


Abstract
Throughout evolution, eukaryotic genomes have been invaded by transposable elements (TEs). Little is known about the factors leading to genomic proliferation of TEs, their preferred integration sites and the molecular mechanisms underlying their insertion. We analyzed hundreds of thousands nested TEs in the human genome, i.e. insertions of TEs into existing ones. We first discovered that most TEs insert within specific ‘hotspots’ along the targeted TE. In particular, retrotransposed Alu elements contain a non-canonical single nucleotide hotspot for insertion of other Alu sequences. We next devised a method for identification of integration sequence motifs of inserted TEs that are conserved within the targeted TEs. This method revealed novel sequences motifs characterizing insertions of various important TE families: Alu, hAT, ERV1 and MaLR. Finally, we performed a global assessment to determine the extent to which young TEs tend to nest within older transposed elements and identified a 4-fold higher tendency of TEs to insert into existing TEs than to insert within non-TE intergenic regions. Our analysis demonstrates that TEs are highly biased to insert within certain TEs, in specific orientations and within specific targeted TE positions. TE nesting events also reveal new characteristics of the molecular mechanisms underlying transposition.
INTRODUCTION
Transposable elements (TEs) are mobile genomic sequences that have played an important role in animal genome evolution. TEs have undergone an enormous expansion in mammals; they constitute 40% of the mammalian genome versus 3–20% of the genomes of nematode, fly, pufferfish and chicken (1–5). TEs are classified by their mode of propagation. Short and long interspersed repeat elements (SINE and LINE, respectively) and retrovirus-like elements with long terminal repeats (LTR) are propagated by reverse-transcription of an RNA intermediate. In contrast, DNA transposons are propagated via a direct ‘cut-and-paste’ mechanism (6,7). Insertion of TEs may cause genomic deletions and duplications (7). More than 25 human genetic diseases are attributed to TE-related rearrangements (8). Since a new TE insertion is potentially harmful, an elaborate cellular counterattack has coevolved with TEs to suppress transposition and retrotransposition events. Expression inhibition through DNA methylation, RNA interference and RNA editing are just a few of the cellular mechanisms used to restrain TE proliferation (7). Another support for the importance of tight cellular regulation over TE activity is the over-expression of retroelements in many types of cancers (9–11). Other works have described the ability of TEs and their related factors to generate new functional genetic elements, such as genes, exons, introns and regulatory sequences (7,12–22).
Analysis of the insertions of TEs into previously transposed elements, also known as nested TEs, has proven highly informative in exploring evolutionary and molecular aspects of the genome. Giordano et al. (23) determined the evolutionary history of mammalian TEs using the simple and elegant rule that newer TEs are able to insert into older TEs, but not vice versa. Several DNA transposons were shown to be active in the primate lineage due to insertion into primate specific TEs (24). Nested TE analysis can also serve as a tool for measuring the conservation of the targeted TE sequence, since a conserved TE will be protected from insertions of other TEs into it. Such analysis indicates that L1 elements are less frequently interrupted in regions displaying X-inactivation than in other genomic regions, supporting L1 role in X inactivation (25). As the pre-integration sequence of a TE can be inferred from the consensus sequences of the targeted TE, Ichiyanagi et al. (26) were able to demonstrate the possible mobility pathways for non-LTR retrotransposons. Non-mammalian genomes have also been studied using nested TEs: Bergman et al. (27) were the first to study TE nesting in flies and Kriegs et al. (28) reconstructed gamebird phylogenetic tree using nested chicken repeats 1 information.
We were interested in finding out, in large scale, whether TEs tend to insert within specific sequences. We constructed a large dataset of human-nested TEs which conserve the original position and sequence of TE insertion within the targeted TE. Using this dataset, we demonstrated that insertions of TEs from different classes occur at ‘hotspots’ within the targeted TEs. Some of the hotspots are intriguing since they represent non-canonical TE integration sites. We also devised a method based on our dataset to identify new sequence motifs favored for integration of MaLR and ERV1 TE families, and to refine the sequence motifs favored by hAT and Alu TE families.
What underlies the mammalian TEs expansion is yet unclear. The identification of favorable TE integration sites within many transposed elements led us to hypothesize that some human TEs used already transposed elements to facilitate their genomic proliferation. Here we demonstrate that in general, young TEs insert randomly in intergenic regions. However, there are hundreds of TEs which show a clear and significant bias to insert within older TEs, and considerably fewer cases of TEs avoiding insertion within TEs. In addition, we demonstrate that certain TEs prefer to insert into specific TEs and in specific orientation. Overall, our analysis provides a deeper understanding of the evolution of human TEs, TE insertion preferences and the molecular mechanisms by which TEs proliferate.
MATERIALS AND METHODS
Construction of exact TE insertions database
The database was implemented using MySql server. We downloaded nestedRepeats table from the UCSC table browser (29) as separate blocks, that is, separated fragments of interrupted repeats. We used only TEs (repeats from classes SINE, LINE, DNA and LTR). For each fragment, we linked start and end positions over its consensus sequence, as was determined by RepeatMasker (rmsk). Two fragments of an interrupted TE were joined together as a targeted TE if they conformed to the following requirements:
the blocks were consecutive (e.g. blocks 1 and 2) with the same repeat name,
the second fragment was the exact continuation of the first fragment over the TE consensus sequence. For example, if the first fragment ended at position 100 over the TE consensus sequence, then the second TE started at position 101 over the TE consensus sequence, and
the distance between the two fragments over the chromosome was maximally 20 000 nt.
For each pair of targeted TE fragments, we associated an inserted TE, which could itself serve as a target for another TE. The positions of the inserted TE were taken from the rmsk table in the UCSC table browser. The inserted TE was the closest downstream TE after the first interrupted fragment and the closest upstream TE before the second interrupted fragment. An inserted TE is serving also as a targeted TE if it was divided into two TE fragments carrying the same TE name.
The result was a database of 296 209 exact TE insertions. In 96% of the exact insertions, the inserted TE was located between the two interrupted parts of the targeted TE without any spacer nucleotides. It should be noted that, on the one hand, this dataset is more extensive than the one used in the next analysis (over-/under-represented nested TEs), since we examined all nesting events in the entire human genome, and not only those of new TEs into old TEs in intergenic regions; but, on the other hand, it is more conservative, since only exact insertions were retained. We did not search for TSDs during the construction of the exact insertion database. However, TSDs as long as 10 bp were identified in the resulting database (identification of TSDs is described below). This seems to be the result of RepeatMasker failing to precisely align the edges of fragmented repeats. TSDs should have been characterized by position overlap within the consensus sequence in the two fragments of the targeted TE (e.g. upstream fragment is aligned to positions 1–100 of the TE and downstream fragment is aligned to positions 90–200 of the TE), instead of having consecutive positions in the two fragments. However, such position overlap (up to 30 bp long) was noticed in only ∼50 000 nested TE events versus ∼300 000 exact insertions events that we compiled, ∼124 000 of these have recognized TSDs longer than 3 bp.
