

Abstract
Recent progress in elucidating the peptide bond formation process on the ribosome has led to notion of a proton shuttle mechanism where the 2'-hydroxyl group of the P-site tRNA plays a key role in mediating proton transfer between the nucleophile and leaving group, whereas ribosomal groups do not actively participate in the reaction. Despite these advances, the detailed nature of the transition state for peptidyl transfer and the role of several trapped water molecules in the peptidyl transferase center remain major open questions. Here, we employ high-level quantum chemical ab initio calculations to locate and characterize global transition states for the reaction, described by a molecular model encompassing all the key elements of the reaction center. The calculated activation enthalpy as well as structures are in excellent agreement with experimental data and point to feasibility of an eight-membered “double proton shuttle” mechanism in which an auxiliary water molecule, observed both in computer simulations and crystal structures, actively participates. A second conserved water molecule is found to be of key importance for stabilizing developing negative charge on the substrate oxyanion and its presence is catalytically favorable both in terms of activation enthalpy and entropy. Transition states calculated both for six- and eight-membered mechanisms are invariably late and do not involve significant charge development on the attacking amino group. Predicted kinetic isotope effects consistent with this picture are similar to those observed for uncatalyzed ester aminolysis reactions in solution.
Keywords: density functional theory, protein synthesis
The peptide bond formation step in protein synthesis is catalyzed by the peptidyl transferase center (PTC) on the large ribosomal subunit. This reaction involves the attack of the α-amino group of an aminoacyl-tRNA molecule bound to ribosomal A-site on the ester carbon of the peptidyl-tRNA in the adjacent P-site of the ribosome. The growing peptide chain is thereby transferred to the A-site tRNA and elongated by one amino acid. Recent years have witnessed considerable progress in our understanding of the peptidyl transfer process due to high-resolution crystallographic structures of the large ribosomal subunit with transition state (TS) analogs (1), as well as kinetic measurements (2–9), mutagenesis data (4, 10–14), and computational studies (15–17). Hence, the current model of the peptidyl transfer reaction is that ribosomal nucleobases are not directly involved in bond making or breaking through acid-base catalysis (4, 10, 11, 13, 18, 19), but that the A76 2′-OH group of the P-site substrate plays a key role in mediating proton transfer from the attacking nucleophile to the leaving 3′ ester oxygen (1, 15, 20–22). Furthermore, it was shown that the lower free energy barrier for the ribosome reaction, compared to an uncatalyzed reference reaction in water, is entirely due to a less negative activation entropy (3, 6, 7). Besides contributions from substrate proximity and alignment effects, computer simulations predicted that a preorganized hydrogen bonding network, involving several water molecules together with ribosomal hydroxyl groups, is a major source of this entropic stabilization on the ribosome (15–17, 23).
A major open question is, however, the detailed nature of the reaction mechanism and its transition states. The latter have not been characterized at the electronic level and it is e.g., still not clear whether the rate-limiting TS is “early” or “late” (Fig. 1). That is, is the highest energy cost associated with forming the new C - O bond or is it rather associated with breaking the C - O ester bond? Strobel and coworkers measured 15N kinetic isotope effects (KIEs) for the reaction with an A-site substrate analog, a CC-puromycin derivative, on the 50S ribosomal subunit and suggested an early TS based on these experiments (24). Subsequently reported Brønstedt plots (25) showed a very weak dependence on nucleophile pKa, compatible with both early and late transition states, and were interpreted in terms of an essentially neutral nucleophile in the TS. Computer simulations employing the empirical valence bond (EVB) method as well as quantum chemical ab initio calculations on a small model system (15–17), on the other hand, indicated a late TS that is mostly associated with elongation of the ester C - O3′ bond.
Fig. 1.

Schematic overview of the peptidyl transfer reaction. The top row shows examples of an early and late TS while the bottom row depicts the proposed six- and eight-membered reaction mechanisms.
