3000 persons. While most remain either asymptomatic or experience a sudden fainting episode, LQTS can present with a sentinel event of sudden cardiac death in infancy, childhood, or adulthood.(14) In particular, defects in the SCN5A-encoded cardiac sodium channel (LQT3) have been implicated both in SIDS and autopsy negative sudden unexplained death (SUD) in childhood.(11) In addition, the ethnic specific common cardiac sodium channel polymorphism, S1103Y-SCN5A, has recently been associated with African American SIDS and sudden cardiac death in young black adults.(15,16)
Until recently, it was thought that LQTS was exclusively a cardiac channelopathy. However, this view has been expanded by recent findings that other proteins associated with the ion channel’s pore-forming subunit may alter their function in such a way that cardiac repolarization is disrupted, producing the phenotype of LQTS. For example, defective ankyrin-B, encoded by ANK2, has been identified as causative for type 4 LQTS.(17) In addition, we have recently demonstrated that secondary disturbances in SCN5A channel function via mutations in CAV3-encoded caveolin-3 mimic the LQT3 cellular phenotype in some patients with LQTS (LQT9).(18)
Given the previous identification of SIDS-predisposing SCN5A mutations(11,12) and the new discovery of LQTS-susceptibility mutations in CAV3(18), we hypothesized that CAV3 mutations causing a functional LQT3-like channelopathy may be responsible for some cases of SIDS. Here, we set out to determine the spectrum and prevalence of CAV3 mutations in a large population-based cohort of SIDS.
METHODS
SIDS Cohort
Between September 1997 and December 2000, frozen necropsy tissue from a population-based cohort of unexplained infant deaths (N = 134, 57 females, 83 white, 50 black, 1 Hispanic, average age = 2.7 months) was submitted for postmortem genetic testing in the Sudden Death Genomics Laboratory. Following the general San Diego definition, SIDS is defined as the sudden unexpected death of an infant < 1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history(1). Infants whose death was due to asphyxia or specific disease were excluded. The institutional review board of the Mayo Foundation approved this anonymous necropsy study. As an anonymous study, only limited medical information was available including sex, ethnicity, and age at death. Time of day, medication use, and position at death were not available. By definition, the infant’s past medical history and family history was negative.
CAV3 Mutation Analysis
Genomic DNA was extracted from frozen myocardium (∼25mg) using the QIAamp DNA Mini Kit (Qiagen, Inc, Valencia, California). The entire coding region of the caveolin-3 gene (CAV3, chromosome 3p25, OMIM 601253), was amplified from genomic DNA by polymerase chain reaction, and open reading frame/splice site mutational analysis was conducted by denaturing high-performance liquid chromatography (DHPLC) and direct DNA sequencing as previously described.(19) Primer sequences, PCR conditions, and DHPLC conditions are available upon request.
To be considered a possible SIDS-predisposing mutation, the genetic variant had to i) be a non-synonymous variant (synonymous single-nucleotide polymorphisms were excluded from consideration), ii) that involved a highly conserved residue, iii) absent among 400 reference alleles from 100 healthy white and 100 healthy black control subjects, and iv) resulted in a functionally altered, pro-arrhythmic cellular phenotype. Control genomic DNA was obtained from the Human Genetic Cell Repository sponsored by the National Institute of General Medical Sciences and the Coriell Institute for Medical Research (Camden, New Jersey). Mutations were annotated using the single letter nomenclature whereby L79R for example denotes a non-synonymous SNP producing a missense mutation involving a substitution of leucine (L) by arginine (R) at amino acid 79.
Functional Analysis of Caveolin-3 Mutations
Cloning, mutatgenesis, and voltage-clamp techniques were performed as previously described.(18) The wild-type (WT) human CAV3 453bp coding sequence was cloned from human cardiac cDNA in order to generate the CAV3-V5-6His fusion protein, as previously described.(18) The V14L-, T78M-, and L79R-CAV3 mutations were introduced into the WT construct using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene: La Jolla, California). Primer sequences used for site-directed mutagenesis are available upon request. The mutated V14L-, T78M-, and L79R-CAV3 clones were then sequenced to ensure the presence of the mutations, as well as the absence of other substitutions introduced by the DNA polymerase.