Statistical analysis of TE hotspots identification
Our null hypothesis was that TEs are inserted in target TEs as expected according to the position distribution of the targeted TEs. This distribution was calculated for each target TE using the nestedRepeat blocks table (fragments of target TEs) joined with rmsk alignment data (see above). The expected number of insertions for each insertion type in each position was calculated as follows:
where Ci is the number of nestedRepeats blocks covering position i of the targeted TE, 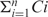 is the total number of blocks covering any position of the targeted TE, and TI is the total number of insertions from the specific insertion type. By using an empirical background model based on only TEs that were actually fragmented, rather than using all TEs in the human genome, we achieved a more accurate model. Our model is undoubtedly better than assuming the same probability of insertion for all targeted TE positions (uniform distribution), as this does not take into account the fact the some target TEs (such as LINEs) tend to be truncated, causing higher genomic prevalence of one TE end (data not shown).
is the total number of blocks covering any position of the targeted TE, and TI is the total number of insertions from the specific insertion type. By using an empirical background model based on only TEs that were actually fragmented, rather than using all TEs in the human genome, we achieved a more accurate model. Our model is undoubtedly better than assuming the same probability of insertion for all targeted TE positions (uniform distribution), as this does not take into account the fact the some target TEs (such as LINEs) tend to be truncated, causing higher genomic prevalence of one TE end (data not shown).
The observed insertions for each insertion type in each position over the target TE were retrieved from the exact insertions database. We then divided the target TE into six equal position bins along the target TE sequence, starting from the first observed position of insertion and ending in the last observed position of insertion. Six was selected as the number of bins since this offered high resolution of insertion hotspots and allowed at least five expected insertions in each bin (χ2 test requirements). The last perquisite is critical especially for the rare insertion types in the genome. We discarded the ends of the target TE consensus sequence as potential insertion sites since we assumed that insertions in such regions would not be properly identified by RepeatMasker, since one of the fragments of the target TE would be too short. The observed and expected insertions numbers in each bin were calculated by summing the values for all positions in the bin position range. We then used χ2 tests to determine whether the observed and expected distributions for each insertion type were similar. We demanded that the expected number of insertions in each bin will be at least five, as part of χ2 test prerequisites. The P-values were corrected for multiple hypotheses testing using FDR (30). Supplementary Figure S6 shows examples for observed and expected exact insertions with their corresponding P-values. The background model for expected insertions considers known features of the target TE, such as the common LINE 5′ truncation (31) (Supplementary Figure S6B). The prerequisite of a minimum of five expected insertion in each bin removed many of the less abundant insertion types. In order to circumvent this, without decreasing the number of bins, we joined together all insertion types with a common targeted TE type and orientation, where the inserted TE type was from the same TE family. Histograms of the observed insertions positions for statistically significant unclustered insertion types and insertion types clustered by inserted TE family are presented in Supplementary Dataset S3 and S4, respectively. The statistical analysis was performed using the R programming language.
Position-specific scoring matrices generation and visualization
The sequences flanking inserted TEs in all exact insertions were downloaded using Galaxy (32). Position-specific scoring matrices (PSSMs) were built for all combinations of an inserted TE, a target TE family and an orientation using a perl script. PSSMs were visualized using pictograms based on the seqLogo library (33) in R. Information content was used as a measure for the strength of the motif in each pictogram, where high information content denotes significant deviation from a background model. The background model in this case is defined as an equal proportion (0.25) for each of the four nucleotides.
Target-site duplication (TSD) identification
The size of the TSD was calculated in a relatively conservative way using a Perl script which searches for the longest possible identical sequences flanking the inserted TE, allowing one mismatch for sequences longer than 3 bp and not allowing any mismatches for sequences equal to or shorter than 3 bp.
Construction of database of nested TEs in unique human intergenic regions
Using Galaxy (32) we retrieved both chainSelf and genomicSuperDups tables from the UCSC table browser (29) of human genome build hg18, which represents alignment of the human genome against itself and duplications of >1000 bases, respectively. These genomic regions were merged to yield the duplicated part of the human genome. The unique part of the genome, corresponding to 2.17 Gb, was extracted by discarding the duplicated region. We then removed known genes from the unique genomic region. This was done by merging positions of all genes taken from the known Gene table (‘UCSC genes’) and subsequently discarding these positions. The result was the unique 1.29 Gb intergenic regions of the genome. We then retrieved all TEs from rmsk table in this region. TEs are rmsk elements from the classes SINE, LINE, LTR and DNA. The intergenic TEs were also divided to those that were up to 10 Kbp from genes and those that were located futher from genes. These TEs were joined with interrupted TEs from nestedRepeats table. We required that the positions of the inserted TE were completely contained in the positions of the interrupted TE. TEs that were inserted within a very large TE (>20 kb) were filtered out, since the interrupted TEs were unreliable based on manual inspection. It should be noted that the resulting database contains all TE nesting events and not just exact insertions. The entire database is located in Dataset S5.
Statistical analysis of insertions of new TEs into old TEs
We divided 360 different human TE types into new and old TEs based on Supplementary Table S2 from Gioradano et al. (23). The first 299 TEs of this table were considered old TEs and the last 61 TEs were considered as new TEs. Although the chronology of these TEs is also based on nested TEs, i.e. a newer TE will insert into older TE but not vice versa, the dating algorithm ignored new TE insertions into non-TE regions and therefore it does not follow that a given new TE would preferentially nest within a given older TE. The division into ‘new TEs’ and ‘old TEs’ was done for several reasons. First, ignoring the age of TEs, some of which were acting in non-overlapping periods, would yield an incorrect excess of under-represented insertion types in large-scale nested TE analysis, in which a certain group of TEs are completely depleted in another group of TEs, with the inserted TEs being older than the targeted TEs. Second, the selected new TEs were active in a recent evolutionary period, during the last 40-45 million years, in the anthropoid primate lineage. In this period, our ancestral genome was already rich in older inactive transposed elements that could have served as potential target sites for new TE insertions. Third, due to the high DNA sequence identity among anthropoids [average 93% human–macaque sequence identity (34)], we assume that the expansion of anthropoid genome resulted primarily from TE integration. Therefore, by removing the new TEs fraction from the current human genome we can estimate the ancestral anthropoid genome structure and size before the proliferation of these elements. Assuming that genome expansion was mainly the result of TE insertions is less reliable for more ancient ancestral genomes (e.g. the ancestral mammalian genome), which should be quite different from the current human genome. Finally, using only relatively new TEs as the inserted elements, allows us to assume that these elements were mainly inserted into older elements rather than being fragmented by other newer or contemporary TEs.
For each insertion type, comprising a new TE, old TE and orientation of insertion, we calculated observed and expected number of insertions. The observed value was simply the number of insertions of the new TE into the old interrupted TE in the specific orientation in the unique intergenic part of the human genome (see above). The expected number of insertions of a new TE into an old TE within the ancestral anthropoid genome (before new TEs were active) was calculated as follows:
where C0 is defined as the specific old TE coverage (the fraction of the genome covered by this TE), CTN is defined as the coverage of all new TEs (so C0/(1 − CTN) represents the old TE coverage in the genome before new TEs activity period), and Total InsertionsN is defined as the total number of the specific new TE insertions inside and outside of other TEs. All three variables above were calculated exclusively for the unique intergenic part of the human genome. A χ2 test was carried out for each insertion type where O1 is observed insertions of new TE in old TE in unique intergenic regions, O2 is observed insertions of new TE in non-old TEs in unique intergenic regions, E1 is expected insertions of new TE in old TE in unique intergenic regions, and E2 is expected insertions of new TE in non-old TE in unique intergenic regions (degrees of freedom = 1). The P-value was corrected for multiple hypotheses using the Bonferroni correction. The strong tendency toward over-representation (see ‘Results’ section) was not sensitive to P-value cutoffs or to fold-change thresholds. Thus, when using fold change of 1.5 as a threshold and correcting P-value with the false discovery rate (FDR) there were 1494 overrepresented insertion types and only 418 under-represented insertion types. The entire statistical analysis was performed using the R programming language. The AluSx star graph was constructed using igraph and tkplot libraries (35).