Second, it remains to be shown how the proton transfer proceeds from the α-amino group to the leaving A76 O3′ of the P-site substrate. Several different reaction routes have been suggested, the most important being a six-membered mechanism where proton shuttling goes through the 2′-hydroxyl group of the P-site A76 (20) and an eight-membered TS involving a water molecule as an additional proton relay (1) (see Fig. 1). The eight-membered mechanism was suggested by Steitz et al. based on the observation of a water molecule in close contact with both the O2' and O3' of the P-site A76 (1), but has never been further explored. The presence of this water molecule had been predicted by molecular dynamics (MD) simulations (15, 17) and was later confirmed in a high-resolution crystal structure of the RAP TS analog bound to the PTC (1). None of the other proposed mechanisms (26, 27) have yielded reasonable energetics in calculations (15, 28).
Third, there is another water molecule that is in a perfect position to stabilize any developing negative charge in the transition state on the ester carbonyl oxygen of the P-site substrate. Simulations have predicted this water molecule to be hydrogen bonded to the carbonyl oxygen (15, 17), and it was subsequently observed experimentally (1, 29). Although it has been recognized for some time that this seems to be the only chemical group directly interacting with the substrate “oxyanion”, its role in the reaction remains obscure.
Finally, the charge development on the amino nitrogen and the carbonyl oxygen in the TS is also of great interest. As mentioned above, Brønstedt plots suggest an essentially uncharged amine in the TS (25), and it is still not clear whether there is an accumulation of negative charge on the ester carbonyl oxygen. That is, can we speak of an “oxyanion” in this reaction? All of these questions can be adressed with high-level ab initio quantum chemical calculations provided that they reproduce both experimental rate measurements and crystal structures. For this reason we set out to build a model of the PTC that is large enough to capture the leading contribution of the ribosomal environment on bond making and breaking but small enough to allow for quantitative quantum chemical calculations.
With a 76 atom subset of the PTC, we are able to find unconstrained transition states of this complex by first fixing and then gradually releasing the nonreactive scaffolding. The idea being to let the inert scaffolding and water molecules settle into a configuration that is consistent with the transition state. Such a configuration can be obtained only after numerous iterations until the transition state optimizations converged on a fully relaxed structure. By virtue of this method the resulting TS complex can be compared directly with high-resolution crystal structures and the energies with experimental energy barriers from rate measurements and kinetic isotope effects. As will be shown, these calculations yield a consistent picture of the peptidyl transfer reaction with unprecedented quantitative agreement with experimentally determined structures and activation enthalpies on the ribosome, thus opening up a window to explore the intrinsic properties of the peptidyl transfer reaction.
Results
The PTC model that was excised from representative snapshots of previous MD simulations (15) and then optimized with ab initio density functional theory (DFT) methods is shown in Fig. 2A. Apart from the reacting species it includes the catalytically important 2′-hydroxyl group of A2451 (13, 30), the 2′-hydroxyl of C2063, which interacts with the water molecule that bridges the 2′- and 3′-oxygens of A76, and five hydrogen bonded water molecules (E. coli numbering is used throughout). In this model the catalytic hydroxyl groups were represented by methanol molecules and the peptidyl-tRNA substrate with a dipeptide (X-Ala), where the first residue was truncated at the α-carbon. The incoming A-site aminoacyl-tRNA was modeled as alanine and terminated at the C3′-atom of the tRNA. These groups were chosen to faithfully represent the environment in the PTC and turn out to give excellent agreement with crystal structures, without using any biasing constraints, thus indicating that the key interactions are indeed taken into account.
Fig. 2.

(A) The small model used for ab initio optimizations forming the eight-membered TS. (B) The eight-membered TS (in green) superimposed on a crystal structure of the RAP transition state analog (in yellow) bound to the peptidyl transferase center (in blue) (1). Crystal waters are shown as spheres whereas ab initio water molecules are shown with hydrogens. The water molecule that prevents the optimization of a six-membered transition state is positioned almost precisely where ordered crystal water appears in the experimental structure. In addition, there is striking agreement between the positioning of the A2451 and C2063 2′-hydroxyl groups and the methanol fragments that model these functional groups.