WT-, V14L-, T78M-, and L79R-CAV3 were sub-cloned into the mammalian expression vector pCDNA3 (Invitrogen, Carlsbad, CA) for transfection and expression in human embryonic kidney (HEK 293) cells. The WT or mutant DNA was transiently co-transfected with green fluorescent protein (GFP) at of 1:10 a ratio to allow for selection of transfected cells. After 24 hours under normal growth conditions, the transfected cells were electrophysiologically studied for macroscopic sodium current measurements by standard whole-cell-patch-clamp method at room temperature using an Axopatch 200B amplifier and pClamp8.0 software (Axon Instruments®, Foster City, CA) under previously verified conditions.(18)
Standard activation, inactivation, and recovery protocols were performed, activation and inactivation data were fitted to a charge-voltage Boltzmann equation, and recovery data was normalized to peak sodium current and fitted to a two-exponential standard equation as previously described.(20) The late INa was measured following a 700 msec depolarization from -140 mV to -40 mV after passive leak subtraction and represented by percentage of late INa to peak INa, as previously described (ref). Clampfit 8.2 (Axon Instruments®, Foster City, CA) and Origin 6.0 software (Northampton, MA) were applied for data fitting and statistical analysis. Goodness of fit was determined both visually and by a sum of squares errors. One-way ANOVA was performed to determine statistical significance among multiple means. Statistical significance was determined by a P value < 0.05.
RESULTS
Postmortem Molecular Analysis of SIDS Cohort
This cohort of 134 sudden infant deaths was comprised of 77 males and 57 females with a racial distribution of 83 white, 50 black, and 1 Hispanic infant. The average age at death was 2.7 ± 2.0 months (2.5 + 1.9 months for white infants and 3.1 + 2.2 months for black infants, p-value = NS). Consistent with prior epidemiologic studies of SIDS, the majority of infant deaths occurred prior to 6 months. Only 12/134 infants (9%) died > 6 months of age. Putative SIDS-conferring mutations in CAV3-encoded caveolin-3 were identified in 3/50 (6%) black infants but none of the 83 white infants (p value = 0.05). These CAV3 mutations were present in 2/12 infants who died > 6 months of age compared to one of the 124 infants who died before 6 months (p = 0.02).
Figure 1 details the molecular characterization of the three missense mutations. An abnormal DHPLC elution profile (Figure 1A) and subsequent DNA sequencing (Figure 1B) led to identification of a nucleotide substitution (nucleotide 40 G > C) producing a V14L substitution in a 6-month-old black male. A nucleotide substitution (nucleotide 233 C > T) causing a T78M missense mutation was identified in a 2-month-old black female. Similarly, a nucleotide substitution (nucleotide 236 T > G) resulting in an L79R missense mutation was identified in an 8-month-old black female. All three mutations involved highly conserved residues and were not detected in 400 (200 ethnic matched) reference alleles. Figure 1C depicts the linear topology of caveolin-3 and the localization of these mutations. The V14L mutation resides in the N-terminus of caveolin-3 and the T78M and L79R mutations are localized to the intra-membrane domain. We discovered the T78M mutation recently in 3 unrelated cases of LQTS with a positive family history yet absent in over 1000 control alleles.(18)
Figure 1.
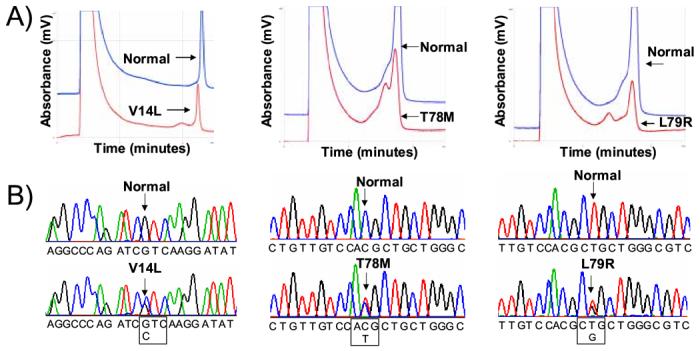

Molecular Characterization of CAV3 Mutations Identified in SIDS Cases. Depicted are the A) DHPLC profiles (normal, blue trace and abnormal, red trace) and B) DNA sequencing chromatograms for SIDS conferring CAV3 mutations V14L, T78M and L79R. C) Illustrated is a topology figure of caveolin 3 that shows location of SIDS-associated mutations in bold, LQT9-associated mutations by *, and myopathy-associated mutations in plain text.