For the purpose of identifying insertion orientation bias (insertions in sense versus antisense), we calculated the number of insertions for each TE family within each TE family in the unique intergenic part of the genome. We ignored self insertions of the same TE type in the sense orientation since it would be difficult to differentiate between these insertions and a TE which was fragmented multiple times (each of the internal fragments maybe mistakenly considered as a self insertion in sense). We also ignored nested repeats from the type ‘RepeatName’-int within ‘RepeatName’, whereas ‘RepeatName’ is a type of an LTR retrotransposon and ‘RepeatName’-int is the alignment of the internal part of the TE type, since these are not real TE nesting events.
Location of Alu elements within genes
Alu elements from rmsk table (elements of repeat family Alu) from UCSC table browser were annotated as located in sense orientation, antisense orientation or none of these, with regard to UCSC known genes table.
RESULTS
Hotspots for TE insertions within targeted TEs
TEs have expanded throughout evolution and currently account for 45% of the human genome. In many cases, a TE was inserted into DNA that originates in previously transposed elements. We hypothesized that the TE nesting phenomenon may occur due to specific sequence motifs, located within the targeted TEs that serve as preferred integration sites for other TEs. As a first step for examining this hypothesis at the global level, we set out to determine whether TEs tend to have specific locations, or hotspots, along the targeted TEs into which they preferentially insert. To address this question, we constructed a large dataset of human-nested TE events which we termed ‘exact TE insertions’. In this dataset, we included all nested TE events in which the two parts of the targeted TE that flank the inserted TE, constituted an exact continuation of each other when aligned against their consensus sequence. An example for a typical exact insertion event versus a non-exact insertion event is illustrated in Supplementary Figure S1. The advantage of using exact TE insertions is that they enable relatively precise reconstruction of the original genomic sequence into which a TE was inserted. We assembled a dataset of nearly 300 000 exact insertion events in the human genome (Supplementary Dataset S1). Table 1 shows the most common exact insertions events, with Alu elements being the most prevalent inserted TEs, whereas L1 sequences are the most prevalent targeted TEs, representing 75 and 46% of the exact insertions, respectively. To analyze this data, we grouped together similar events, in two steps. First, exact insertion events with shared inserted TE type, targeted TE type, and insertion orientation were joined together into groups named ‘insertion types’. The insertion orientation was defined as ‘sense’ when the two TEs shared the same DNA strand orientation and as ‘antisense’ otherwise. An example for an insertion type is the group of 1046 AluSx (an Alu subtype) exact insertions into MIRb in sense (Supplementary Table S1). Next, insertion types were clustered by common inserted TE families (a TE family contains several different TE types). The latter step was performed to increase the robustness and power of our statistical analysis, based on the reasoning that many TEs from the same TE family share the same mechanism of transposition. An example for a clustered insertion type is the group of 2957 Alu (from all subtypes) exact insertions into MIRb in sense (Supplementary Table S2). For each clustered insertion type, we assessed whether the insertions of the inserted TE occurred according to an empirical background model, based on χ2 tests corrected for multiple testing (see ‘Materials and Methods’ section, and Supplementary Tables S1 and S2).
Table 1.
Twenty major exact TE nesting events in the human genome
| Inserted TE family | Targeted TE family | Number of appearances in the human genome |
|---|---|---|
| Alu | L1 | 104 308 |
| Alu | Alu | 18 821 |
| Alu | MaLR | 18 537 |
| Alu | L2 | 18 158 |
| Alu | ERV1 | 14 583 |
| Alu | MER1_type | 13 522 |
| MaLR | L1 | 12 190 |
| Alu | MIR | 11 071 |
| Alu | MER2_type | 9814 |
| L1 | L1 | 6698 |
| Alu | ERVL | 5133 |
| ERV1 | L1 | 4140 |
| MaLR | L2 | 3751 |
| MER2_type | L1 | 3295 |
| MaLR | MaLR | 3207 |
| MER1_type | L1 | 2453 |
| MaLR | MIR | 2367 |
| MaLR | ERVL | 1842 |
| MER1_type | L2 | 1776 |
| Alu | CR1 | 1746 |
All TEs belonging to the same TE family (according to rmsk) are grouped.
The majority, 82% (654/799) of the insertion types clustered by common inserted TE family were significantly different from their expected distribution. These insertion types included members of all four TE classes (DNA, LTR, SINE, LINE). The same trend was observed for the unclustered insertion types, mostly consisting of insertions of different Alu subtypes into other TEs: 85% of these were significantly different from their expected distribution. Clear ‘hotspots’ for the insertion of certain TEs were detected. For example, AluSx (Figure 1A) tended to insert immediately after positions 72, 183–186, 5824 and 6142, and 102 of MER5A1, MLT1C, L1MB3 and MER45A, respectively (the first three in the sense orientation and the last one in the antisense orientation). These hotspots do not necessarily correspond with known integration sites. For example, the known Alu integration site, 5′-TTAAAA-3′, does not form part of MER5A1 and MLT1C consensus sequence (Supplementary Table S3). The same hotspots were also utilized when other types of Alus inserted within these targeted TE (Supplementary Dataset S3). Additionally, as can be seen for insertions of both MaLR (Figure 1B) and MER1 (Figure 1C) inserted TE family-clustered insertion types, the hotspots for TE nesting are not restricted to Alu insertions. Interestingly, insertions of MER1 (hAT transposons) use the same hotspots, positions 154 and 160, when inserted into MIR3 in both sense and antisense orientations (Figure 1C, the two left bar graphs).
Figure 1.

Several statistically significant hotspots for TE nesting events involving different types of TEs.In all figures the X axis represents positions over the consensus sequence (the position just upstream to the insertion) of the targeted TE and the Y axis represents the number of exact insertions in those positions within the human genome, (A) AluSx (Alu retrotransposons) insertions (B) MaLR (LTR retrotransposons) insertions and (C) MER1 (DNA transposons) insertions.
Recently, Abrusan et al. (25) showed that most Alus tend to harbor insertions of other Alus in the polyA stretch of the linker region between the two Alu arms. Our analysis supports these results (Figure 2A) and also revealed a more striking phenomenon. Insertions of Alu elements within Alus tend to occur (i) within a single, specific prominent hotspot, directly after the A-rich linker (position 133 within the consensus sequence of most of the Alu subfamilies), and (ii) in the sense orientation. Due to this prominent hotspot, Alu self insertions deviated most significantly from the expected insertion distribution (Supplementary Tables S1 and S2). This result was further supported by alignment of the sequences flanking the inserted Alu, obtained from all 15 759 Alu self insertions in sense in the human genome. Visualization of this alignment via a pictogram (Figure 2B, lower part) revealed a striking similarity to the Alu consensus sequence (Figure 2B, upper part) in the region downstream to the A-rich linker, clearly confirming that this single position is favored for Alu self insertions. It is noteworthy that the entire Alu consensus sequence does not contain the canonical Alu integration site (Supplementary Table S3). The selection of this hotspot by Alu is explained using a revised molecular model for Alu integration (see ‘Discussion’ section).
Figure 2.

