

Abstract
Two-component signal transduction systems are widespread in prokaryotes and control numerous cellular processes. Extensive investigation of sensor kinase and response regulator proteins from many two-component systems has established conserved sequence, structural, and mechanistic features within each family. In contrast, the phosphatases which catalyze hydrolysis of the response regulator phosphoryl group to terminate signal transduction are poorly understood. Here we present structural and functional characterization of a representative of the CheC/CheX/FliY phosphatase family. The X-ray crystal structure of Borrelia burgdorferi CheX complexed with its CheY3 substrate and the phosphoryl analogue  reveals a binding orientation between a response regulator and an auxiliary protein different from that shared by every previously characterized example. The surface of CheY3 containing the phosphoryl group interacts directly with a long helix of CheX which bears the conserved (E - X2 - N) motif. Conserved CheX residues Glu96 and Asn99, separated by a single helical turn, insert into the CheY3 active site. Structural and functional data indicate that CheX Asn99 and CheY3 Thr81 orient a water molecule for hydrolytic attack. The catalytic residues of the CheX·CheY3 complex are virtually superimposable on those of the Escherichia coli CheZ phosphatase complexed with CheY, even though the active site helices of CheX and CheZ are oriented nearly perpendicular to one other. Thus, evolution has found two structural solutions to achieve the same catalytic mechanism through different helical spacing and side chain lengths of the conserved acid/amide residues in CheX and CheZ.
reveals a binding orientation between a response regulator and an auxiliary protein different from that shared by every previously characterized example. The surface of CheY3 containing the phosphoryl group interacts directly with a long helix of CheX which bears the conserved (E - X2 - N) motif. Conserved CheX residues Glu96 and Asn99, separated by a single helical turn, insert into the CheY3 active site. Structural and functional data indicate that CheX Asn99 and CheY3 Thr81 orient a water molecule for hydrolytic attack. The catalytic residues of the CheX·CheY3 complex are virtually superimposable on those of the Escherichia coli CheZ phosphatase complexed with CheY, even though the active site helices of CheX and CheZ are oriented nearly perpendicular to one other. Thus, evolution has found two structural solutions to achieve the same catalytic mechanism through different helical spacing and side chain lengths of the conserved acid/amide residues in CheX and CheZ.
Keywords: CheX, CheY, two-component systems, dephosphorylation, convergent evolution
Two-component signal transduction systems control diverse cellular processes (ranging from pathogenesis to morphological development) in prokaryotes, fungi, and plants (1–4). To date, genes for more than 50,000 proteins that function in two-component systems have been identified by genome sequencing projects (www.p2cs.org). In two-component systems, a sensor kinase autophosphorylates on a histidyl residue. The phosphoryl group is then transferred to an aspartyl residue on the receiver domain of a cytoplasmic response regulator protein, which activates the response regulator to execute a cellular response to a stimulus. Deactivation of the response regulator occurs by hydrolysis of the phosphoryl group, sometimes via an auxiliary phosphatase.
In contrast to sensor kinases and response regulators [see refs. (5–7) for recent reviews], the mechanisms by which auxiliary phosphatases operate are poorly characterized. At least five distinct families of response regulator phosphatases have been identified to date: CheC/CheX/FliY (8), CheZ (9), Rap (10), sensor kinases (11, 12), and Spo0E (13). However, the active site has been definitively identified and a reaction mechanism established for only the CheZ class of phosphatases. In principle, structural, mechanistic, or amino acid sequence characteristics which are shared between families of response regulator phosphatases could be used to predict which proteins from genomic databases possess phosphatase activity toward response regulators. Here we use structural and enzymatic methods to investigate a representative of the CheC/CheX/FliY class of phosphatases.
The CheC/CheX/FliY family spans diverse phyla within bacteria and archaea (8, 14). X-ray crystal structures of Thermotoga maritima chemotaxis proteins CheX and CheC (15) reveal similar folds which reflect an internal sequence pseudosymmetry (16). A conserved (E-X2-N) motif is present twice in CheC due to the pseudosymmetry, but CheX contains only the second copy of the motif. For each motif, the conserved glutamate and asparagine are one helical turn apart on a solvent exposed alpha helix. Mutagenesis studies (15, 17, 18) suggest that the (E-X2-N) motif mediates catalysis. The (E-X2-N) motifs in CheC and CheX appear comparable to the structurally unrelated CheZ phosphatase, whose active site acid and amide residues Asp143 and Gln147 are also separated by a single helical turn (9). However, attempts to superimpose a catalytic motif of CheC onto its counterpart in CheZ suggested that the mode of interaction of the CheC/CheX/FliY phosphatases with CheY differs from CheZ (15).
Here, we present the X-ray crystal structure of a complex containing Borrelia burgdorferi CheX and its CheY3 substrate bound to the phosphoryl analogue 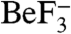 and Mg2+. CheX and CheY3 operate in the two-component system which mediates chemotaxis in B. burgdorferi (19–21), the causative agent of Lyme disease. The structure reveals a unique mode of binding between a response regulator and an auxiliary protein, but a catalytic mechanism which is virtually identical to that used by the structurally unrelated CheZ, providing a striking example of convergent evolution. The data also suggest a possible CheX regulatory mechanism through dissociation of the CheX homodimer.
and Mg2+. CheX and CheY3 operate in the two-component system which mediates chemotaxis in B. burgdorferi (19–21), the causative agent of Lyme disease. The structure reveals a unique mode of binding between a response regulator and an auxiliary protein, but a catalytic mechanism which is virtually identical to that used by the structurally unrelated CheZ, providing a striking example of convergent evolution. The data also suggest a possible CheX regulatory mechanism through dissociation of the CheX homodimer.
Results
Overall Structure of 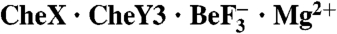 .
.
