

Abstract
Activation of the Gαs-coupled EP2 receptor for prostaglandin E2 (PGE2) promotes cell survival in several models of tissue damage. To advance understanding of EP2 functions, we designed experiments to develop allosteric potentiators of this key prostaglandin receptor. Screens of 292,000 compounds identified 93 that at 20 μM (i) potentiated the cAMP response to a low concentration of PGE2 by > 50%; (ii) had no effect on EP4 or β2 adrenergic receptors, the cAMP assay itself, or the parent cell line; and (iii) increased the potency of PGE2 on EP2 receptors at least 3-fold. In aqueous solution, the active compounds are largely present as nanoparticles that appear to serve as active reservoirs for bioactive monomer. From 94 compounds synthesized or purchased, based on the modification of one hit compound, the most active increased the potency of PGE2 on EP2 receptors 4- to 5-fold at 10 to 20 μM and showed substantial neuroprotection in an excitotoxicity model. These small molecules represent previously undescribed allosteric modulators of a PGE2 receptor. Our results strongly reinforce the notion that activation of EP2 receptors by endogenous PGE2 released in a cell-injury setting is neuroprotective.
Keywords: excitotoxicity, neuronal injury, prostaglandin E2, time-resolved fluorescence resonance energy transfer, ultra high-throughput screening
Cyclooxygenase-2 (COX2), which is constitutively expressed at low to moderate levels in both neuronal cell bodies and dendritic spines in the hippocampus, is regulated by synaptic activity (1) and is rapidly induced in neurons after a seizure (2) or cerebral ischemia (3, 4). Studies in rodents have demonstrated that COX2 activation by ischemia or status epilepticus generally contributes to neuronal injury (5–8), but multiple downstream COX2 signaling pathways suggest that the mechanisms promoting and opposing brain injury are complex. Among the enzymatic products of COX2 is prostaglandin E2 (PGE2), which can activate four G protein-coupled receptors (GPCRs): EP1, EP2, EP3, and EP4. Activation of the EP2 receptor by PGE2 appears to be neuroprotective after ischemia (3, 4), whereas EP1 activation promotes neurodegeneration (8). The yin-yang nature of the PGE2 receptor family demonstrates the value of a neuroprotection strategy involving modulation of a specific prostanoid receptor rather than generic block of the entire COX2 signaling cascade.
EP2 is a Gαs-coupled receptor that, when activated by PGE2, stimulates adenylate cyclase, resulting in elevation of cytoplasmic cAMP (cAMP) level. In addition to its suspected neuroprotective role, EP2 activation has also been shown to promote spatial learning (9), to improve survival of epithelial cells after radiation injury (10), to accelerate bone healing after fracture (11), and to improve renal function in a HgCl2 model of chronic renal failure (12). On the other hand, EP2 activation in microglia could promote inflammation and neurotoxicity in some neurodegenerative disease models, such as Alzheimer’s disease and amyotrophic lateral sclerosis (13, 14). Two selective agonists for EP2 are known: butaprost, which has a prostaglandin-like structure, and CP-533536, a pyridyl sulfonamide (11). An approach to developing highly selective compounds that has been successful for ligand-gated ion channels and other GPCRs is to focus on allosteric regulators of receptor function (15, 16). Allosteric modulators provide spatial and temporal context dependence because they only affect receptor activation in the presence of the agonist. Allosteric regulators typically bind to a site on the receptor different from the agonist binding site and, thus, do not target the often highly conserved agonist binding pocket. Allosteric regulators are also insensitive to the concentration of agonist and, therefore, are effective across the whole range of concentrations of released prostaglandins expected during and after tissue injury.
We have developed a suite of six high-throughput TR-FRET (time-resolved fluorescence resonance energy transfer) assays to identify selective allosteric potentiators of the human EP2 receptor, and report a series of modulators that act by enhancing the potency with which the endogenous agonist, PGE2, activates the receptor. In aqueous solution these compounds are largely present as spherical nanoparticles (150–300 nm diameter). Our results suggest that the colloidal nanoparticles act as active reservoirs to release soluble compound near the cell surface. These EP2 allosteric potentiators show low cellular toxicity and are neuroprotective against NMDA-induced excitotoxicity on cultured hippocampal neurons.