Alu insertions within Alu DNA in the human genome. (A) Bar graph describing all exact insertions of Alu family members into AluJo in sense (left) and antisense (right). The internal polyA stretch is located between positions 118 and 136. The X axis describes the exact insertion position (the position just upstream to the integration site) over the AluJo consensus sequence. The Y axis describes the number of exact insertions in the human genome. (B) In the bottom is a pictogram of the flanking sequences of the inserted Alu. The flanking sequences are located over the targeted Alu. The height of each letter is proportional to the frequency of the corresponding base at the given position. The nucleotides in each position appear from top to bottom, sorted by descending frequency. The pictogram was created using all 15 759 self Alu insertions in sense orientation in the human genome. The cyan dashed vertical line denotes the Alu insertion position. The sequence upstream of this line is upstream of the insertion site and the sequence downstream of the line is downstream of the insertion site. A sequence alignment of the pictogram consensus sequence to the AluJo consensus sequence [retrieved from Repbase (52)] is denoted above the pictogram. The TSD surrounding the insertion position tends to be ‘TT’ or ‘TTA’, yielding a somewhat degenerate sequence upstream of the insertion (e.g. T or A in position−1).
Identification of novel TE integration sequence motifs
The existence of a hotspot for insertion of a certain TE within a targeted TE sequence may be due to existence of a preferred TE integration site in the hotspot position. By examining the insertion sites of the same TE within different targeted TEs of completely different DNA sequence, we sought to infer the favored DNA integration motifs for particular inserted TEs. To obtain high-confidence integration sites, we constructed ‘insertion profiles’ for each specific inserted TE as follows. Similar to our approach above, we created groups named ‘combined insertion types’ that included all exact insertion events for a particular inserted TE type, targeted TE family (containing several targeted TE types) and an insertion orientation. An example for a combined insertion type is the group of 24 873 exact genomic insertions of AluSx (a specific TE type) into L1 (a TE family) in the sense orientation. We filtered out all low-confidence combined insertion types that occurred fewer than 10 times within the genome. We next extracted the 20 nucleotides upstream and downstream of the insertion sites, and constructed PSSM for each combined insertion type. The PSSM represents the frequency of each of the four possible nucleotides (A, C, T and G) at each position within the 40 nucleotides flanking the insertion. For each inserted TE, we then generated a new PSSM, which we termed ‘insertion profile’ by unifying the PSSMs of combined insertion types of the same inserted TE. This was done by averaging the information content of each position across the PSSMs of the relevant combined insertion types. An example for insertion profile construction is depicted in Figure 3. We used only high confidence insertion profiles derived from at least three different combined insertion types of a particular inserted TE. Thus, these insertion profiles reflect the sequence motifs into which a TE preferentially integrates. The insertion profiles are highly informative as they are based on many TE insertion events into several types of targeted TE sequences in different orientations. The requirement for multiple targeted TE sequences in the insertion profile reduces the bias derived from any preference of the inserted TE for a specific targeted TE. We used information content as a measure of the informativeness of a motif (see ‘Materials and Methods’ section).
Figure 3.

Example of an insertion profile construction. PSSMs were constructed for the exact insertions of MER33 (a DNA transposon) into different types of TE families in both orientations. The arrows from MER33 point to six different pictograms representing PSSMs of combined insertion types. The number in parenthesis denotes the number of occurrences of the combined insertion types within the genome (e.g. 36 exact insertions of MER33 into L1 in sense). Only combined insertion types of more than 10 genomic occurrences were considered. The height of each nucleotide in the pictogram is proportional to its information content in the given position. The cyan dashed vertical line denotes the MER33 insertion position. Following construction of the PSSMs, we constructed the insertion profile by averaging all PSSMs of a given inserted TE. The unified insertion profile of MER33 is shown on the bottom.
This analysis uncovered several known and unfamiliar TE integration sites. An example of a known integration site that was precisely reconstituted by our analysis is the Tc1-Mariner integration site. The members of this superfamily strongly prefer to insert adjacent to 5′-TA-3′, generating TA target site duplication (TSD) (36). Our insertion profiles for different members of this superfamily indeed revealed flanking ‘TA’s upstream and downstream of the TE insertion site (Supplementary Figure S2).
Examination of insertion profiles of additional TE types yielded novel DNA integration sites and characteristics. Members of the hAT transposons superfamily are known to have a ‘NTCTAGAN’ 8-bp-TSD (37,38). We computationally identified flanking TSDs (see ‘Materials and Methods’ section) and confirmed that 8-bp-TSDs were the most prevalent TSD for hAT (Supplementary Figure S3). Furthermore, the insertion profiles of hAT family members with 8-bp-TSD (Figure 4A) reveal that: (i) the central 5′-TA-3′ at positions 4 and 5 of the TSD is the most conserved part of the TSD and (ii) the TSD is flanked by an AT-rich motif in positions −11 to −10 and a complementary motif in positions 10 to 11, relative to the inserted TE (or positions −3 to −2 and 2 to 3 relative to the edges of the TSD).
Figure 4.

Common sequence motifs of hAT, ERV1, MaLR integration sites. All pictograms in this figure are insertion profiles of fixed-size TSD created as described in Figure 3. The height of each nucleotide in the pictogram is proportional to its information content in the given position. The number in parenthesis in the title of each pictogram denotes the number of different insertion types (insertions into different target sequences) on which the insertion profile is based. Cyan dashed vertical lines denote the TE insertion position. Orange rectangles denote the TSD area. (A) Insertion profiles of five different members of the hAT superfamily (DNA transposons). The black dashed vertical lines are flanking the complementary AT-rich motif in positions −11 to −10 and positions 10 to 11. The orange dashed vertical lines are flanking the ‘TA’ dinucleotide in positions −5 to −4 and positions 4 to 5. (B) Insertion profiles of five different members of the ERV1 family (LTR retrotransposons). The black dashed vertical lines are flanking the common complementary motif in positions −6 and 6 or −7 and 7. (C) Insertion profiles of five different members of the MaLR family (LTR retrotransposons). The black dashed vertical lines are flanking the common AT-rich motif in positions −8 and 8.
We next identified novel integration sequence motifs common to two different LTR retrotransposons families. LTR retrotransposons are known to be flanked by TSDs of 4–6 nts (36). ERV1 insertions are characterized by 4-bp-TSD (Supplementary Figure S3). This TSD was flanked by T and a complementary A in positions −6 and 6, or −7 and 7, relative to the inserted TE, respectively (Figure 4B). Similarly, MaLR insertions, characterized by a 5-bp−TSD (Supplementary Figure S3), had the same nucleotide bias in position −8 and 8 relative to the inserted TE, but this motif is more degenerate sequence (AT-rich) than ERV1 motif (Figure 4C). We also noted that TEs belonging to MLT2 subfamily from the ERVL family (e.g. MLT2A1 and MLT2B3 in Figure 4C) share the same nucleotide bias with different MaLR family members, indicating that they may share the same retrotransposition mechanism.
Insertions of Alu sequences are dependent on L1 endonuclease nicking the antisense DNA at 5′-TTTT|AA-3′ (where ‘|’ denotes cleavage position) and a second nick occurring in the sense strand within 15–16 bp downstream (39). These are also characterized by a TSD of variable length. Our data confirms all previous results (Figure 5 and Supplementary Figure S3) and provides further information for the resulting TSD length. The 15–16 bp between the two nicking sites are divided into a variably sized polyA stretch followed by a degenerate TSD sequence that is weakly characterized by a T/G in its first position. We found an inverse correlation between the length of the polyA stretch and the length of the TSD (Spearman Rho = −0.21 P ≈ 0 for TSD ≥ 3). This observation led us to discover that the size of the TSD is roughly equal to the distance between the two nicks (15–16 bp) minus the length of the polyA stretch (Figure 5). Based on this finding, we suggest a revised model for Alu retrotransposition (see ‘Discussion’ section).