Functional Analysis of SIDS-Associated Missense Mutations in Caveolin-3
When wild-type (WT) CAV3 and the V14L-, T78M-, and L79R-mutant CAV3 were transiently transfected into an HEK cell line stably expressing the most common (Q1077del) variant of SCN5A, robust sodium current (INa) traces were recorded and were not obviously different than SCN5A alone (Figure 2, Table 1). Current density, activation, inactivation, and recovery inactivation were studied by standard protocols. Co-expression with the WT and mutant CAV3 tended to have a more negative midpoint of activation than with SCN5A alone, but this did not reach statistical significance by ANOVA. Steady-state inactivation and recovery from inactivation were studied by standard two-pulse protocols. For inactivation, the two CAV3 mutants tended to have more negative midpoints of inactivation than SCN5A alone, but once again the differences did not reach statistical significance. Recovery from inactivation after a 1 second conditioning pulse showed no difference in the two components of recovery (Table 1).
Figure 2.

Whole Cell Sodium Current (INa) Traces. Whole cell INa traces were recorded with test potentials of 24 ms duration from -120 to +60 mV from a holding potential of -140 mV. Representative INa traces were recorded from pcDNA3(panel A), WT Cav3 (panel B), V14L (panel C), T78M (Panel D) and T79R (panel E) transiently expressed in NaV1.5 stable cell lines
Table 1.
Kinetic parameters for Nav1.5 with or without WT Cav3 and with three Cav3 mutants
| pcDNA3 | Cav3 | V14L | T78M | T79R | |
|---|---|---|---|---|---|
| INa density | |||||
| pA/pF | −330±96 | −439±74 | −506±74 | −351 ±42 | −451±33 |
| Activation | |||||
| V½, mV | −44±1 | −50±5 | −53±6 | −40±1 | −51±8 |
| Slope factor | 4 | 4 | 4 | 4 | 4 |
| Inactivation | |||||
| V½, mV | −83±1 | −85±1 | −90±4 | −84 ±4 | −86±3 |
| Recovery | |||||
| ιf, ms | 2.3±0.2 | 2.7±0.2 | 3.0±0.6 | 3.1±1.3 | 3.1±0.4 |
| ιs, ms | 49±4 | 51±4 | 50±8 | 47±16 | 56±12 |
| Af | 79%±2 | 76%±1 | 76%±2 | 77 %±5 | 81%±4 |
| n | 8 | 6 | 6 | 6 | 4 |
Table 1. The fitted kinetic parameters and INa density from n experiments were averaged and are reported as means ± SD. pcDNA3(An expression vector), WT Caveolin3 (Cav3) and three Cav3 mutants (V14L, T78M and T79R) were transiently expressed in a mammalian cell line stably expressing SCN5A. All parameters were analyzed by one-way ANOVA across pcDNA3, WT Cav3 and three Cav3 mutants. There is no statistically significant for INa density, activation, inactivation from recovery and rate of recovery when pcDNA3, WT Cav3 or the three Cav3 mutants was expressed in NaV1.5 stable cell lines.
Late INa was measured as the leak subtracted inward current remaining at the end of a 775 ms long depolarization (Figure 3A), and then it was normalized and expressed as a percentage of the peak INa for that cell. Examples of current traces for a depolarization to -40 mV (Figure 3B) show INa on an expanded y-axis where the peak INa is off scale. SCN5A alone had the smallest late INa, followed by increasing amounts of late INa for WT CAV3, then the three CAV3 mutants. Summary data (Figure 3B) shows a two-fold increase in late INa for WT CAV compared to SCN5A alone that did not reach statistical significance, and a statistically significant 5-fold increase in late INa for all of the CAV3 mutations found in SIDS. This increase in late INa is comparable to the increases observed for the previously identified LQTS-causing CAV3 mutations and for both LQT3-causing and SIDS-associated mutations in SCN5A.(11, 18, 20,21) This shared molecular phenotype supports the concept that the pathogenic mechanism in these cases of SIDS may have been a LQTS-precipitated, fatal ventricular dysrhythmia.
Figure 3.