As the initial positions were gradually relaxed to a local minimum with DFT (B3LYP/6-31G*), a water molecule that starts out hydrogen bonded to the C2063 2′-OH persistently finds an entrapped location above the A76 ribose moiety (Fig. S1). Poised in contact with the A76 2′-hydroxyl group, the water molecule then straddles the O3′ and the C2063 O2′ in the TS and forms a nearly equilateral triangle in the plane ecliptic to the reacting heavy atoms. This is almost precisely the configuration in which a water molecule is found in the high-resolution crystal structure of the RAP TS analog bound to the PTC (Fig. 2B). Interestingly, the configuration was found even though the starting position for the optimization was taken from an MD trajectory that was somewhat different from the 1VQP structure in this respect. In fact, attempting to put water molecules anywhere in the vicinity of the O2′ and O3′ would result in this geometry. Thus DFT seems to confirm the position of this crystal water, as well as the included 2′-hydroxyl groups of the C2063 and A2451 residues. In the presence of this entrapped water molecule, the ab initio search algorithms persistently optimize the geometry to an eight-membered transition state where this water acts as a proton shuttle. Although the six-membered TS was initially of more interest, it could only be found when all explicit solvent molecules and the C2063 2′-OH group were removed from this region altogether (see Fig. S1), a solution that inevitably produces higher reaction barriers due to desolvation of the 2′- and 3′-oxygens of the P-site A76 (vide infra).
To find out whether this eight-membered reaction mechanism is concerted or stepwise ground state potential surfaces were calculated with partially relaxed geometry scans (B3LYP/6-31G). The potential surface of the eight-membered mechanism is a function of at least four distances (see inset of Fig. 3A): That between the ester carbonyl carbon and the leaving O3′; the polar amine hydrogen and the O2′; the A76 H2′ and the water bridge oxygen; the water bridge hydrogen and the leaving O3′. A total number of 265 points were calculated on this surface with as many geometry optimizations (Fig. S2) and a subset of the surface containing the approximate TS is shown in Fig. 3A. From these contour plots it can be seen that the transition state is late as both C - O bond cleavage and protonation of the leaving group lag behind proton shuttling via the 2′-oxygen. In addition, a scan of the energy surface for the six-membered mechanism was carried out in order to explore the possibility of an early TS occurring at larger N - C distances. That such a transition state does not exist is evident both from surface of Fig. S3 and from the fact that the reaction path from the reactants to the global TS is barrierless (see below). Corresponding scans of the six-membered transition state with implicit solvent have also been reported by Sharma et al. (16).
Fig. 3.

(A) The ground state potential surface of the four reactive distances shown in the inset. Each vertex in the grid corresponds to a partially optimized structure using DFT (B3LYP/6-31G). At these C-O3′ distances the carbonyl tetrahedral center forms spontaneously and the N-C bond is at an equilibrium. A global saddle point (TS) is located close to the encircled vertex and the reaction progresses from reactant (R) to product (P) in the direction of the arrows. (B–C) The minimum energy path found by following the intrinsic reaction coordinate (IRC) from the transition states (inset figures) towards the reactants and products for the six- (B) and eight-membered (C) mechanisms. Energy decompositions are shown along these paths with MP2//B3LYP energies, corrections to the vibrational enthalpy and the experimental reaction barrier. The final energy estimates are thus given by 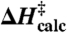 .
.
Following these scans, the Berny algorithm could optimize unconstrained six- and eight-membered global transition states at the B3LYP/6-311G** level by the iterative approach outlined in the introduction. The resulting electronic energy barriers are shown in Fig. 3B, C together with subsets of the minimum energy paths connecting the TS with the reactants and products. Since the reactant and transition states from these calculations are in local minima of the ground state potential with respect to all degrees of freedom besides the reaction coordinate, it is straightforward to obtain the vibrational eigenmodes for these states. These in turn permit the calculation of enthalpies and kinetic isotope effects. The effect of vibrational enthalpies along the minimum energy path around the transition states is also shown in Fig. 3B, C. It can immediately be seen that the calculated 18 kcal/mol activation enthalpy of the eight-membered mechanism is in excellent agreement with the ribosomal enthalpy barriers of about 17 kcal/mol (3, 7), while the six-membered transition state is about 12 kcal/mol higher in energy. However, these calculations do not necessarily rule out a six-membered mechanism because the higher reaction barrier may be due to incomplete solvation of the O2′ and O3′ (see e.g. ref. 16), and the auxiliary water molecule was also found to stabilize a six-membered TS in earlier EVB simulations (15). The detailed geometries and atomic partial charges from natural population analysis (NPA) of these transition states are shown in Fig. 4A, B. It is evident from these structures that the new N - C bond is essentially formed in the TS, with a bond distance of ∼1.5 Å, and that it is the C - O ester bond that is significantly elongated in the TS with a length of about 2.1 Å. Hence, both mechanisms definitely have a late transition state.