Figure 5.

Alu insertion profile reveals negative correlation between the size of an A-rich stretch and downstream TSD size. The insertion profiles of different Alu sequences from different ages (AluJ–AluY) are presented with four variable sizes of TSD. The dark blue arrow indicates a length of 16 bp, which is the nearly constant distance between the two nicking positions of L1 endonuclease, one in sense and the other in antisense. To the left side of the arrow is TT|AAAA, complementary to the TTTT|AA canonical L1 endonuclease nicking site in the antisense strand. Cyan dashed vertical lines denote the TE insertion position. Orange rectangles denote the TSD region. The number in parenthesis in the title of each pictogram denotes the number of different insertion types (insertions into different target sequences) on which the insertion profile is based.
The preferential sequences that we describe here are indicative of specific molecular mechanisms underlying transposition, such as enzymatic recognition sites (see ‘Discussion’ section). The informative insertion profiles of 125 and 241 different TE types when ignoring the TSD and when using fixed-size TSD in each profile, respectively, are shown in Dataset S2. Several additional integration sequence motifs of TEs are presented in Table S4.
Increased tendency for young TEs to insert within old TEs
Since DNA originated in TEs may contain potential target sequences for insertion of other TEs, we hypothesized that TE proliferation throughout human evolution may be driven by a tendency for new TEs to insert within existing ones. We were therefore interested in assessing whether TEs exhibited a tendency to insert within existing ones. Our hypothesis was that younger TEs are overrepresented within older TEs. The null hypothesis was that a given young TE inserts within the genome randomly and is not affected by whether the integration site originated in an old TE or not. Significant deviations from the null hypothesis would be indicative of enrichment or depletion of TE target sites within previously transposed TEs which can encourage or reject further transposition events, respectively.
We used a set of 360 human TE types known to interact with numerous TE types (23). We divided this set into 61 relatively ‘new TEs’ and 299 ‘old TEs’ according to a previously established chronology (23). The new TEs that we used were active during the last 40–45 million years, in the anthropoid primate lineage (24,40). The division into ‘new TEs’ and ‘old TEs’ was done for several reasons, which are elaborated in the ‘Materials and Methods’ section, all aiming to yield an accurate model for random insertion of TEs. Our null hypothesis predicts that each new TE insertion in the human genome occurred within an older TE or outside a TE, and the probability of insertion was determined based on the fraction of ancestral anthropoid genome covered by the old TE. We also assumed that concurrent or newer TE insertions into new TEs were negligible since only 3% of new TE insertions in our dataset occurred within another new TE. Another requirement of the null hypothesis was that the region of TE insertion would be free of strong purifying pressure. Since insertion of TEs into introns is presumably under purifying selection (25,41–43), we only considered TE nesting events found in intergenic regions. In order to avoid contamination of our data with nested TEs that were duplicated as part of larger genomic rearrangements, we discarded all genomic areas which are annotated as duplicated. The result of this filtration process yielded a genomic region of 1.29 gigabases, constituting ∼40% of the human genome (see ‘Materials and Methods’ section for complete details).
TE nesting events types found were classified based on three variables: the identity of the new inserted TE (n = 61), the identity of the old targeted TE (n = 299), and their relative orientation. This analysis yielded 36 478 (61 × 299 × 2) combinations, that we again named ‘insertion types’. For each insertion type, we counted the number of times it occurred in the intergenic part of the genome, and also calculated an expected number of occurrences based on the prevalence of targeted TEs in the ancestral anthropoid genome. A highly significant correlation (Figure 6A, Spearman Rho = 0.68, P ≈ 0) was obtained between observed and expected values, suggesting that, in general, TEs tend to insert randomly in the genome.
Figure 6.

Insertion pattern of new TEs into older TEs in intergenic regions of the human genome. (A) Correlation between observed and expected values of the different insertion types. Insertion types with more than 10 observed insertions are presented. (B) The volcano plot shows the observed/expected insertion ratios for insertions of new TEs into old TEs and the statistical significance of these ratios. Each point represents insertions of a new TE into an old one in a single orientation (sense/antisense). Points in the upper right rectangle are considered overrepresented with P < 1.37 × 10−6 (P < 0.05 after Bonferroni correction for 36 478 tests) and observed/expected ratio >2. Points in the upper left rectangle are considered under-represented with Bonferroni adjusted P < 1.37 × 10−6 and observed/expected ratio <0.5. Points of P < 10−50 were considered, for visualization purposes, as P = 10−50. (C) Insertion pattern of AluSx into old TEs. In this star graph, AluSx is the central vertex and is connected by edges to 299 vertices, representing the distinct old TE types into which AluSx could have inserted in unique intergenic regions. All TEs from the same family have a common vertex color and all families belonging to the same repeat class have close colors (e.g. SINEs are purple-pink). The edge color represents whether the insertion type was roughly as expected, under-represented or overrepresented in intergenic regions. Edge color also indicates the number of orientations in which over-/under-representation exists: sense, antisense or both. Sig. indicates statistically significant; underrep. indicates under-represented; overrep. indicates overrepresented.
To obtain a more detailed picture at the level of the insertion type, we next assigned each insertion type a P-value and a fold change. The former was calculated based on Bonferroni (multiple testing) corrected χ2 tests, and the latter was based on the observed/expected ratio. Based on these two measures, insertion types were classified as belonging into one of three groups: overrepresented (P < 1.37 × 10−6 and fold-change>2), under-represented (P < 1.37 × 10−6 and fold-change<0.5), or as expected (all other events). Figure 6B shows the observed/expected insertions ratios for insertions of new TEs into old TEs and the statistical significance of these ratios. The majority of insertion types (n = 35 846) are observed in the genome roughly as expected under the null hypothesis, meaning that they insert randomly in intergenic regions; however, there are 4-fold more overrepresented insertion types (n = 516) than under-represented insertion types (n = 116). This strong tendency toward over-representation was not sensitive to P-value cutoffs or to fold-change thresholds (see ‘Materials and Methods’ section). The observed and expected insertion patterns for all 36 478 insertion types are presented in Supplementary Table S5. Our results also demonstrate that the tendency toward over-representation of nested TEs is independent of the location of the intergenic TE relative to genes (see Supplementary Data) and the tendency toward overrepresented insertion types is stronger closer to genes than far, in accordance with the reported higher frequency of TE interruptions close to genes (25). The observed and expected insertion patterns for all 36 478 insertion types close to or far from genes are presented in Supplementary Table S6.
Table 2 provides an overview of the frequently overrepresented and under-represented new and old TEs. New Alu retrotransposons show a mixed trend: These elements are overrepresented within some old TEs but are under-represented within others. LTR retrotransposons are well overrepresented in nested TE events as both newly inserted TEs (THE1A, THE1B, LTR10C, LTR10F) and older targeted TEs (MER31-int, MER4B-int). We also noticed a phenomenon of ‘TE nepotism’ by which new MaLRs from THE1A/B types are overrepresented in old MaLRs from MLT1 family (Table S5). The MER2B and MER33 DNA transposons served as targets for insertions of different TEs, whereas old SINEs (MIRb, MIR3, AluJb, AluJo) seem to reject insertion of newer TEs.