The crystal structure of B. burgdorferi  was determined at a resolution of 1.96 Å (Table S1). The asymmetric unit contained one molecule each of CheX and CheY3. The 2Fo-Fc electron density map displayed clear density for residues 4–158 of CheX (160 residues total) with the exception of weak density for several short stretches (residues 33–40, 109–113, and 125–136), all of which are distant from the interaction surface with CheY3. There was clear electron density for residues 13–146 of CheY3 (146 residues total), as well as the
was determined at a resolution of 1.96 Å (Table S1). The asymmetric unit contained one molecule each of CheX and CheY3. The 2Fo-Fc electron density map displayed clear density for residues 4–158 of CheX (160 residues total) with the exception of weak density for several short stretches (residues 33–40, 109–113, and 125–136), all of which are distant from the interaction surface with CheY3. There was clear electron density for residues 13–146 of CheY3 (146 residues total), as well as the 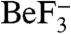 and Mg2+-ions bound to the CheY active site. Figure 1A shows the asymmetric unit and illustrates that the interaction between the proteins brings the conserved residues Glu96 and Asn99 of CheX proximal to the
and Mg2+-ions bound to the CheY active site. Figure 1A shows the asymmetric unit and illustrates that the interaction between the proteins brings the conserved residues Glu96 and Asn99 of CheX proximal to the 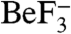 moiety in CheY3.
moiety in CheY3.
Fig. 1.

Overall topology of 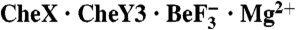 . (A) Ribbon representation of the asymmetric unit. CheX is cyan and CheY3 is green. CheY3 Asp79 (orange),
. (A) Ribbon representation of the asymmetric unit. CheX is cyan and CheY3 is green. CheY3 Asp79 (orange), 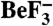 (purple), CheX Glu96 (red, Upper), and CheX Asn99 (red, Lower) are in stick representation. Mg2+ is a magenta sphere. (B).Overlay of B. burgdorferi CheX (cyan) and T. maritima CheX chain B (pdb 1XKO, tan). Regions of B. burgdorferi CheX with high disorder (Cα thermal B-factors > 70) are blue. B. burgdorferi CheX residues 37–40 are sketched as blue dots. T. maritima CheX residues corresponding to the disordered regions of B. burgdorferi CheX are red.
(purple), CheX Glu96 (red, Upper), and CheX Asn99 (red, Lower) are in stick representation. Mg2+ is a magenta sphere. (B).Overlay of B. burgdorferi CheX (cyan) and T. maritima CheX chain B (pdb 1XKO, tan). Regions of B. burgdorferi CheX with high disorder (Cα thermal B-factors > 70) are blue. B. burgdorferi CheX residues 37–40 are sketched as blue dots. T. maritima CheX residues corresponding to the disordered regions of B. burgdorferi CheX are red.
B. burgdorferi CheX Is Not Present As a Homodimer in the Cocrystal.
Superposition of B. burgdorferi 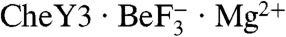 and E. coli
and E. coli 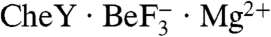 revealed highly similar backbones (rmsd = 0.8 Å for Cα atoms within secondary structure elements), active site geometries, and rotameric positions of residues which change upon activation, indicating that CheY3 was in a fully active conformation in the cocrystal (Fig. S1). CheY3 residues 13–26, part of an extended amino terminus absent in other CheYs, formed an ordered random coil which did not interact with other residues in CheY3, but made crystal contacts with a CheX chain from a neighboring asymmetric unit. Overlay of the CheX chains from
revealed highly similar backbones (rmsd = 0.8 Å for Cα atoms within secondary structure elements), active site geometries, and rotameric positions of residues which change upon activation, indicating that CheY3 was in a fully active conformation in the cocrystal (Fig. S1). CheY3 residues 13–26, part of an extended amino terminus absent in other CheYs, formed an ordered random coil which did not interact with other residues in CheY3, but made crystal contacts with a CheX chain from a neighboring asymmetric unit. Overlay of the CheX chains from 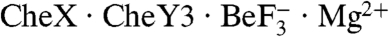 and T. maritima CheX (Fig. 1B) showed good backbone alignment for a majority of the secondary structure elements (rmsd = 0.9 Å for Cα atoms). However, several regions showed poor alignment. Notably, a continuous stretch of B. burgdorferi CheX residues (125–136) were disordered with crystallographic B-factors for Cα carbons of 70–100, resulting in poor alignment with T. maritima CheX. These residues correspond to the β1′-βx′ loop, βx′, and the βx′-β2′ loop in T. maritima CheX2, which comprise a large portion of the intradimer surface. There was also weak electron density for residues 37–40 of B. burgdorferi CheX, which could not be modeled into the structure (Fig. 1B). These residues correspond to the βx strand in T. maritima CheX, which interacts directly with βx′. Therefore the regions of disorder in B. burgdorferi CheX are localized to the regions which mediate intersubunit interactions in the T. maritima CheX2 structure.
and T. maritima CheX (Fig. 1B) showed good backbone alignment for a majority of the secondary structure elements (rmsd = 0.9 Å for Cα atoms). However, several regions showed poor alignment. Notably, a continuous stretch of B. burgdorferi CheX residues (125–136) were disordered with crystallographic B-factors for Cα carbons of 70–100, resulting in poor alignment with T. maritima CheX. These residues correspond to the β1′-βx′ loop, βx′, and the βx′-β2′ loop in T. maritima CheX2, which comprise a large portion of the intradimer surface. There was also weak electron density for residues 37–40 of B. burgdorferi CheX, which could not be modeled into the structure (Fig. 1B). These residues correspond to the βx strand in T. maritima CheX, which interacts directly with βx′. Therefore the regions of disorder in B. burgdorferi CheX are localized to the regions which mediate intersubunit interactions in the T. maritima CheX2 structure.