Electrophysiological Phenotype of the Cardiac Sodium Channel when CoExpressed with V14I-CAV3, T78M-CAV3, or L79R-CAV3 Mutations. Late INa was measured with a testing -40 mV potential of 700 ms duration for a holding potential of -140 mV. A) Demonstrates the persistent and increased sodium channel late INa current associated with coexpressed wild-type SCN5A and mutant CAV3. B) Summarized is the late INa current represented as a percent (% ± SD) of peak INa. All the values were analyzed by one-way ANOVA across pcDNA3 (SCN5A alone), WT Cav3 (SCN5A + Cav3) and three Cav3 mutants (SCN5A + mutant Cav3). *Statistically significant values (p<0.05). The amount of late current is comparable to other functionally characterized LQT3-associated SCN5A mutations (i.e. ∆KPQ-SCN5A, white box, Nagatomo et al. Am J Physiol 1998; 275(6 Pt 2):H2016-2024)
DISCUSSION
Here, we present the first study demonstrating a novel mechanism of altered cardiac sodium channel (SCN5A) function via mutations in CAV3 that are pathogenic in 2% of SIDS infants studied including 6% of the black infants and none of the white infants. Postmortem genetic testing was performed on a large prospectively collected 3-year, population-based cohort of autopsy negative SIDS identifying 3 (2 novel) non-synonymous mutations that were absent in ethnic matched controls. We previously discovered the T78M mutation in 3 unrelated cases of LQTS with a positive family history. T78M was not seen in over 1000 control alleles.(18) Along with V14I and L79R, this T78M mutation has been functionally characterized for the first time in this study with all 3 mutations showing an SCN5A “gain-of-function” phenotype when co-expressed with wild-type SCN5A. Whether these mutations represent familial or sporadic de novo pathogenic mutations is indeterminate due to the anonymous nature of this postmortem study.
In addition to ethnic-specificity suggested in this study with CAV3 mutations found only in black SIDS infants, we have also observed that two of the CAV3 mutations were discovered among the 12 “older” SIDS cases. Over 90% of “classical” SIDS occurs before 6 months as evident in this cohort of SIDS cases as well. It is possible that infant deaths occurring after this vulnerable window may stem from fundamentally distinct substrates. Nevertheless, the designation of SIDS would be applied since the death occurred prior to the infant’s first birthday. Including the previously published channel mutations, we have now found primary or secondary channelopathy-causing mutations for one-third of these older infants compared to less than 5% of those infants who died before 6 months.
The functional alteration of SCN5A resulting from mutation of CAV3 is presumed to be the cause of SCD in these infants because of the shared molecular phenotype of increased late INa that has been well described for mutations in SCN5A that cause type 3 long QT syndrome (LQT3). Increased late INa has also been implicated previously as a mechanism for SIDS for the mutations A997S and R1826H in SCN5A(11), and also late INa has been associated with an increased risk of SIDS for the common polymorphism S1103Y found in blacks.(16) Moreover, we recently found mutations in CAV3 (including the T78M-CAV3 mutation described here) in LQTS, and functional studies of these LQTS-associated mutations demonstrated that coexpression of CAV3 mutations with normal SCN5A resulted in an increase in the late sodium current consistent with the sustained sodium current found in patients with LQT3 and cases of SIDS with SCN5A mutations.(11, 18)
Recent evidence has shown that caveolin-3 co-localizes with the cardiac sodium channel (18, 22) and interacts with the dystrophin-glycoprotein complex (DGC) to facilitate proper membrane localization of ion channels such as SCN5A.(23) CAV3 co-localizes with SCN5A and plays an important role in the regulation of cardiac sodium channel current amplitude.(18, 22) CAV3 has also been found to mediate the sympathetic response via binding to G-proteins, activating cAMP-dependent protein kinase A (PKA) and phospholipase C (PLC)-activated protein kinase C (PKC), which both directly phosphorylate SCN5A and regulate its function.(24) CAV3 plays a role in prolonging action potential by binding to calmodulin (CaM), which binds SCN5A and increases its slow-inactivation kinetics in response to regional increase in the concentration of calcium.(25) These findings all indicate that CAV3 plays an important role in the functional regulation of the cardiac sodium channel and thus may be involved in the pathogenic mechanisms causing sudden death.