Fig. 4.

Bond lengths and NPA charges of the four transition states optimized here. (A) The six-membered transition state. (B–D) Eight-membered transition states with (B) a single screening oxyanion water, (C) a naked and (D) a doubly screened oxyanion.
To examine the effect of the oxyanion bound water molecule on this reaction, the eight-membered TS was used as input guesses for two additional TS optimizations: One of an unscreened oxyanion with no interacting water molecule and the other with the inclusion of an extra screening water molecule. Their resulting TS structures and partial charges are shown in Fig. 4C, D. Whereas the N - C bond is seen to be fully formed in all three cases, the addition of solvent screening on the oxyanion gradually reduces bond cleavage between the leaving O3′ and carbonyl carbon. Judging from the bond lengths there is a significant qualitative relation between oxyanion screening and C - O bond length in the TS. Solvent polarization of the oxyanion due to these water molecules was examined by comparing the TS NPA charges of the amino nitrogen, the leaving carbonyl carbon, and the oxyanion with the corresponding reactant state charges (Fig. 5A). As negative charge is building up on the carbonyl oxygen, it is diminishing on the leaving O3′. This could be compared with the amine nitrogen charge which is more or less unaffected by this screening. The impact on the enthalpy barrier is shown in the same graph. As screening water molecules are added to the carbonyl oxygen the activation enthalpies go down. A longstanding conundrum in ribosomal peptidyl transfer is why the activation enthalpy is higher on the ribosome than in the solution reaction. One screening water molecule is seen to give an enthalpy comparable to rate measurements on the ribosome. Adding an additional water molecule lowers the enthalpy barrier, moving it closer to the solution reaction, and removing screening water altogether increases the activation enthalpy by 3 kcal/mol. Indeed the presence of one such ordered oxyanion screening water molecule was predicted by our earlier MD simulations (15, 17) and later observed in crystal structures of TS analogs (1, 29). Natural bond orbital (NBO) occupancies and the corresponding Lewis structures (accounting for ∼98.8% of the total occupancy) and Wiberg natural atomic orbital (NAO) bond orders of the 3′-oxygen, carbonyl and amino group are shown in Fig. 5B. The quantum chemical effect of having these water molecules screening the carbonyl oxygen is that the leaving O3′ loses a formal negative charge to the carbonyl oxygen thus transforming it into a formal oxyanion. In the NBO picture, solvent screening is stabilizing the extra oxyanion lone pair orbital by allowing a transfer of populations from the leaving O3′ lone pair orbital to the oxyanion. The corresponding orbitals are plotted in the inset of Fig. 5A.
Fig. 5.

(A) Activation enthalpies (left ordinate) and NPA charges (right ordinate) for the eight-membered TSs with no, one and two screening waters on the oxyanion (abscissa). The charges are given with respect to the reactant state (unfilled markers) and the TSs (filled markers) for the oxyanion (circles), the leaving O3′ (squares) and the attacking nitrogen atom (triangles) as given in the inset scheme. (B) Natural Lewis structures for the three TSs following the same order as in (A). Natural bond orbital populations are given within parantheses and Wiberg bond orders on the NAO basis are given in plain text. Oxyanion π natural bonding and lone pair orbitals are also shown as insets in (A), in purple and blue respectively. As the oxyanion is screened, populations are gradually transferred from the π bonding C = O orbital to an additional oxygen lone pair orbital ending in a formally charged oxygen species. The opposite behavior is observed with the leaving O3′ ester oxygen, thus making this oxygen pair an effective charge relay in this reaction.
The oxyanion screening water molecule is clearly stabilizing the transition state and affecting its qualitative features. It should be noted however that there is a nontrivial entropy cost involved when positioning water molecules in this fashion. An accurate evaluation of entropies requires extensive sampling of the configuration space in a more complete model of the PTC where these groups are being correctly positioned by the ribosome. This is a task best suited for molecular dynamics simulations and is beyond the scope of this study. However, the vibrational entropy terms that are directly associated with the hydrogen bond interactions can be readily obtained from the calculated eigenmodes in the harmonic approximation (Table S1). From these values the relative entropy cost (-TΔΔS‡) of adding one screening water is only 0.3 kcal/mol at room temperature, whereas it is 3 kcal/mol higher when adding the second water molecule. It is also interesting to note that the eight-membered TS with one oxyanion water is about 2 kcal/mol entropically more favorable than the six-membered mechanism.