T. maritima CheX forms a dimer in both the crystal and solution (15). Although the asymmetric unit of the 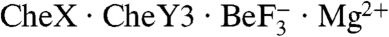 structure contained only one chain of CheX, a homodimer could potentially be formed from two CheX chains contributed from different asymmetric units. However, generation of all of the neighboring asymmetric units did not yield a CheX monomer in a position correlating to the second subunit in T. maritima CheX2, nor was a dimer with an alternate surface of interaction implicated. The mobility of
structure contained only one chain of CheX, a homodimer could potentially be formed from two CheX chains contributed from different asymmetric units. However, generation of all of the neighboring asymmetric units did not yield a CheX monomer in a position correlating to the second subunit in T. maritima CheX2, nor was a dimer with an alternate surface of interaction implicated. The mobility of 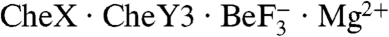 on an analytical gel filtration column was also consistent with a complex which contained one molecule each of CheX and CheY3 (Fig. S2).
on an analytical gel filtration column was also consistent with a complex which contained one molecule each of CheX and CheY3 (Fig. S2).
The observation that CheX is not present as a homodimer in the 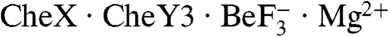 complex was unexpected because gel filtration analysis indicated that purified B. burgdorferi CheX is dimeric in solution (20). Because gel filtration can give misleading results for some proteins, we used a combination of Rayleigh light scattering and refractometry to determine the associative state of CheX. This analysis gave a polydispersity index of 1.000 ± 0.003, indicating homodispersity of the CheX sample, and a molecular weight of 36,980 (cf. CheX2 theoretical molecular weight of 37,760), confirming CheX2 dimer formation. Therefore, it appears that binding
complex was unexpected because gel filtration analysis indicated that purified B. burgdorferi CheX is dimeric in solution (20). Because gel filtration can give misleading results for some proteins, we used a combination of Rayleigh light scattering and refractometry to determine the associative state of CheX. This analysis gave a polydispersity index of 1.000 ± 0.003, indicating homodispersity of the CheX sample, and a molecular weight of 36,980 (cf. CheX2 theoretical molecular weight of 37,760), confirming CheX2 dimer formation. Therefore, it appears that binding  to CheX results in dissociation of the CheX2 dimer. The disorder in the portions of B. burgdorferi CheX which correspond to the intradimer surface in T. maritima CheX2 are likely due to the loss of stabilizing interchain interactions within the CheX2 dimer.
to CheX results in dissociation of the CheX2 dimer. The disorder in the portions of B. burgdorferi CheX which correspond to the intradimer surface in T. maritima CheX2 are likely due to the loss of stabilizing interchain interactions within the CheX2 dimer.
Novel Orientation of CheY3 and CheX Within 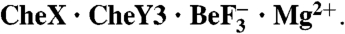
In the  structure, CheY3 is oriented relative to CheX so that the α1 helix in CheY3 is essentially perpendicular to the α1′ helix in CheX which bears the conserved (E-X2-N) sequence motif (Figs. 1A, 2). Residues throughout the entire length of CheX α1′, as well as the CheX α3-α1′ loop and the β1′ strand, interact with residues on all of the CheY3 βα loops except β2α2, as well as the N-terminal two turns of α1 (Fig. S3). The area of the interaction surface between CheX and CheY3 is 896 Å2 and contains five interchain hydrogen bonds and one salt bridge interspersed with patches of hydrophobic interactions (Fig. S3). Table S2 lists intermolecular interactions within the
structure, CheY3 is oriented relative to CheX so that the α1 helix in CheY3 is essentially perpendicular to the α1′ helix in CheX which bears the conserved (E-X2-N) sequence motif (Figs. 1A, 2). Residues throughout the entire length of CheX α1′, as well as the CheX α3-α1′ loop and the β1′ strand, interact with residues on all of the CheY3 βα loops except β2α2, as well as the N-terminal two turns of α1 (Fig. S3). The area of the interaction surface between CheX and CheY3 is 896 Å2 and contains five interchain hydrogen bonds and one salt bridge interspersed with patches of hydrophobic interactions (Fig. S3). Table S2 lists intermolecular interactions within the 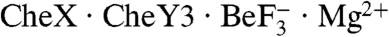 asymmetric unit.
asymmetric unit.
Fig. 2.

Comparison of the relative orientation of CheX and CheY3 with that of YPD1 (a histidyl phosphotransferase) and SLN1-R1 (a receiver domain) within  (pdb 2R25). For
(pdb 2R25). For 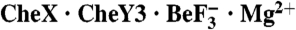 (Upper), CheX is gray except for α1′, which is cyan. CheY3 is green except α1 is blue and α5 is red. For
(Upper), CheX is gray except for α1′, which is cyan. CheY3 is green except α1 is blue and α5 is red. For  (Lower), YPD1 is gray except for αC (teal). SLN1-R1 is pale yellow except α1 is blue and α5 is red.
(Lower), YPD1 is gray except for αC (teal). SLN1-R1 is pale yellow except α1 is blue and α5 is red.
The relative orientation of CheY3 and CheX in the cocrystal structure contrasts that of the three available high resolution structures of a complex containing a receiver domain and a protein which mediates phosphoryl group chemistry (Fig. 2). These structures are E. coli CheY complexed with the CheZ phosphatase (9) and two complexes between a receiver domain and a histidyl phosphotransferase (22, 23), a protein which mediates phosphotransfer between its own histidyl residue and the receiver domain aspartate. In all three structures, a surface comprised of one or two near-parallel alpha helices of the partner protein interacts with the active site region of the receiver domain but the receiver domains are tilted so that the α1 helix is at an angle of about 30° with respect to the dominant interacting helix on the partner protein, in contrast to the 90° angle in  . The receiver domains are also rotated about 40° around an internal axis roughly central and parallel to their β-strands relative to CheY3 (Fig. 2).
. The receiver domains are also rotated about 40° around an internal axis roughly central and parallel to their β-strands relative to CheY3 (Fig. 2).
The Interaction Surface Between CheY3 and CheX Includes the Catalytic Site.