Results
Ultra-High-Throughput Screening and Hit-Triaging Counterscreens.
The natural agonist of EP2 receptors is PGE2, and EP2 activation stimulates adenylate cyclase activity, resulting in elevated cAMP level. Using the optimized cell-based cAMP TR-FRET assay (Fig. S1), a library of 292,000 small molecules was screened in singlicate for compounds that decrease the TR-FRET signal in the presence of an EC15 of PGE2 (typically 0.3 nM) in C6 glioma (C6G) cells that overexpress EP2. Another EP2 cell line, human colon tumor (HCT-15), was developed to ensure that results were not dependent on cell type. The results of the primary screen are shown in Fig. 1A. A total of 2,352 compounds potentiated the response of C6G-EP2 cells to an EC15 of PGE2 by at least 50%, and 2,002 of these were selected for testing in a suite of six secondary assays designed to eliminate false-positives and evaluate selectivity (Fig. 1B).
Fig. 1.

Ultra-high-throughput screening and hit confirmation strategy. (A) Raw data from the primary screen of 292,000 compounds, each at 20 μM. (B) Hit triage design. Five secondary assays are each tested in triplicate at 20-μM compound; a compound passes assay (i) if it causes > 40% reduction in TR-FRET ratio, and passes assays (ii–v) if it causes < 15% reduction. In addition, each compound is tested for its ability to shift the PGE2 dose-response curve to the left by at least 3-fold. The 93 compounds that survived all six tests were grouped into three well-populated structural clusters. (C) Structures and secondary assay results for representative members of two clusters. The substance identification number (SID) in PubChem is shown.
The first five of these assays were carried out in triplicate at 20-μM compound concentration: (i) repeat of the primary assay for potentiating EP2; (ii) potentiation of EP4 receptor activation by an EC15 of PGE2; (iii) potentiation of β2-adrenergic receptor activation by an EC15 of isoproterenol; (iv) reduction of the TR-FRET signal in a cell-free system in the presence of an EC15 of cAMP; and (v) reduction of the TR-FRET signal itself in the parent C6G cells. Compounds had to be positive in the first assay (defined as > 40% reduction of the TR-FRET signal) and negative in assays ii to v (< 15% reduction) to proceed. Assays ii and iii are designed to examine selectivity because, like EP2, both EP4 and β2-adrenergic receptors couple through Gαs to stimulate adenylate cyclase; a negative response in these assays also minimizes the possibility that a compound elevates cAMP by inhibiting phosphodiesterase. Inactivity in assay iv demonstrates that the compound does not interfere with the assay itself, for example by quenching the TR-FRET signal. Inactivity in assay v demonstrates that the compound does not activate an unidentified Gαs-coupled receptor in C6G cells. Thus, compounds that pass these five assays are very likely to be selective potentiators of EP2 receptors. A total of 813 compounds passed all five assays.
We then tested 2,002 primary hits for their ability to increase the potency of PGE2 at least 3-fold. An 8-point concentration-response curve for PGE2 was carried out in triplicate, in the presence and absence of 6 μM and 20 μM of each compound. A total of 143 compounds passed this test, and 93 compounds passed all six tests.
Clustering analysis was performed just after the identification of the 2,002 active compounds in the EP2 primary screen. We used the Jarvis-Patrick method (17), with a threshold value of 0.4 to generate the cluster sets, and analyzed the results in LeadScope (18). Out of this, 728 singles were identified, while 140 compound clusters were rejected because their membership was no greater than two compounds or because members in that cluster showed activity in the counterscreens. Eleven compounds from the top three populated clusters of confirmed actives and four singletons were rescreened to ensure activity. Two of the top populated structural clusters contained a thiophene amide functionality. Representative members of two clusters are shown in Fig. 1C, along with their behavior in the five secondary assays. Analogs of hit compound (PubChem SID 14735057) were synthesized to explore structure-activity relationships (Tables 1 and 2 and Figs. S2 and S3).