Table 2.
Frequently overrepresented or under-represented new and old TEs
| New TEs frequently overrepresented |
Old TEs frequently overrepresented |
New TEs frequently under-represented |
Old TEs frequently underrepresented |
||||
|---|---|---|---|---|---|---|---|
| TE name | Insertion types involved | TE name | Insertion types involved | TE name | Insertion types involved | TE name | Insertion types involved |
| AluSx | 51 | L1M | 16 | AluSx | 48 | MIRb | 15 |
| AluY | 35 | MER2B | 13 | AluSq | 12 | AluJb | 13 |
| AluSg | 24 | MER31-int | 13 | AluSg | 11 | AluJo | 8 |
| AluSp | 24 | AluJ | 12 | AluY | 11 | MIR3 | 8 |
| AluSq | 21 | MER33 | 10 | FRAM | 5 | L1PA13 | 6 |
| THE1B | 18 | HAL1 | 9 | L1PA7 | 5 | L1PA16 | 5 |
| AluSc | 17 | L1MEc | 9 | AluSg/x | 4 | L2 | 5 |
| LTR10F | 17 | MER4B-int | 9 | HERVH | 4 | L1PA15 | 4 |
| LTR10C | 15 | L1ME3B | 8 | AluSc | 3 | L1PB1 | 4 |
| THE1A | 15 | L1ME4a | 8 | AluSp | 3 | L1PREC2 | 4 |
Each list is sorted in descending order according to the number of insertion types in which the relevant TE is involved.
The insertion orientation also plays an important role in dictating whether a TE will be inserted into another TE. Over-representation or under-representation of an insertion type was generally found in a single orientation, while the insertions in the other orientation were as expected. No insertion type was overrepresented in one orientation and under-represented in the other orientation. Table 3 presents nested TEs of the most prominent insertion orientation bias. The orientation-dependent insertion pattern of 292 combinations of inserted and targeted TE families is presented in Supplementary Table S7.
Table 3.
Nested TEs demonstrating inserted orientation bias
| Inserted TE family | Targeted TE family | Insertions in sense | Insertions in antisense | Fold change sense/antisense |
|---|---|---|---|---|
| Preferential insertion in sense | ||||
| Alu | Alu | 3419 | 1296 | 2.63812 |
| MuDR | ERVL | 50 | 20 | 2.5 |
| Alu | L1 | 44 751 | 20 044 | 2.23264 |
| L1 | L1 | 21 559 | 9899 | 2.1779 |
| L1 | Alu | 222 | 103 | 2.15534 |
| ERVL | ERVL | 1080 | 535 | 2.01869 |
| MIR | RTE (L4) | 51 | 30 | 1.7 |
| ERVL | ERV1 | 215 | 138 | 1.55797 |
| Alu | CR1 | 544 | 352 | 1.54545 |
| RTE (L4) | MaLR | 49 | 32 | 1.53125 |
| Preferential insertion in antisense | ||||
| L2 | ERV1 | 20 | 38 | 0.526316 |
| Alu | L2 | 3783 | 6895 | 0.548658 |
| MIR | MER2_type | 29 | 50 | 0.58 |
| Tip100 | ERVL | 36 | 61 | 0.590164 |
| MaLR | Tc2 | 28 | 47 | 0.595745 |
| L2 | RTE (L4) | 23 | 37 | 0.621622 |
| MaLR | RTE (L4) | 51 | 82 | 0.621951 |
| Tip100 | MIR | 37 | 59 | 0.627119 |
| Alu | ERVK | 27 | 43 | 0.627907 |
Intergenic nested TEs of more than 50 genomic occurrences and fold change higher than 1.5 or lower than 0.66 are shown. TE family names are those given by rmsk.
We demonstrate the complex pattern of TE interactions using an insertion graph for AluSx (Figure 6C). AluSx is the most prevalent anthropoid-specific TE, responsible for 32% of all new TEs insertions into old TEs. Insertions of AluSx into DNA transposons, such as those of Charlie family, are well overrepresented. This preferential insertion can be explained by the enrichment of Alu integration sites within the Charlie consensus sequence: 5-fold more than on the average TEs (Supplementary Table S3).
The insertions of AluSx into the different Alu and L1 family members had a striking dependence on the insertion orientation (Supplementary Figure S4). AluSx specifically, and Alus in general, were inserted into Alu/L1 over twofold more times in sense rather than in antisense orientation. Such orientation bias was described before for tandemly inserted Alu sequences (44). This clear insertion orientation bias was seen also in L1 insertions into Alu/L1 (Table 3) and it can be explained mechanistically by intrinsic sequence features of the L1/Alu sequences (see Supplementary Data). We suggest that this insertion bias serves as a mode of inactivation of the targeted Alu/L1 (see Supplementary Data).
To conclude, our analysis shows that although TEs are usually inserted randomly in intergenic regions, there are hundreds of exceptions to this rule. These exceptions have a strong tendency to be overrepresented rather than under-represented, meaning that some new TEs tend to insert within older TEs. We provide examples for TE types that are enriched or depleted within other TE types. Insertion orientation also plays an important role in dictating whether a TE will insert into another.
DISCUSSION
Our analysis of TE nesting events at various levels provides several conclusions. Through evaluation of roughly 300 000 exact TE insertions, we discovered that TEs insert in specific positions along the targeted TE target sequence. As the single prominent hotspot for Alu self insertions demonstrates, the hotspot can be a non-canonical integration site. Using the same dataset and a different method, we were able to uncover previously unknown integration sequence preferences of MaLR and ERV1 TE families and to refine the known integration motifs of Alu and hAT TE families. These integration motifs may improve the understanding of the molecular mechanisms underlying transposition. Finally, we devised a simple and relatively accurate model to identify TE nesting events that are over- or under-represented. Our model demonstrates that most intergenic anthropoid-specific TEs inserted randomly in intergenic regions, without preferring or avoiding older transposed TEs in this regions. Yet, there are hundreds of significant exceptions in which TE nesting is over- or under-represented, with overrepresented insertion types 4-fold more common than under-represented insertion types. The insertion pattern of TEs varies as a function of the inserted TE type, the targeted TE type, their relative orientation and the distribution of integration sequences within the targeted TE.
We noted that most TEs were inserted in an uneven manner along the target TE. The identified insertion hotspots could not be predicted by existence of known integration sites only, as the latter ones are often absent from the consensus sequence of the targeted TEs. Several unusual TE integration events in the human genome were described previously [e.g. tailless SINEs (45)]. Analysis of inserted TEs with previously unknown integration sites, such as MaLR retrotransposons, also demonstrated insertion hotspots in certain types of targeted TEs. Further exploration of the common properties of these unique sites may shed light on the mechanism by which these TEs are integrating into the genome.