Inspection of the CheY3 active site region (Fig. 3A) reveals that the two strictly conserved CheX residues, Glu96 and Asn99, which reside one helical turn apart on CheX α1′, are adjacent to the CheY3 active site to form a region with an extensive network of hydrogen bonds. CheX Glu96 OE2 forms a salt bridge with conserved residue CheY3 Lys129 NZ as well as a hydrogen bond with CheY3 Thr37 OG1. The CheX Asn99 side chain is pointed toward the 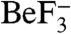 moiety on CheY3 so that Asn99 ND2 forms a hydrogen bond with an ordered water molecule (water B162) which is located in a position close to that expected for in-line attack of the phosphoryl group—3.4 Å from the Be atom and
moiety on CheY3 so that Asn99 ND2 forms a hydrogen bond with an ordered water molecule (water B162) which is located in a position close to that expected for in-line attack of the phosphoryl group—3.4 Å from the Be atom and 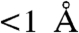 from a direct in-line position. Water B162 forms an additional hydrogen bond with CheY3 Thr81 OG, implicating roles for both CheX Asn99 and CheY3 Thr81 in orienting a water molecule for nucleophilic attack in CheX-mediated catalysis of the dephosphorylation reaction.
from a direct in-line position. Water B162 forms an additional hydrogen bond with CheY3 Thr81 OG, implicating roles for both CheX Asn99 and CheY3 Thr81 in orienting a water molecule for nucleophilic attack in CheX-mediated catalysis of the dephosphorylation reaction.
Fig. 3.

Active site structure and kinetic characterization of CheX and CheY3. (A) Close-up view of the active site. Residues are green for CheY3 and cyan for CheX. The ordered water molecule (Wat B162) is a blue sphere and Mg2+ is a light green sphere. Hydrogen bonds revealed by the structure are represented by black-dashed lines. The red-dashed line between Wat B162 and the beryllium atom (distance of 3.4 Å) is nearly collinear with the bond connecting the beryllium atom and Asp79 OD1. (B) CheX phosphatase activities. The CheY3 concentration is 15 μM. Reactions contained wild-type CheX/wild-type CheY3 (closed squares), CheX 96EA/wild-type CheY3 (closed triangles), CheX 99NA/wild-type CheY3 (closed diamonds), or wild-type CheX/ CheY3 81TA (closed circles). Rates measured in the absence of CheY3 for wild-type CheX (open squares), CheX 96EA (open triangles) and CheX 99NA (open diamonds) reflects nonspecific phosphatase activity due to trace contaminants, and was subtracted out. (C) Schematic model for transition state stabilization in CheX -catalyzed dephosphorylation of CheY3. The bipyramidal transition state is colored black with dashed lines representing partial bonds. Stabilizing interactions are represented with red-dashed lines with the assumption that interactions observed in 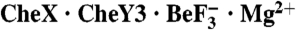 persist in the transition state. One of several possible hydrogen bonding arrangements between CheY3 T81 or CheX N99 and the water is shown. Residues from CheY3 (green) and CheX (cyan) are indicated.
persist in the transition state. One of several possible hydrogen bonding arrangements between CheY3 T81 or CheX N99 and the water is shown. Residues from CheY3 (green) and CheX (cyan) are indicated.
The active site configuration observed in the 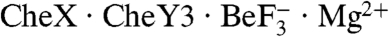 complex is directly comparable to that observed in E. coli
complex is directly comparable to that observed in E. coli 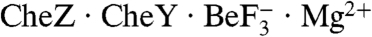 where an acid residue on CheZ (Asp143) forms a salt bridge with CheY Lys109 and an amide residue on the next turn of the CheZ helix (Gln147) inserts into the CheY active site. However, in the
where an acid residue on CheZ (Asp143) forms a salt bridge with CheY Lys109 and an amide residue on the next turn of the CheZ helix (Gln147) inserts into the CheY active site. However, in the  structure, there was not sufficient resolution to assign density to ordered water molecules and the residue analogous to CheY3 Thr81, CheY Asn59, was not implicated as catalytically important based on the structure. However, the analogous residue of phosphatases within the haloacid dehalogenase family, which possess similar folds and active site geometries as response regulators, is conserved as an aspartate and has been proposed to act as a general base toward the nucleophilic water for hydrolysis of the aspartyl-phosphate intermediate (24).
structure, there was not sufficient resolution to assign density to ordered water molecules and the residue analogous to CheY3 Thr81, CheY Asn59, was not implicated as catalytically important based on the structure. However, the analogous residue of phosphatases within the haloacid dehalogenase family, which possess similar folds and active site geometries as response regulators, is conserved as an aspartate and has been proposed to act as a general base toward the nucleophilic water for hydrolysis of the aspartyl-phosphate intermediate (24).
Enzymatic and Genetic Assays Support Mechanistic Roles for CheX and CheY3 Residues Implicated by the Cocrystal Structure.
Potential roles for CheX Glu96, CheX Asn99 and CheY3 Thr81 in the CheX-stimulated dephosphorylation of CheY3 were assessed by enzyme kinetic analysis (Fig. 3B). For wild-type CheY3 in the absence of CheX, the rate of inorganic phosphate (Pi) release (using phosphoimidazole as a phosphodonor) reflects CheY3 autodephosphorylation and displayed a rate constant (7.8 ± 0.1 × 10-4 s-1), similar to measurements obtained by using [32P]CheY3-P (20). Increasing concentrations of wild-type CheX enhanced the Pi release rate in a linear fashion and gave an activity of 3.3 ± 0.3 × 10-2 μM Pi/s·μM CheX. In contrast, CheX N99A and CheX E96A displayed activities of 3.3 ± 1.4% and 9.7 ± 4.4% of wild-type CheX activity, respectively, consistent with catalytic and/or binding roles for both CheX residues. CheY3 T81A exhibited an autodephosphorylation rate constant which was decreased about threefold compared to wild-type CheY3. Moreover, titration of CheY3 T81A with wild-type CheX indicated only ∼1–2% of the CheX sensitivity of wild-type CheY3. Thus, Thr81 plays a modest role in CheY3 autodephosphorylation and a major role in CheX-mediated dephosphorylation. Taken together (see Fig. 3C for a model of proposed transition state and stabilizing interactions), the structural and kinetic data indicate that the CheX Asn99 and CheY3 Thr81 side chains participate directly in catalysis by orienting the nucleophilic water. It is assumed that the proton lost by the attacking water in the reaction is picked up by a solvent water molecule. The primary role of Glu96 appears to be in binding the CheY3-P substrate, based on its involvement in both a hydrogen bond and salt bridge with CheY3 residues (Figure 3A), and consistent with diminished CheY binding affinity of CheC mutants containing substitutions at the analogous acid residues (17).