Table 1.
Concentration-dependent, selective allosteric potentiation of EP2 receptors
| Fold left-shift in EC50 |
|||||
| EP2 |
EP4 | β2-AR | |||
| Compound | 5 μM | 10 μM | 20 μM | 20 μM | 20 μM |
| 1 | 1.2 ± 0.2 (9) | 2.1 ± 0.4 (8) | 4.2 ± 0.6 (14) | 0.9 ± 0.2 (5) | 1.1 ± 0.1 (4) |
| 2 | 1.4 ± 0.3 (4) | 1.9 ± 0.4 (4) | 3.8 ± 0.9 (8) | 1.0 ± 0.3 (5) | 1.0 ± 0.1 (4) |
| 3 | 1.5 ± 0.7 (3) | 2.3 ± 0.8 (3) | 3.0 ± 0.9 (5) | 0.7 ± 0.2 (6) | 1.0 ± 0.1 (4) |
| 4 | 1.4 ± 0.2 (3) | 1.2 ± 0.6 (3) | 2.2 ± 1.1 (6) | 1.5 ± 0.4 (5) | 1.0 ± 0.1 (4) |
Data presented as mean ± SEM (n).
Table 2.
Structural determinants of biological and physical properties of EP2 allosteric potentiators
| Compound | 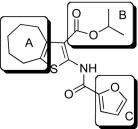 |
EP2potentiation* | IC50 for cyto-toxicity(μM)† | Solub-ility(μM)‡ | Nano-particle-diameter(nm)§ | Percent of compound- as soluble monomer║ |
| 1 | 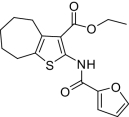 |
4.2 ± 0.6(14) | 248(5) | < 25 | 410 ± 7.9(4) | 1.86 ± 0.50(5) |
| 2 | 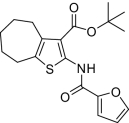 |
3.8 ± 0.9(8) | 50(5) | NM | 427 ± 15.2(4) | 0.83 ± 0.18(3) |
| 3 | 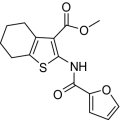 |
3.0 ± 0.9(5) | 4,420(3) | < 25 | 634 ± 20.2(4) | 7.82 ± 2.75(3) |
| 4 | 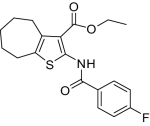 |
2.2 ± 1.1(6) | 107(3) | < 25 | 591 ± 12.9(4) | 1.2 |
| 5 | 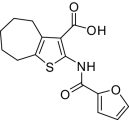 |
0.8 ± 0.1(5) | NM | > 800 | UD | 96.8 ± 1.24(4) |
| 6 | 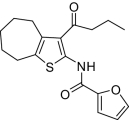 |
1.8 ± 0.3(6) | 1,090(3) | < 25 | 2,560 ± 477(4) | NM |
| 7 | 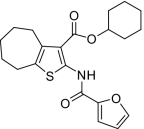 |
1.2 ± 0.1(5) | NM | < 25 | 568 ± 32.7(4) | 2.05 ± 0.66(3) |
| 8 | 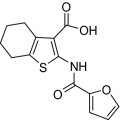 |
1.3 ± 0.2(6) | 483 (3) | NM | NM | NM |
| 9 | 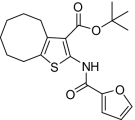 |
1.4 ± 0.1(6) | 36(3) | NM | NM | NM |
| 10 | 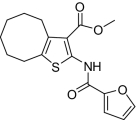 |
1.9 ± 0.5(5) | NM | < 25 | 625 ± 80.5(4) | NM |
| 11 | 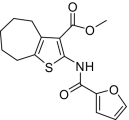 |
3.9 ± 0.5(5) | NM | < 25 | 678 ± 37.6(4) | NM |
| 12 | 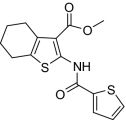 |
1.7 ± 0.3(5) | NM | < 25 | 610 ± 67.9(4) | NM |
| 13 | 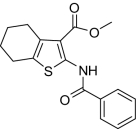 |
2.2 ± 0.3(5) | NM | < 25 | 676 ± 17.5(4) | NM |
| Doxorubicin | NM | 0.35(3) | NM | UD | NM |
NM, not measured; UD, undetectable. Data presented as mean ± SEM (n).