By virtue of the iterative TS search-and-relax procedure and the resulting vibrational normal modes, the kinetic isotope effects for these transition states can be estimated. The KIEs from substituting the α-amino nitrogen, A76 3′-oxygen, leaving carbonyl carbon and oxyanion are shown in Table 1. As expected from the calculated TS geometries these KIEs are all indicative of a late transition state dominated by C - O bond fission. That is, the 14N/15N KIE is inverse which reflects an increase of bond order to the nitrogen in the TS compared to reactants, while the leaving group 16O/18O effect is normal and relatively large which is consistent with a significant degree of C - O bond breaking.
Table 1.
Kinetic isotope effects (KIE) at 310 K of the different reactive atoms.
| Eight-membered | |||||
| Atom | Isotope | Six-membered | O = C < | H2O·O = C < | (H2O)2·O = C < |
| α-amino N | 15N | 0.979 | 0.981 | 0.989 | 0.989 |
| Leaving O3′ | 18O | 1.023 | 1.038 | 1.037 | 1.036 |
| Carbonyl C | 13C | 1.025 | 1.013 | 1.019 | 1.028 |
| Carbonyl O | 18O | 1.001 | 1.002 | 1.004 | 1.010 |
The calculated 14N/15N KIEs are also largely consistent with measurements of similar reactions in aqueous solution, such as 0.996 for the acylation of aniline (31) and 0.990 for the hydrazinolysis of methyl formate (32), indicating that the fundamental reaction mechanism in this ribosome model is similar to the uncatalyzed process. Since the reported isotope effects in Table 1 refer to a reactant state with associated substrates they should strictly speaking be compared to experimental KIEs on kpep (the peptidyl transfer rate) rather than V/K measured in (24), which also includes the substrate binding step. However, by eliminating low frequency vibrations, corresponding to intermolecular substrate interactions (33), from the reactant state in the isotope effect calculations the KIEs with respect to isolated A- and P-site substrates can be estimated. This does not change the qualitative picture from Table 1 and, for instance, the corresponding KIEs for the eight-membered TS with the single oxyanion water molecule are 0.980, 1.021, and 1.016 for the amino, leaving group O3′ and ester carbon, respectively (an additional check with optimization of an isolated A-site substrate surrounded by four water molecules also yielded an inverse 14N/15N KIE). Hence, we can conclude that all KIEs are consistent with a late TS that is mainly associated with C - O bond fission.
Discussion
The aim of this work is to describe the detailed chemical mechanism of the peptidyl transfer reaction and to answer two longstanding questions: Whether the transition state is early or late, and what the precise role of water molecules in the peptidyl transferase center is for the catalysis of the reaction. It seems beyond doubt that the proton transfer in this reaction has to occur via the O2′ of the P-site substrate A76 ribose (22). The easiest way in which this could happen is through a six-membered transition state where the O2′ acts as a proton shuttle (15, 16, 20, 28). Although this mechanism has been the working hypothesis in our previous investigations (15, 17), the high-level ab initio theory employed here consistently predicts two findings; namely that a water molecule migrates to a position that bridges the O2′ and O3′ of the A76 ribose and that an eight-membered transition state is formed involving this water molecule as an auxiliary proton shuttle.
Such a mechanism was suggested by Steitz et al. based on their high-resolution crystal structure of the RAP transition state TS analog bound to the large subunit of the ribosome (1). Superimposing the crystal structure (PDB code: 1VQP) with the DFT transition state complex (Fig. 2B), one can see that ab initio theory through our iterative optimization procedure arrives at exactly the same conclusion. In addition, the calculated activation enthalpy of 18 kcal/mol for the eight-membered mechanism is in near perfect agreement with experimental rate measurements that yield ΔH‡ = 17 kcal/mol (3, 7). The six-membered transition state reported here has a higher barrier, but this is most likely because the 2′- and 3′-oxygen atoms could not be adequately solvated and EVB simulations (15, 17) show that the auxiliary water also stabilizes a six-membered TS. Regardless of the proton shuttle mechanism, the transition states all have an essentially uncharged amine, which is in agreement with Brønstedt plots for this reaction (25), and they are all late. It thus takes more energy to break the C - O bond than for the amine to approach the ester carbonyl carbon. Judging from the ground state potential surfaces in Fig. 3 and the ab initio study of Sharma et al. (16) an early transition state would not be feasible in either a six- or eight-membered mechanism. The reason for this is that the barrier for forming the transient tetrahedral intermediate is very low and this was also seen even in a minimal gas-phase model (SI Text to ref. 24). An O2′ proton shuttle mechanism with an early TS would therefore have to assume that the ribsome environment increases such a barrier beyond that associated with C - O bond fission, which apppears highly unlikely.