An independent experiment measuring wild-type CheX activity as a function of wild-type CheY3 concentration demonstrated that the CheX activity determined under the conditions used for Fig. 3B represented a maximal velocity. Thus, CheX enhances the rate of dephosphorylation of CheY3 by a factor of ≈40 (3.3 × 10-2 s-1/7.8 × 10-4 s-1), similar to the enhancement of CheY dephosphorylation by CheZ of ≈100-fold (25).
Cells containing plasmids with the cheX E96A and cheX N99A mutations gave the genetic phenotype predicted for defective phosphatase activity in a complementation assay. Cells with a disabled cheX gene exhibit a constant flexing swimming phenotype due to increased levels of phosphorylated CheY3 (20). A vector expressing a wild-type copy of the cheX gene restored the swimming behavior of the mutant B. burgdorferi cells to wild-type behavior (a combination of run, pause, reverse and flex events), but cells which were transformed with plasmids containing cheX E96A or cheX N99A failed to complement the cheX mutant phenotype and constitutively flexed.
CheX and CheZ Catalytic Mechanisms Are Virtually Identical Despite Distinct Interaction Modes.
The preceding structural and functional analysis demonstrates that two conserved residues on CheX- Glu96 and Asn99- interact directly with the active site of CheY3 and play important roles in catalysis. This is directly comparable to the E. coli chemotaxis phosphatase CheZ where conserved residues Asp143 and Gln147 insert into the CheY active site and possess binding and/or catalytic roles (9). Superposition of the CheY and CheY3 molecules in the  and
and 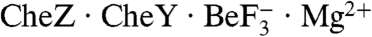 cocrystal structures reveals that whereas the CheX and CheZ structures overlay poorly (Fig. 4A), the catalytic residues assume virtually identical locations with respect to the CheY/CheY3 active site geometry (Fig. 4B). Moreover, the functional atoms within the residues (the carboxylic acid oxygen atom or the amide nitrogen atom) are themselves essentially superimposable between CheX and CheZ. Thus, CheX and CheZ mediate dephosphosphorylation of their partner CheYs by essentially identical mechanisms. It is striking that, despite the near superimposability of the active sites, the alpha helices bearing the catalytic residues in CheX and CheZ are oriented nearly perpendicular with respect to each other (Fig. 4B), a manifestation of the distinct overall orientation of CheY3 relative to CheX discussed above.
cocrystal structures reveals that whereas the CheX and CheZ structures overlay poorly (Fig. 4A), the catalytic residues assume virtually identical locations with respect to the CheY/CheY3 active site geometry (Fig. 4B). Moreover, the functional atoms within the residues (the carboxylic acid oxygen atom or the amide nitrogen atom) are themselves essentially superimposable between CheX and CheZ. Thus, CheX and CheZ mediate dephosphosphorylation of their partner CheYs by essentially identical mechanisms. It is striking that, despite the near superimposability of the active sites, the alpha helices bearing the catalytic residues in CheX and CheZ are oriented nearly perpendicular with respect to each other (Fig. 4B), a manifestation of the distinct overall orientation of CheY3 relative to CheX discussed above.
Fig. 4.

Two structural solutions achieve the same catalytic end. (A) Ribbon representation of B. burgdorferi 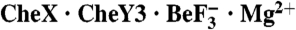 and E. coli
and E. coli 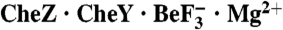 (pdb 1KMI) with superposition of CheY3 (green) and CheY (light green). CheX is cyan and CheZ is orange. CheX Glu96 (Left) and Asn99 (Right) are in cyan sticks. CheZ Asp143 (Left) and Gln147 (Right) are in orange sticks. The
(pdb 1KMI) with superposition of CheY3 (green) and CheY (light green). CheX is cyan and CheZ is orange. CheX Glu96 (Left) and Asn99 (Right) are in cyan sticks. CheZ Asp143 (Left) and Gln147 (Right) are in orange sticks. The 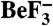 moiety is in purple sticks. (B) Close-up of region within the indicated square from Panel (A). Select active site groups from B. burgdorferi CheY3 (green) or E. coli CheY (light green) are shown. The residues are sticks and the divalent cations (Mg2+ for CheY3 and Mn2+ for CheY) are spheres.
moiety is in purple sticks. (B) Close-up of region within the indicated square from Panel (A). Select active site groups from B. burgdorferi CheY3 (green) or E. coli CheY (light green) are shown. The residues are sticks and the divalent cations (Mg2+ for CheY3 and Mn2+ for CheY) are spheres.
How can the two distinct orientations of an alpha helix observed with CheX and CheZ result in near identical placement of two catalytically essential residues which are, in both proteins, separated by a single turn of an alpha helix? A conspicuous difference between the catalytic pair in CheX (Glu96 and Asn99) and CheZ (Asp143 and Gln147) is their separation by two other residues in CheX and three residues in CheZ. As a result, the Cα atoms of the two catalytic residues are separated by different distances in CheX (4.79 Å) and CheZ (6.25 Å) and the side chains project from the helices at different angles. Hence, a different orientation of the helix bearing these residues is necessary to get the functional atoms of side chains in the same approximate positions in space, and a near perpendicular backbone orientation achieves this goal. Finally, the positions of the functional atoms are fine-tuned by the different lengths of the side chains. The longer side chain of the acid residue (Glu96) and shorter side chain of the amide residue (Asn99) in CheX versus CheZ (Asp143 and Gln147, respectively) places the functional atoms in nearly identical positions (Fig. 4B).