Caveolin-3 (CAV3) is the major component of caveolae which are omega-shaped invaginations of the plasma membrane in both cardiac and skeletal muscle.(26) Besides this present study and our previous study detailing CAV3 as a novel LQTS-susceptibility gene(18), five pathogenic mutations in CAV3 have been identified previously.(26,27) These mutations were associated with extreme phenotypic variability in association with phenotypes of autosomal dominant limb girdle muscular dystrophy (LGMD1C), persistent elevated levels of serum creatine kinase (hyperCKemia) without muscle weakness, distal myopathy, and rippling muscle disease, of which cardiac abnormalities including atrioventricular (AV) conduction disturbances and arrhythmias may be observed.(28-32) Our current findings suggest that sudden infant death due to LQTS should be added to this compendium of phenotypic variability observed in CAV3 mutation carriers. A clinical cardiologic assessment including an electrocardiogram may be warranted in patients with a caveolin-3 mediated muscle disease.
While age-dependent muscle weakness and/or myopathy manifestation may be observed in some individuals with CAV3 mutations, by definition these SIDS infants had a negative autopsy and thus no gross or microscopic evidence of skeletal myopathy. In addition, the patients with LQTS with LQT3-like conferring CAV3 mutations reportedly had no evidence of skeletal myopathy or muscle weakness.(18) However, Ng et. al. reported that cardiac arrhythmias may serve as presenting symptoms in young patients with unrecognized limb girdle muscular dystrophy.(31) The muscle pathological picture in caveolin-mediated myopathy may be relatively benign and show mild or moderate myopathic features.(33)
The present study characterizes the molecular phenotype by expression of the ion channels and associated proteins in a non-cardiac muscle cell line, which does not necessarily recapitulate all the key components of the macromolecular complex found in the more native environment. Although this experimental method has been the standard for defining function of ion channel abnormalities in hundreds of previous studies, the limitation of this method must be kept in mind, and the possibility of different or additional abnormalities might be described when these proteins and the mutations residing therein are examined in native cardiac cells.
CONCLUSION
This large population-based autopsy series of SIDS has now provided molecular and functional evidence to implicate CAV3 as a novel candidate gene in the pathogenesis of SIDS, particularly among older infants (> 6 months). Together with our previous reports establishing long QT syndrome (LQTS)-conferring cardiac channel mutations as a pathogenic basis of SIDS, we now estimate that 5-10% of SIDS cases and up to one-third of “older” SIDS may be attributed to underlying mechanisms associated with potentially lethal arrhythmogenic disorders such as LQTS.
Footnotes
LBC, BY, TK, and DJT are co-equal first authors.
Financial Support: Dr. Ackerman’s research program is supported by a Mayo Foundation Clinical Research Award, the Dr. Scholl Foundation, the CJ Foundation for SIDS, an Established Investigator Award from the American Heart Association, and the National Institutes of Health (HD42569).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, Cutz E, Hanzlick R, Keens TG, Mitchell EA. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–238. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- 2.American Academy of Pediatrics AAP Task Force on Infant Positioning and SIDS: Positioning and SIDS. Pediatrics. 1992;89:1120–1126. [PubMed] [Google Scholar]
- 3.Willinger M, Hoffman HJ, Hartford RB. Infant sleep position and risk for sudden infant death syndrome: report of meeting held January 13 and 14, 1994; National Institutes of Health, Bethesda, MD. Pediatrics. 1994; pp. 814–819. [PubMed] [Google Scholar]
- 4.Gibson E, Cullen JA, Spinner S, Rankin K, Spitzer AR. Infant sleep position following new AAP guidelines. American Academy of Pediatrics. Pediatrics. 1995;96:69–72. [PubMed] [Google Scholar]
- 5.Dwyer T, Ponsonby AL, Blizzard L, Newman NM, Cochrane JA. The contribution of changes in the prevalence of prone sleeping position to the decline in sudden infant death syndrome in Tasmania. Jama. 1995;273:783–789. [PubMed] [Google Scholar]
- 6.Martin JA, Kochanek KD, Strobino DM, Guyer B, MacDorman MF. Annual summary of vital statistics--2003. Pediatrics. 2005;115:619–634. doi: 10.1542/peds.2004-2695. [DOI] [PubMed] [Google Scholar]