The innate ability of the ester carbonyl oxygen to acquire charge when it is in contact with hydrogen bond donors is apparent in Fig. 5A, which substantiates the notion of an “oxyanion”. Its considerable polarizability is also seen to affect the whole transition state structure in Fig. 4B–D. These structures demonstrate that the transition state indeed depends on the positioning of hydrogen bonds, in this case water molecules that are hydrogen bonded to the oxyanion. The natural Lewis structures in Fig. 5B also depend on how many water molecules that are being kept in the right position during the reaction. Specifically, the leaving O3′ and the ester carbonyl oxygen are seen to act as a charge relay, switching between a formal charge on the O3′ in the absence of oxyanion screening and a formal charge on the oxyanion in the presence of screening. Positioning of water molecules also affects the reaction barriers. As shown in Fig. 5A, inclusion of additional solvent to the oxyanion in the TS lowers the activation enthalpy closer to that in solution. At the same time it is noteworthy that the inclusion of the first screening oxyanion water yields a very low activation entropy effect, while the second water molecule causes an increase in -TΔS‡ of 3 kcal/mol. The presence of precisely one water molecule near the oxyanion may thus be optimal not only for steric reasons, but also in terms of the extra entropic cost of preorganizing additional water molecules, and the higher activation enthalpy on the ribosome than in solution may partly reflect the differences in oxyanion screening.
Kinetic isotope effects are often not intuitive, especially not in concerted reactions. For example, the suggestion (24) that a complete deprotonation of the nucleophile would result in a normal KIE for the peptidyl transfer reaction is at odds with the finding that a TS N-H distance of 1.6 Å (Fig. 4D) still yields a corresponding inverse 14N/15N KIE of 0.989 (Table 1). That is, although the nitrogen nucleophile is almost completely deprotonated its KIE is still inverse. This is a consequence of a nearly fully formed peptide bond in the TS, judging from the short internuclear distances shown in Fig. 4 and Wiberg bond orders of 0.9 in Fig. 5B. It was also reported that ab initio calculations yielded a normal isotope effect (24), but the barrier from which that KIE was calculated pertains to the formation of a tetrahedral intermediate only. The reported TS (24) is not a transition state of the reaction, but merely a shallow barrier of 1.6 kcal/mol, that occurs when a nucleophilic ammonia is brought together with the carbonyl carbon of a protonated formic acid, which is far from the experimental activation enthalpy of ∼17 kcal/mol. Whereas such a hypothetic transition state seems hard to reconcile with the concept of the ribosome as an entropy trap (3) the transition states presented here reinforces this notion by explaining the precise role of the nearest preorganized hydrogen bond donors in this reaction. These TSs are all late with inverse KIEs on the amino nitrogen, which suggests that the peptidyl transfer mechanism is similar to the reaction in solution.
From an evolutionary viewpoint it is of interest not only that the reacting chemical groups have no interactions with ribosomal proteins, which has led to the ribozyme view of the ribosome, but also that there appears to be a clear division of tasks within the PTC between rRNA, substrates and water molecules. That is, the rRNA is folded into a rather inert scaffold that does not directly participate in the chemistry, but that provides key interactions to those groups that are directly involved in the reaction. It might appear somewhat surprising that the active groups are limited to only the A76 2'-OH and two “trapped” water molecules, but this could possibly be an ancient feature of the peptidyl transfer reaction. In this respect, the hydrogen bonded network holding these groups in place appears to be of key importance and may reflect the original minimal requirements for catalysis.