Discussion
Identical Catalytic Strategies Achieved Through Distinct Binding Modes.
CheX and the structurally unrelated CheZ both use a pair of functional groups—an acid and an amide—as a bidentate prong which inserts into their respective CheY active sites. Differences in residue spacing on the primary sequence and side chain length are compensated for by disparate binding orientations of CheX and CheZ with the end result of the acid and amide groups positioned in virtually identical locations within the CheY active site. Examples of such “mechanistic” convergent evolution amongst enzyme active sites have been documented (26, 27). However, the mechanistic convergence demonstrated by CheX and CheZ is distinct from that of conventional enzymes which catalyze reactions of small molecules. CheX and CheZ do not possess a typical active site pocket, but instead bind a protein substrate and contribute additional functional groups to enhance the activity of a preexisting active site. Thus, CheX and CheZ have evolved both the ability to bind their protein substrate with appreciable affinity [Fig. S2, (25)] and to provide the appropriate functional groups in the right locations at the protein interface to mediate catalysis. CheX and CheZ achieve these dual abilities quite differently. CheX contains one protein surface which mediates both binding and catalysis whereas CheZ separates the binding and catalytic functions into two separate domains (9).
Could the Conserved Catalytic Mechanism Used by CheX and CheZ Be Utilized by Structurally Different Classes of Response Regulator Phosphatases?
The identical geometries of the active sites in CheX·CheY3 and CheZ·CheY raises the possibility that the conserved catalytic strategy represents a universal solution for stimulation of response regulator dephosphorylation. The Spo0E family of response-regulator phosphatases form short helical hairpins with a conserved (-SQELD-) motif (28), containing an exposed Asp/Gln pair. However, whereas the identities of the Asp/Gln pair match CheZ, they are in reverse order and their separation by two residues is reminiscent of CheX. Mutation of the conserved Spo0E aspartate has a greater detrimental affect on catalytic activity than mutation of the glutamine, leading to the proposal that the aspartate interacts with the nucleophilic water (29). It is possible that the Spo0E helix binds to its substrate with the same directionality as observed for both CheX and CheZ. Based on the separation of the glutamine and aspartate by two residues, it is predicted that the helix bearing the catalytic dyad would be oriented similarly to the CheX helix. This interaction would put the Spo0E glutamine in the position to form a hydrogen bond with the response regulator lysine and the Spo0E aspartate in the position to interact with the attacking water molecule. This model, if shown to be correct, would suggest that a common fundamental catalytic strategy could be modified by adjusting the chemical properties of similarly located residues.
Implications for Interaction of CheY with CheC.
The mode of binding between phosphorylated CheY and CheX described here should be applicable to CheC and FliY, two other CheY phosphatases which are closely related to CheX. CheC has a similar topology as CheX but is monomeric with two active site motifs—one on α1′ (as in CheX) and one on α1 (15, 17). We generated a reasonable model for a  complex (Fig. S4) by overlaying a stretch of ten residues centered around the conserved Glu/Asn for each catalytic motif in CheC with the homologous region in CheX. CheC and the deamidase, CheD, form a heterodimer which mediates reciprocal regulation of enzymatic activities (30), but the mechanism by which CheD activates CheC is not known. Superposition of CheC from T. maritima CheC·CheD (pdb 2F9Z) with CheC within the modeled CheC·
complex (Fig. S4) by overlaying a stretch of ten residues centered around the conserved Glu/Asn for each catalytic motif in CheC with the homologous region in CheX. CheC and the deamidase, CheD, form a heterodimer which mediates reciprocal regulation of enzymatic activities (30), but the mechanism by which CheD activates CheC is not known. Superposition of CheC from T. maritima CheC·CheD (pdb 2F9Z) with CheC within the modeled CheC·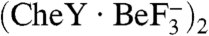 complex reveals that CheD is, at its closest,
complex reveals that CheD is, at its closest, 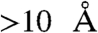 removed from CheY (Fig. S4), inconsistent with direct interaction between CheD and CheY in a ternary complex. Therefore CheD does not appear to act to expand the surface of interaction with CheY, and may instead act as an allosteric effector of CheC.
removed from CheY (Fig. S4), inconsistent with direct interaction between CheD and CheY in a ternary complex. Therefore CheD does not appear to act to expand the surface of interaction with CheY, and may instead act as an allosteric effector of CheC.
CheY3-Dependent Dissociation of CheX2: Possible Mechanism and Function.
The mechanism by which binding  (and presumably CheY3–P) induces the dissociation of the stable CheX2 dimer remains to be determined. The apparent limited degree of overlap between CheY3 and CheX interaction surfaces on the CheX monomer (Fig. S5) suggests that
(and presumably CheY3–P) induces the dissociation of the stable CheX2 dimer remains to be determined. The apparent limited degree of overlap between CheY3 and CheX interaction surfaces on the CheX monomer (Fig. S5) suggests that 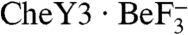 might bind directly to the intact CheX2 dimer which could somehow induce dissociation of CheX2. Alternatively, the CheX2 dimer could be in equilibrium with a small amount of monomer.
might bind directly to the intact CheX2 dimer which could somehow induce dissociation of CheX2. Alternatively, the CheX2 dimer could be in equilibrium with a small amount of monomer.  could selectively bind monomeric CheX, thus "pulling" the dimer/monomer equilibrium toward the monomeric state. The functional significance of dissociation of the CheX2 dimer in the CheX-mediated dephosphorylation of CheY3 also remains to be determined. One possibility is that CheX2 dimer dissociation could function to expose a surface on CheX which allows interaction with an auxiliary regulatory protein, as seen with CheC and the Rap family of response regulator phosphatases.
could selectively bind monomeric CheX, thus "pulling" the dimer/monomer equilibrium toward the monomeric state. The functional significance of dissociation of the CheX2 dimer in the CheX-mediated dephosphorylation of CheY3 also remains to be determined. One possibility is that CheX2 dimer dissociation could function to expose a surface on CheX which allows interaction with an auxiliary regulatory protein, as seen with CheC and the Rap family of response regulator phosphatases.