- 7.Guntheroth WG. Theories of cardiovascular causes in sudden infant death syndrome. J Am Coll Cardiol. 1989;14:443–447. doi: 10.1016/0735-1097(89)90200-3. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, Grancini F, Marni ED, Perticone F, Rosti D, Salice P. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709–1714. doi: 10.1056/NEJM199806113382401. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, Richard TA, Berti MR, Bloise R. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343:262–267. doi: 10.1056/NEJM200007273430405. [DOI] [PubMed] [Google Scholar]
- 10.Hunt CE, Hauck FR. Sudden infant death syndrome. CMAJ Canadian Medical Association Journal. 2006;174:1861–1869. doi: 10.1503/cmaj.051671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ackerman MJ, Siu BL, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. Jama. 2001;286:2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- 12.Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. doi: 10.1016/j.cardiores.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Crotti L, Arnestad M, Insolia R, Pedrazzini M, Rognum T, PJ S. The Role of Long QT Syndrome in Sudden Infant Death Syndrome. Eur Heart J. 2005;26:127. [Google Scholar]
- 14.Ackerman MJ, Clapham DE. Ion channels--basic science and clinical disease. N Engl J Med. 1997;336:1575–1586. doi: 10.1056/NEJM199705293362207. [DOI] [PubMed] [Google Scholar]
- 15.Burke A, Creighton W, Mont E, et al. Role of SCN5A Y1102 polymorphism in sudden cardiac death in blacks. Circulation. 2005;112:798–802. doi: 10.1161/CIRCULATIONAHA.104.482760. [DOI] [PubMed] [Google Scholar]
- 16.Plant LD, Bowers PN, Liu QY, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. Journal of Clinical Investigation. 2006;116:430–435. doi: 10.1172/JCI25618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohler PJ, Schott J-J, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 18.Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 19.Ackerman MJ, Tester DJ, Jones G, Will MK, Burrow CR, Curran M. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clinic Proceedings. 2003;78:1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 20.Nagatomo T, Fan Z, Ye B, et al. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T DeltaKPQ Na+ channels. Am J Physiol. 1998;275:H2016–2024. doi: 10.1152/ajpheart.1998.275.6.H2016. [DOI] [PubMed] [Google Scholar]
- 21.Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 22.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude.[see comment] Circulation Research. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- 23.Head BP, Patel HH, Roth DM, et al. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. Journal of Biological Chemistry. 2005;280:31036–31044. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 24.Feron O, Kelly RA. Gaining respectability: membrane-delimited, caveolar-restricted activation of ion channels. Circ Res. 2002;90:369–370. doi: 10.1161/01.res.0000012911.90134.ef. [DOI] [PubMed] [Google Scholar]
- 25.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Galbiati F, Razani B, Lisanti MP. Caveolae and caveolin-3 in muscular dystrophy. Trends in Molecular Medicine. 2001;7:435–441. doi: 10.1016/s1471-4914(01)02105-0. [DOI] [PubMed] [Google Scholar]
- 27.Vorgerd M, Ricker K, Ziemssen F, et al. A sporadic case of rippling muscle disease caused by a de novo caveolin-3 mutation. Neurology. 2001;57:2273–2277. doi: 10.1212/wnl.57.12.2273. [DOI] [PubMed] [Google Scholar]
- 28.Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nature Genetics. 1998;18:365–368. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- 29.Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology. 1373;54:1373–1376. doi: 10.1212/wnl.54.6.1373. [DOI] [PubMed] [Google Scholar]
- 30.Tateyama M, Aoki M, Nishino I, et al. Mutation in the caveolin-3 gene causes a peculiar form of distal myopathy.[erratum appears in Neurology 2002 Mar 12;58(5):839 Note: Itoyoma Y [corrected to Itoyama Y]] Neurology. 2002;58:323–325. doi: 10.1212/wnl.58.2.323. [DOI] [PubMed] [Google Scholar]
- 31.Ng W, Lau CP. Cardiac arrhythmias as presenting symptoms in patients with limb-girdle muscular dystrophy. International Journal of Cardiology. 1997;59:157–160. doi: 10.1016/s0167-5273(96)02911-7. [DOI] [PubMed] [Google Scholar]
- 32.van der Kooi AJ, de Voogt WG, Barth PG, et al. The heart in limb girdle muscular dystrophy. Heart. 1998;79:73–77. doi: 10.1136/hrt.79.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fulizio L, Chiara Nascimbeni A, Fanin M, et al. Molecular and muscle pathology in a series of caveolinopathy patients. Human Mutation. 2005;25:82–89. doi: 10.1002/humu.20119. [DOI] [PubMed] [Google Scholar]