Methods
The quantum chemical calculations are described in detail in the SI Text. In brief, the ab initio models were excised from MD snapshots of the PTC (15, 17) and relaxed into a local minimum with partial optimizations (B3LYP/6-31G*). Then the ground state potential surface of the eight-membered reaction was calculated by scanning the four reacting distances shown in the inset of Fig. 3A (B3LYP/6-31G). This approximate TS was the starting point of an iterative partial TS optimization (B3LYP/6-311G**) that resulted in a fully relaxed TS structure (shown in Fig. 2A). This TS was in turn used to optimize transition states without and with a second oxyanion bound water molecule (see Fig. 4 and Fig. S1). A six-membered TS was optimized in a similar fashion. The fully relaxed TS and reactant structures were all validated with frequency calculations and intrinsic reaction coordinate following.
IRCMax theory (MP2/6-311G**//B3LYP/6-311G**) was used to address the inherent limitations of semilocal DFT in bond making and breaking calculations, as discussed in e.g. (34). Vibrational eigenmodes from the reactant and TS states were used to calculate the enthalpy of activation at 298 K and also the KIEs at 310 K as given by the Bigeleisen-Wolfsberg equation. The final reaction enthalpies were calculated with the composite method shown in Eq. 1 where ΔB3LYP‡ is the B3LYP/6-311G** barrier, ΔδH‡, ΔΔMP2‡ and ΔΔB3LYP++‡ are the corrections to the vibrational enthalpy, the MP2/6-311G** and the B3LYP/6-311 ++G(2df,pd) level of theory respectively.
| [1] |
These values (Table S1) are based on the IRCMax TS structures in all cases but one (the eight-membered TS with two screening water molecules) where instead the B3LYP TS structure was used.
Supplementary Material
Acknowledgments.
This work was supported by the Swedish Research Council (VR). We thank Prof. Måns Ehrenberg for useful discussions.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0914192107/DCSupplemental.
References
- 1.Schmeing TM, et al. Structural insights into the roles of water and the 2' hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Katunin VI, et al. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol Cell. 2002;10:339–346. doi: 10.1016/s1097-2765(02)00566-x. [DOI] [PubMed] [Google Scholar]
- 3.Sievers A, et al. The ribosome as an entropy trap. Proc Natl Acad Sci USA. 2004;101:7897–901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youngman EM, et al. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- 5.Brunelle JL, et al. The interaction between C75 of tRNA and the A loop of the ribosome stimulates peptidyl transferase activity. RNA. 2006;12:33–39. doi: 10.1261/rna.2256706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroeder GK, Wolfenden R. The rate enhancement produced by the ribosome: An improved model. Biochemistry. 2007;46:4037–4044. doi: 10.1021/bi602600p. [DOI] [PubMed] [Google Scholar]
- 7.Johansson M, et al. The kinetics of ribosomal peptidyl transfer revisited. Mol Cell. 2008;30:589–598. doi: 10.1016/j.molcel.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Wohlgemuth I, et al. Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem. 2008;283:32229–32235. doi: 10.1074/jbc.M805316200. [DOI] [PubMed] [Google Scholar]
- 9.Pavlov MY, et al. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci USA. 2009;106:50–4. doi: 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson J, et al. Analysis of mutations at residues A2451 and G2447 of 23S rRNA in the peptidyltransferase active site of the 50S ribosomal subunit. Proc Natl Acad Sci USA. 2001;98:9002–9007. doi: 10.1073/pnas.151257098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polacek N, et al. Ribosomal peptidyl transferase can withstand mutations at the putative catalytic nucleotide. Nature. 2001;411:498–501. doi: 10.1038/35078113. [DOI] [PubMed] [Google Scholar]
- 12.Beringer M, et al. Essential mechanisms in the catalysis of peptide bond formation on the ribosome. J Biol Chem. 2005;280:36065–36072. doi: 10.1074/jbc.M507961200. [DOI] [PubMed] [Google Scholar]