Materials and Methods
Expression Plasmids, Protein Expression and Protein Purification.
Plasmid derivatives of pQE30 (Qiagen Inc.) encoding His-tagged CheX and CheY3 have been described (20). Plasmids encoding CheX E96A and CheX N99A were generated from the wild-type cheX plasmid by site directed mutagenesis (QuikChange, Stratagene). Plasmids were transformed into M15/pREP4 cells for overexpression as described (20). For incorporation of seleno-methionine (Se-Met) into CheX for crystal growth, the M15/pREP4.pQE30 CheX strain was grown in minimal media supplemented with 19 amino acids (all except methionine). L-seleno-methionine (100 mg per liter) was added immediately before overnight induction at 30 °C. His-tagged CheX and CheY3 were purified from crude lysates by using Ni2+-NTA affinity chromatography according to manufacturer’s instructions (Qiagen Inc.) followed by gel filtration chromatography (Superose 12, GE Biosciences) in buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl. Protein concentration was determined by absorbance at 280 nm. Extinction coefficients (0.237 (mg/mL)-1 cm-1 for CheX and 0.255 (mg/mL)-1 cm-1 for CheY3) were estimated by using the Protparam utility (http://ca.expasy.org).
Crystallization and Data Collection.
Crystals were prepared in a sitting drop format by using Microbridges (Hampton Research). Drops contained 2 μL of reservoir buffer (0.1 M sodium citrate, pH 5.6 and 1.5 M ammonium sulfate) and 2 μL of a mixture of CheY3 (2.6 mg/mL), SeMet-CheX (2.5 mg/mL), 20 mM MgCl2, 0.75 mM BeCl2, and 20 mM NaF. The protein mix also contained Tris buffer (23 mM) and NaCl (69 mM) due to contribution from protein stock solutions. Crystals required 1–2 weeks to grow to maximal size (200 μm × 90 μm × 90 μm). Crystals were cryoprotected by using Fomblin Y LVAC 14/6 perfluoropolyether vacuum pump oil (Aldrich) and flash frozen in liquid nitrogen. A single anomalous diffraction (SAD) dataset was collected at the peak wavelength of Se (0.97923 Å) to a resolution of 1.96 Å at the Advanced Photon Source at Argonne National Labs, Beamline 22-ID (SER-CAT consortium).
Structural Determination and Refinement.
The dataset was processed with the HKL-2000 suite (31) and the structure was solved by using ShelXD (32) and RESOLVE (33, 34). Four of the six Se-Met residues were located and initial models were created manually by using E. coli CheY·BeF3·Mn2+ (pdb 1FQW) and T. maritima CheX (pdb 1XKO) based on the location of the Se atoms. SWISS-MODEL homology modeling program (35) (http://swissmodel.expasy.org/) was then used to create an updated model which contained the B. burgdorferi CheY3 and CheX sequences. We incorporated the new model, by using rigid body refinement in Refmac5 from CCP4 (36), to orient it in the high resolution SAD maps and then initiated refinement. Manual model building was performed by using Coot (37) and further restrained refinement was carried out by using Refmac5 in CCP4 (36). The final stages of model building and refinement were carried out by using O (38) and Crystallography and NMR System (39), respectively. Crystallization parameters are in Table S1.
Measurement of CheX Phosphatase Activity.
Inorganic phosphate release was measured in reactions containing CheY3, CheX, and the phosphodonor monophosphoimidazole by using a spectroscopic enzyme-linked assay (EnzChek Pi, Invitrogen), as previously described (25).
Molecular Weight Determination.
His-tagged CheX was eluted off the Ni2+-NTA agarose column, dialyzed at 4 °C versus 2 × 500 mL of buffer (20 mM Hepes, pH 7.0, 200 mM NaCl, and 1 mM EDTA), concentrated to ∼2 mg/mL, and filtered. The sample (100 μL) was then chromatographed on a Superdex 200 10/30 column (GE Healthcare) at 0.5 mL/ min in the same buffer used for dialysis. The column eluent was fed directly into a DAWN EOS multiangle light scattering spectrometer, followed by an OptiLab rEX Refractive Index detector, and a quasi-elastic light scattering module Wyatt QELS (Wyatt Technology Corporation). The light scattering and refractive index outputs were recorded continuously and the data combined to determine the polydispersity and the number average molecular weight.
Genetic Complementation Experiments.
Shuttle vectors expressing CheX E96A and CheX N99A were constructed and transformed into B. burgdorferi cheX::kan cells as described for wild-type CheX (20). Western blots demonstrated that cells transformed with the mutant and wild-type cheX genes expressed similar levels of CheX. Dark field microscopy was carried out as described in ref. (20).
Supplementary Material
Acknowledgments.
We are indebted to Robert Bourret for expert review of the manuscript and Brenda Temple, Laurie Betts, and Shawn Williams for advice with structural solution and analysis. We thank Ashutosh Tripathy (UNC Macromolecular Interactions Facility) for assistance with light scattering and the Advanced Photon Source at Argonne National Labs for X-ray data collection. This work was supported by National Institutes of Health Grants GM050860 (to Robert B. Bourret), AR054582 (to M. A. M.), AI29743 (to N. W. C) and GM080334 (to R. Z.).
Note.
In the recent structure of a T. maritima histidine kinase/receiver domain complex (40), the orientation of the receiver domain and the kinase resembles that of the three previously solved receiver domain-containing complexes discussed here.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911185107/DCSupplemental.