Our analyses reveal novel insights regarding retrotransposition of Alu elements. Upstream to Alu insertions there is an A-rich region, the length of which is negatively correlated with the length of the immediately downstream TSD. The total length of the A-rich region and the TSD sequence tends to be around 15–16 bp, precisely the distance between the first and second nicking by L1 endonuclease (39). This observation leads us to suggest a revised model for Alu integration (Figure 7). Initially, the L1 endonuclease processes the target DNA with the first nick usually occurring in the consensus site TTTT|AA in the antisense strand and the second nicking occurs 15–16 bp downstream and in the sense strand in a TNTN|AA site (39). The 5′ portion of this 15–16 bp long spacer is often A-rich (Figure 5) and its antisense polyT stretch serves as an excellent template for binding of the polyA tail of the Alu RNA during the subsequent target-primed reverse transcription (TPRT) (46). Following TPRT, the polyT stretch is maintained as part of the antisense sequence to the terminal polyA of the inserted Alu. The size of polyT stretch dictates the size of the resulting variable TSD, which is equal to the distance between the two nicks (15–16 bp) minus the length of the polyT stretch. Next, the DNA is ligated and the single-stranded parts are replicated using the DNA template to yield a dsDNA. This model can explain the observed hotspot for self Alu insertion. We noticed that the nearly 19 000 genomic occurrences of self Alu insertions contain mainly a single hotspot located between the internal polyA stretch and Alu right arm in the Alu sense strand (Figure 2). A careful look revealed that this is the best location for Alu insertion within the Alu sequence. There is a suboptimal L1 endonuclease recognition site just between the Alu left arm and the polyA linker (in the antisense strand): TTTT|AG. Between the polyA linker and the Alu right arm in the sense strand, there is another suboptimal L1 endonuclease recognition site AATT|AG. Between these two sites, there is an exactly 15–16 nucleotide A-rich stretch, the polyA linker itself, with its antisense polyT stretch nearly fully used for reverse transcription, leaving a very short (if any) TSD (Figure 2). Alu self insertions in antisense tend to be under-represented (Supplementary Table S5) probably because of the absence of a polyT template for reverse transcription.
Figure 7.

Revised model for Alu integration explaining the resulting variable TSD size. The L1 endonuclease first nicks the consensus site TTTT|AA in the antisense strand and then 15–16 bp downstream in the sense strand in a TNTN|AA site. The 5′ portion of the 15–16 bp spacer is often A-rich. The antisense polyT stretch serves as a template for the polyA tail of the Alu RNA during the subsequent TPRT. The size of polyT stretch dictates the size of the resulting variable TSD, which is equal to the distance between the two nicks (15–16 bp) minus the length of the polyT stretch. Finally, the DNA is ligated and the single-stranded parts are replicated using the DNA template to create a dsDNA flanked by an upstream polyA stretch and TSD and a downstream TSD.
TEs can be used for human gene therapy and functional genomics. By using a TE with a specific integration site, one can knockout specific mutated genes or explore the function of a gene of interest without damaging other genomic regions. Specific TEs may also be used to safely insert new genes into cells and cure genetic diseases. The TEs that can be used for these purposes may be from a different species or may even be TEs that are no longer active today. However, the integration-site preferences of TEs, which are critically important for these uses, are poorly understood. Our database of exact insertions enabled us to observe ‘fossilized’ sequences flanking inserted TEs which are probably the original integration sequences for the inserted TE. These sequences have probably been somewhat modified over the course of evolution and are therefore, more difficult to reconstruct when the TE was inserted into non-TE genomic regions. We identified common integration sequence motifs of TEs that can be generalized out of the scope of nested TEs. hAT transposons occupy 1.5% of the human genome (47) and some of them were shown to be active in the primate lineage (24). We discovered that the motif probably recognized by the hAT transposase is the nearly palindromic sequence 5′-W1W1NNTYTARANNW2W2-3′ (Figure 4A), where W denotes A or T and W1 and W2 are complementary nucleotides. The ‘TA’ center of this palindrome is its most conserved part. Subsequent nicking of the motif between positions 3 and 4 in sense and positions 11 and 12 in antisense yields an 8-bp TSD (Supplementary Figure S5).
ERV1 and MalR LTR retrotransposons occupy 2.89% and 3.65% of the human genome, respectively (47). We identified a strong signal flanking the TSD: a T was present in positions −2 (or −3), and −3 of ERV1 and MaLR relative to the TSD, respectively, and a complementary A was observed in positions 2 (or 3) and 3 of ERV1 and MaLR, respectively. This implies that the ERV1 integrase enzyme recognizes the integration sequence 5′-TN6A-3′, where N represents any nucleotide. The integrase nicks this motif between positions 2 and 3 in sense and positions 6 and 7 in antisense, yielding a 4-bp TSD (Figure 4B). For some ERV1s, the central 2-bp of the motif are AT-rich. The MaLR integrase, on the other hand, recognizes the integration sequence 5′-TN9A-3′, with interchangeable A and T (Figure 4C); it then nicks this motif between positions 3 and 4 in sense and positions 8 and 9 in antisense, yielding a 5-bp TSD.
Over-representation of insertion types may result from several factors. The most intuitive factor is increased density of integration sites for the inserted TE within the targeted TE. When a target TE is rich in integration sites for other TEs, we would expect this TE to be frequently targeted by such TEs. Thus, some TEs, once transposed into the genome, supplied new potential integration sites for contemporary and future TEs and thereby assisted in their proliferation. However, the density of known L1/Alu integration sequence (48) in the different targeted TE consensus sequences (Supplementary Table S3) is only weakly positively correlated with the number of observed insertions of young L1/Alu elements into old TEs (Spearman Rho = 0.12, P < 10−40). Moreover, for most TEs, the preferred integration sequence is unknown and therefore it is impossible to use this data in a general model. In addition, incorporation of the density of known TE integration sites into our model, did not improve the correlation between observed and expected insertion events (data not shown). This suggests that the density of TE integration sequences over the targeted TE is not the only factor affecting TE nesting.
High abundance of new TEs within old TEs may also result from a purifying selection-driven process. For example, if the targeted TE is harmful for the cell in its intact version, disruption by a neutral TE can be advantageous. It is also plausible that the intergenic areas, which we assumed to be affected by little or no purifying selection, do contain important regulatory signals. Highly conserved elements are enriched in stable gene deserts (49) and may serve necessary functions. TE insertion into these sites may result in their inactivation and may, therefore, be detrimental to organism fitness. By favoring insertions of TEs into regions which are already marked as genomic ‘junk’ (e.g. areas of transposed elements), the harmful potential of these new TEs is reduced. Supporting this, H3 lysine 9 methylation, which is generally associated with gene silencing, is enriched at Alu elements (50). Therefore, insertions of Alus into other TEs, or vice versa, may lead to silencing of both the inserted and the targeted TE and would therefore be advantageous.
The less frequent under-represented insertion types may also be the result of purifying selection. For example, the tendency of the mammalian-wide interspersed repeat MIRb elements to be conserved in its intact form and not to serve as a targeted TE (15 insertion types where it is under-represented versus zero insertion types where it is overrepresented) may suggest that this intergenic TE has a yet unknown cellular function. Supporting this idea, it was suggested that certain MIRb elements acquired a similar regulatory role near all known receptor-related genes of the reelin signaling pathway (51). MIR3 and MIRm are other examples of MIR types which are avoided from some TE insertions without being favorable by others. However, since MIR elements are relatively old and short, they are barely detected by RepeatMasker alignment to the MIR consensus sequence. Therefore, we expect that there were more TE insertions into MIR elements than were identified as nested MIRs. Thus, the conclusion that TE insertions into MIR are under-represented may be wrong because of this technical bias.