*The fold shift to the left of the PGE2 EC50 in 20 μM compound.
†The mean IC50 for toxicity in C6G cells after 48 h incubation, doxorubicin as positive control.
‡Compound solubility in PBS determined by nephelometry.
§Diameter of nanoparticle formed by compound (20 μM), estimated by dynamic light scattering (DLS).
‖% monomer remaining in a 20 μM solution in 0.2% DMSO after centrifugation at 100,000 g for 10 min, determined by LC-MS.
Allosteric Mode of Action of EP2 Potentiators.
The primary assay was designed to search for compounds that enhance the response of EP2 receptors to a low (EC15) concentration of PGE2. Such compounds could act by increasing the maximum response or the potency of PGE2. The maximum response of PGE2 for activating EP2 receptors in both C6G-EP2 and HCT-15-EP2 cells was as large as that of 10-μM forskolin (Fig. S4), precluding evaluation of the effect of compounds on efficacy. However, in both cell lines tested all compounds produced a clear leftward shift in the PGE2 concentration-response curve, as illustrated by the action of compounds 1 and 2 in Fig. 2 A–C. The effect of these modulators was concentration-dependent (Table 1 and Fig. 2 B and C). We used independent cell lines expressing two related Gαs-coupled receptors to evaluate selectivity: EP4, which is a family member of EP2, also activated by PGE2, and β2-adrenergic receptors activated by isoproterenol. All compounds were highly selective as illustrated by compound 2, which was unable to potentiate the effects of PGE2 on EP4 receptors at 20 μM (Fig. 2D). We also demonstrated that these compounds increased the potency of butaprost at EP2 receptors to approximately the same degree (Fig. S5), so their EP2 modulation is not agonist-specific.
Fig. 2.

Selective allosteric potentiation of EP2 receptor activation. (A) Compound 1 (10 μM) shifts the PGE2 concentration-response curve to the left. Control, EC50 = 3.18 nM; 10 μM 1, EC50 = 0.61 nM. (B) Concentration dependence of compound 1 for EP2 potentiation (Left axis, n = 8–14) and cellular toxicity in C6G cells (Right axis, n = 5). (C) Compound 2 causes a concentration-dependent potentiation of EP2 receptor activation. Control, EC50 = 10.9 nM; 10 μM 2, EC50 = 5.0 nM; 20 μM 2, EC50 = 2.2 nM. (D). No effect of compound 2 on EP4 receptors (Control, EC50 = 3.9 pM; 20 μM 2, EC50 = 3.1 pM).
Four members of the thiophene carboxylate family (cluster 45) were chosen to evaluate in more detail their selectivity for EP2 in comparison with EP4 and β2-adrenergic receptors. None of the compounds, when tested up to 20 μM, increased the potency of agonists for EP4 or β2-adrenergic receptors (Table 1). These results demonstrate a high degree of selectivity of the thiophene carboxylates for EP2 receptors, and effectively rule out an action upstream of the EP2 receptor (e.g., chemical or physical stabilization of PGE2) or downstream (e.g., phosphodiesterase inhibition, direct activation of Gαs or adenylate cyclase, interference with the TR-FRET assay).
Thiophene Carboxylate Potentiators of EP2 Form Nanoparticles.