- 13.Erlacher MD, et al. Chemical engineering of the peptidyl transferase center reveals an important role of the 2'-hydroxyl group of A2451. Nucleic Acids Res. 2005;33:1618–1627. doi: 10.1093/nar/gki308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang K, et al. The role of 23S ribosomal RNA residue A2451 in peptide bond synthesis revealed by atomic mutagenesis. Chem Biol. 2008;15:485–492. doi: 10.1016/j.chembiol.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Trobro S, Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci USA. 2005;102:12395–400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma PK, et al. What are the roles of substrate-assisted catalysis and proximity effects in peptide bond formation by the ribosome? Biochemistry. 2005;44:11307–14. doi: 10.1021/bi0509806. [DOI] [PubMed] [Google Scholar]
- 17.Trobro S, Åqvist J. Analysis of predictions for the catalytic mechanism of ribosomal peptidyl transfer. Biochemistry. 2006;45:7049–7056. doi: 10.1021/bi0605383. [DOI] [PubMed] [Google Scholar]
- 18.Beringer M, et al. The G2447A mutation does not affect ionization of a ribosomal group taking part in peptide bond formation. RNA. 2003;9:919–922. doi: 10.1261/rna.5600503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bieling P, et al. Peptide bond formation does not involve acid-base catalysis by ribosomal residues. Nat Struct Mol Biol. 2006;13:423–428. doi: 10.1038/nsmb1091. [DOI] [PubMed] [Google Scholar]
- 20.Dorner S, et al. Molecular aspects of the ribosomal peptidyl transferase. Biochem Soc Trans. 2002;30:1131–1137. doi: 10.1042/bst0301131. [DOI] [PubMed] [Google Scholar]
- 21.Dorner S, et al. Mononucleotide derivatives as ribosomal P-site substrates reveal an important contribution of the 2'-OH to activity. Nucleic Acids Res. 2003;31:6536–6542. doi: 10.1093/nar/gkg842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinger JS, et al. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 23.Trobro S, Åqvist J. Role of ribosomal protein L27 in peptidyl transfer. Biochemistry. 2008;47:4898–4906. doi: 10.1021/bi8001874. [DOI] [PubMed] [Google Scholar]
- 24.Seila AC, et al. Kinetic isotope effect analysis of the ribosomal peptidyl transferase reaction. Biochemistry. 2005;44:4018–4027. doi: 10.1021/bi047742f. [DOI] [PubMed] [Google Scholar]
- 25.Kingery DA, et al. An uncharged amine in the transition state of the ribosomal peptidyl transfer reaction. Chem Biol. 2008;15:493–500. doi: 10.1016/j.chembiol.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rangelov MA, et al. The syn-oriented 2-OH provides a favorable proton transfer geometry in 1,2-diol monoester aminolysis: Implications for the ribosome mechanism. J Am Chem Soc. 2006;128:4964–4965. doi: 10.1021/ja060648x. [DOI] [PubMed] [Google Scholar]
- 27.Gindulyte A, et al. The transition state for formation of the peptide bond in the ribosome. Proc Natl Acad Sci USA. 2006;103:13327–13332. doi: 10.1073/pnas.0606027103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strajbl M, Florian J, Warshel A. Ab initio evaluation of the potential surface for general base-catalyzed methanolysis of formamide: A reference solution reaction for studies of serine proteases. J Am Chem Soc. 2000;122:5354–5366. [Google Scholar]
- 29.Schmeing TM, et al. An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature. 2005;438:520–524. doi: 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- 30.Erlacher MD, et al. Efficient ribosomal peptidyl transfer critically relies on the presence of the ribose 2'-OH at A2451 of 23S rRNA. J Am Chem Soc. 2006;128:4453–4459. doi: 10.1021/ja0588454. [DOI] [PubMed] [Google Scholar]
- 31.Kaminski ZJ, Paneth P, O'Leary MH. Nitrogen kinetic isotope effects on the acylation of aniline. J Org Chem. 1991;56:5716–5718. [Google Scholar]
- 32.Marlier JF, et al. Heavy-atom isotope effects on the hydrazinolysis of methyl formate. J Am Chem Soc. 1997;119:8838–8842. [Google Scholar]
- 33.Hazra MK. Inter-molecular vibrational frequencies of doubly hydrogen-bonded complexes involving 2-pyridone: Reliability of few selected theoretical level of calculations. Chem Phys Lett. 2009;473:10–16. [Google Scholar]
- 34.Perdew JP, et al. Some fundamental issues in ground-state density functional theory: A guide for the perplexed. J Chem Theory Comput. 2009;5:902–908. doi: 10.1021/ct800531s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.