References
- 1.Laub MT, Goulian M. Specificity in two-component signal transduction pathways. Annu Rev Genet. 2007;41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 2.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 3.Calva E, Oropeza R. Two-component signal transduction systems, environmental signals, and virulence. Microb Ecol. 2006;51:166–176. doi: 10.1007/s00248-005-0087-1. [DOI] [PubMed] [Google Scholar]
- 4.Bahn YS. Master and commander in fungal pathogens: the two-component system and the HOG signaling pathway. Eukaryot Cell. 2008;7:2017–2036. doi: 10.1128/EC.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao R, Mack TR, Stock AM. Bacterial response regulators: Versatile regulatory strategies from common domains. Trends Biochem Sci. 2007;32:225–234. doi: 10.1016/j.tibs.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao R, Stock AM. Biological insights from structures of two-component proteins. Annu Rev Microbiol. 2009:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci. 2001;26:369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 8.Kirby JR, et al. CheC is related to the family of flagellar switch proteins and acts independently from CheD to control chemotaxis in Bacillus subtilis. Mol Microbiol. 2001;42:573–585. doi: 10.1046/j.1365-2958.2001.02581.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E coli chemotaxis phosphatase CheZ. Nat Struct Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 10.Perego M, et al. Multiple protein-aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in B. subtilis. Cell. 1994;79:1047–1055. doi: 10.1016/0092-8674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 11.Porter SL, Roberts MA, Manning CS, Armitage JP. A bifunctional kinase-phosphatase in bacterial chemotaxis. Proc Natl Acad Sci USA. 2008;105:18531–18536. doi: 10.1073/pnas.0808010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Qin L, Yoshida T, Inouye M. Phosphatase activity of histidine kinase EnvZ without kinase catalytic domain. Proc Natl Acad Sci USA. 2000;97:7808–7813. doi: 10.1073/pnas.97.14.7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohlsen KL, Grimsley JK, Hoch JA. Deactivation of the sporulation transcription factor Spo0A by the Spo0E protein phosphatase. Proc Natl Acad Sci USA. 1994;91:1756–1760. doi: 10.1073/pnas.91.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muff TJ, Ordal GW. The diverse CheC-type phosphatases: chemotaxis and beyond. Mol Microbiol. 2008;70:1054–1061. doi: 10.1111/j.1365-2958.2008.06482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park SY, et al. Structure and function of an unusual family of protein phosphatases: the bacterial chemotaxis proteins CheC and CheX. Mol Cell. 2004;16:563–574. doi: 10.1016/j.molcel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 16.Szurmant H, Muff TJ, Ordal GW. Bacillus subtilis CheC and FliY are members of a novel class of CheY-P-hydrolyzing proteins in the chemotactic signal transduction cascade. J Biol Chem. 2004;279:21787–21792. doi: 10.1074/jbc.M311497200. [DOI] [PubMed] [Google Scholar]
- 17.Muff TJ, Ordal GW. The CheC phosphatase regulates chemotactic adaptation through CheD. J Biol Chem. 2007;282:34120–34128. doi: 10.1074/jbc.M706432200. [DOI] [PubMed] [Google Scholar]
- 18.Muff TJ, Foster RM, Liu PJ, Ordal GW. CheX in the three-phosphatase system of bacterial chemotaxis. J Bacteriol. 2007;189:7007–7013. doi: 10.1128/JB.00896-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charon NW, Goldstein SF. Genetics of motility and chemotaxis of a fascinating group of bacteria: the spirochetes. Annu Rev Genet. 2002;36:47–73. doi: 10.1146/annurev.genet.36.041602.134359. [DOI] [PubMed] [Google Scholar]
- 20.Motaleb MA, et al. CheX is a phosphorylated CheY phosphatase essential for Borrelia burgdorferi chemotaxis. J Bacteriol. 2005;187:7963–7969. doi: 10.1128/JB.187.23.7963-7969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge Y, Charon NW. Molecular characterization of a flagellar/chemotaxis operon in the spirochete Borrelia burgdorferi. FEMS Microbiol Lett. 1997;153:425–431. doi: 10.1111/j.1574-6968.1997.tb12606.x. [DOI] [PubMed] [Google Scholar]
- 22.Varughese KI, Tsigelny I, Zhao H. The crystal structure of beryllofluoride Spo0F in complex with the phosphotransferase Spo0B represents a phosphotransfer pretransition state. J Bacteriol. 2006;188:4970–4977. doi: 10.1128/JB.00160-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X, Copeland DM, Soares AS, West AH. Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J Mol Biol. 2008;375:1141–1151. doi: 10.1016/j.jmb.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, et al. Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic "snapshots" of intermediate states. J Mol Biol. 2002;319:421–431. doi: 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- 25.Silversmith RE, Levin MD, Schilling E, Bourret RB. Kinetic characterization of catalysis by the chemotaxis phosphatase CheZ. Modulation of activity by the phosphorylated CheY substrate. J Biol Chem. 2008;283:756–765. doi: 10.1074/jbc.M704400200. [DOI] [PubMed] [Google Scholar]
- 26.Gherardini PF, Wass MN, Helmer-Citterich M, Sternberg MJ. Convergent evolution of enzyme active sites is not a rare phenomenon. J Mol Biol. 2007;372:817–845. doi: 10.1016/j.jmb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 27.Doolittle RF. Convergent evolution: The need to be explicit. Trends Biochem Sci. 1994;19:15–18. doi: 10.1016/0968-0004(94)90167-8. [DOI] [PubMed] [Google Scholar]
- 28.Grenha R, et al. Structural characterization of Spo0E-like protein-aspartic acid phosphatases that regulate sporulation in bacilli. J Biol Chem. 2006;281:37993–38003. doi: 10.1074/jbc.M607617200. [DOI] [PubMed] [Google Scholar]
- 29.Diaz AR, et al. Functional role for a conserved aspartate in the Spo0E signature motif involved in the dephosphorylation of the Bacillus subtilis sporulation regulator Spo0A. J Biol Chem. 2008;283:2962–2972. doi: 10.1074/jbc.M709032200. [DOI] [PubMed] [Google Scholar]
- 30.Chao X, et al. A receptor-modifying deamidase in complex with a signaling phosphatase reveals reciprocal regulation. Cell. 2006;124:561–571. doi: 10.1016/j.cell.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collection in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: Aweb-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 36.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 39.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.