Taken together, the analysis we describe here of human-nested TE events has provided insight on both evolutionary and molecular aspects underlying proliferation of TEs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Israel Science Foundation (ISF 61/09); Joint Germany-Israeli Research Program (ca-139); Deutsche-Israel Project (DIP MI-1317); and European Alternative Splicing Network (EURASNET). S.S. is a fellow of the Edmond J. Safra bioinformatic program at Tel Aviv University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Arian Smit and Dr Gustavo Glusman from the Institute for Systems Biology, Seattle, (Washington, USA) for their suggestions and ideas, and for their assistance in interpreting RepeatMasker output. We also thank Dr Jerzy Jurka from GIRI for his ideas regarding our Alu insertion model. This work was performed in partial fulfillment of the requirements for M.Sc and PhD degree of A.L. and S.S., respectively, Sackler Faculty of Medicine, Tel Aviv University, Israel.
REFERENCES
- 1.Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 2.Biemont C, Vieira C. Genetics: junk DNA as an evolutionary force. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- 3.Consortium ICGS. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 4.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 5.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 6.Hedges DJ, Batzer MA. From the margins of the genome: mobile elements shape primate evolution. Bioessays. 2005;27:785–794. doi: 10.1002/bies.20268. [DOI] [PubMed] [Google Scholar]
- 7.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 8.Callinan PA, Batzer MA. Retrotransposable elements and human disease. Genome Dyn. 2006;1:104–115. doi: 10.1159/000092503. [DOI] [PubMed] [Google Scholar]
- 9.Asch HL, Eliacin E, Fanning TG, Connolly JL, Bratthauer G, Asch BB. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol. Res. 1996;8:239–247. [PubMed] [Google Scholar]
- 10.Depil S, Roche C, Dussart P, Prin L. Expression of a human endogenous retrovirus, HERV-K, in the blood cells of leukemia patients. Leukemia. 2002;16:254–259. doi: 10.1038/sj.leu.2402355. [DOI] [PubMed] [Google Scholar]
- 11.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, Fodinger D, Seppele H, Schanab O, Magin-Lachmann C, et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735–8741. [PubMed] [Google Scholar]
- 12.Ast G. How did alternative splicing evolve? Nat. Rev. Genet. 2004;5:773–782. doi: 10.1038/nrg1451. [DOI] [PubMed] [Google Scholar]
- 13.Corvelo A, Eyras E. Exon creation and establishment in human genes. Genome Biol. 2008;9:R141. doi: 10.1186/gb-2008-9-9-r141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim E, Goren A, Ast G. Alternative splicing: current perspectives. Bioessays. 2008;30:38–47. doi: 10.1002/bies.20692. [DOI] [PubMed] [Google Scholar]
- 15.VolffJ N, Brosius J. Modern genomes with retro-look: retrotransposed elements, retroposition and the origin of new genes. Genome Dyn. 2007;3:175–190. doi: 10.1159/000107611. [DOI] [PubMed] [Google Scholar]
- 16.Barbosa-Morais NL, Carmo-Fonseca M, Aparicio S. Systematic genome-wide annotation of spliceosomal proteins reveals differential gene family expansion. Genome Res. 2006;16:66–77. doi: 10.1101/gr.3936206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sela N, Mersch B, Gal-Mark N, Lev-Maor G, Hotz-Wagenblatt A, Ast G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu's; unique role in shaping the human transcriptome. Genome Biol. 2007;8:R127. doi: 10.1186/gb-2007-8-6-r127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conley AB, Miller WJ, Jordan IK. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet. 2008;24:53–56. doi: 10.1016/j.tig.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Conley AB, Piriyapongsa J, Jordan IK. Retroviral promoters in the human genome. Bioinformatics. 2008;24:1563–1567. doi: 10.1093/bioinformatics/btn243. [DOI] [PubMed] [Google Scholar]
- 20.Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 21.Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE. 2007;2:e203. doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piriyapongsa J, Marino-Ramirez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giordano J, Ge Y, Gelfand Y, Abrusan G, Benson G, Warburton P. Evolutionary history of mammalian transposons determined by genome-wide defragmentation. PLoS Comput. Biol. 2007;3:e137. doi: 10.1371/journal.pcbi.0030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pace JK, II, Feschotte C. The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Res. 2007;17:422–432. doi: 10.1101/gr.5826307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrusan G, Giordano J, Warburton PE. Analysis of transposon interruptions suggests selection for L1 elements on the X chromosome. PLoS Genet. 2008;4:e1000172. doi: 10.1371/journal.pgen.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichiyanagi K, Nakajima R, Kajikawa M, Okada N. Novel retrotransposon analysis reveals multiple mobility pathways dictated by hosts. Genome Res. 2007;17:33–41. doi: 10.1101/gr.5542607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergman CM, Quesneville H, Anxolabehere D, Ashburner M. Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 2006;7:R112. doi: 10.1186/gb-2006-7-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kriegs JO, Matzke A, Churakov G, Kuritzin A, Mayr G, Brosius J, Schmitz J. Waves of genomic hitchhikers shed light on the evolution of gamebirds (Aves: Galliformes) BMC Evol. Biol. 2007;7:190. doi: 10.1186/1471-2148-7-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. 1995;57:289–300. [Google Scholar]
- 31.Grimaldi G, Skowronski J, Singer MF. Defining the beginning and end of KpnI family segments. EMBO J. 1984;3:1753–1759. doi: 10.1002/j.1460-2075.1984.tb02042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bembom O. seqLogo: An R package for plotting DNA sequence logos; 2007. [Google Scholar]
- 34.Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 35.Csárdi G, Nepusz T. The igraph software package for complex network research. InterJournal. 2006 Complex Systems, 1696. [Google Scholar]
- 36.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 37.Kempken F, Windhofer F. The hAT family: a versatile transposon group common to plants, fungi, animals, and man. Chromosoma. 2001;110:1–9. doi: 10.1007/s004120000118. [DOI] [PubMed] [Google Scholar]
- 38.Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc. Natl Acad. Sci. USA. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl Acad. Sci. USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan H, Smit A, Boissinot S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006;16:78–87. doi: 10.1101/gr.4001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sironi M, Menozzi G, Comi GP, Cereda M, Cagliani R, Bresolin N, Pozzoli U. Gene function and expression level influence the insertion/fixation dynamics of distinct transposon families in mammalian introns. Genome Biol. 2006;7:R120. doi: 10.1186/gb-2006-7-12-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simons C, Pheasant M, Makunin IV, Mattick JS. Transposon-free regions in mammalian genomes. Genome Res. 2006;16:164–172. doi: 10.1101/gr.4624306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van de Lagemaat LN, Medstrand P, Mager DL. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol. 2006;7:R86. doi: 10.1186/gb-2006-7-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenger JE, Lobachev KS, Gordenin D, Darden TA, Jurka J, Resnick MA. Biased distribution of inverted and direct Alus in the human genome: implications for insertion, exclusion, and genome stability. Genome Res. 2001;11:12–27. doi: 10.1101/gr.158801. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz J, Churakov G, Zischler H, Brosius J. A novel class of mammalian-specific tailless retropseudogenes. Genome Res. 2004;14:1911–1915. doi: 10.1101/gr.2720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 47.Mandal PK, Kazazian HH., Jr SnapShot: vertebrate transposons. Cell. 2008;135:191–192.e1. doi: 10.1016/j.cell.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 48.Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 49.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kondo Y, Issa JP. Enrichment for histone H3 lysine 9 methylation at Alu repeats in human cells. J. Biol. Chem. 2003;278:27658–27662. doi: 10.1074/jbc.M304072200. [DOI] [PubMed] [Google Scholar]
- 51.Lowe CB, Bejerano G, Haussler D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc. Natl Acad. Sci. USA. 2007;104:8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.