The solubility of compounds 1, 2, 3, and similar esters as measured by nephelometry was quite low (< 25 μM), while the free acid analog 5 was freely soluble at > 800 μM (Table 2). Small molecules with low solubility could form colloid-like particles in aqueous solution, and recent work suggests that such aggregates cause nonspecific inhibition of target proteins (19). Employing dynamic light scattering (DLS), we demonstrated compound 1 (20 μM in HBSS) formed nano-sized particles with predicted z-average diameter of 410 nm (n = 4) (Fig. 3A). These nanoparticles were clearly visualized by transmission electron microscopy (TEM) (Fig. 3B). The particle size estimated by DLS is typically about half the diameter estimated by TEM because TEM visualizes the spherical core of the particle, whereas DLS measures the particle’s hydrodynamic diameter. Most of the thiophene carboxylates formed nanoparticles at 20-μM bulk concentration except the free acid analog 5, which was more soluble than the corresponding ester analogs (Table 2). Centrifuging the nanoparticles eliminated EP2 potentiation by the supernatant (Fig. 3C), indicating the nanoparticles are somehow involved in the pharmacologic effect on EP2 receptors.
Fig. 3.

Thiophene carboxylate EP2 allosteric potentiators form nanoparticles in aqueous solution, which are essential for activity. (A) 20 μM 1 in HBSS buffer forms nanoparticles with diffusion-predicted diameter 410 ± 7.8 nm detected by DLS. (B) 1 (20 μM in HBSS) visualized by TEM. HBSS alone was devoid of particles. (C) Supernatant from centrifugation (100,000 × g, 10 min) of 20 μM 1 in HBSS failed to increase the potency of PGE2 on EP2 receptors. (D) Increasing DMSO concentration (from 0.2, 2 to 5%) in the compound 1 solution increased both the monomer percentage (from 1.9, 3.9 to 16.3%) and the allosteric activity (from 2.2, 3.0 to 3.5) of the compound. (E) 20 μM 1 in 0.2% DMSO shifted the PGE2 dose-response curve leftward by 2.2-fold (Left) compared to a 3.5-fold shift in 5% DMSO (Right). (F) BSA (0.1 mg/mL) did not reduce the allosteric activity of the compounds 1 and 3 (20 μM). (G) Three strong nanoparticle-forming compounds—Congo Red, tetraiodophenolphthalein, and K252c (20 μM)—did not increase the potency of PGE2 on EP2 receptors. Shown are the mean ± SEM of quadruplicate measurements in one experiment; the experiment was repeated at least three times with similar results.
To determine whether the soluble monomer or the nanoparticle is the active form of EP2 allosteric compounds, we carried out three experiments. First, the allosteric activity of 20-μM compound 1 was measured in various concentrations of DMSO. Increasing DMSO from 0.2 to 5% partially solubilized the compound, raising the monomer percentage from 1.9 to 16.3%, and concomitantly increased the leftward shift of the PGE2 dose-response curve from 2.2- to 3.5-fold (Fig. 3 D and E). This is opposite of the result expected if the nanoparticles themselves mediate potentiation. Second, promiscuous effects of nanoparticle aggregates are destroyed by coating them with adjuvants like BSA (20). However, BSA (0.1 mg/mL) did not reduce the allosteric activity of compounds 1 and 3 (20 μM) on EP2 (Fig. 3F). Third, three strong nanoparticle-forming compounds—Congo Red, tetraiodophenolphthalein, and staurosporine aglycone (K252c) (20 μM) (19, 20)—did not shift the PGE2 dose-response curve leftward (Fig. 3G). These results taken together strongly suggest that although the nanoparticles play a role they are not themselves the direct mediators of allosteric potentiation of EP2.
Structure-Activity Relationships.
Compounds were designed and synthesized to examine three main regions of the hit structure of cluster 45 as determinants of compound potency: the aliphatic ring A, the ester linkage B, and the furan ring C (Table 2). The ring size of sector A is critical for activity of these EP2 potentiators. For example, 6- or 7-membered ring compounds were active, but not the 8-membered ring derivatives (compare 2 to 9 and 10 to 11). Methyl, ethyl, and t-butyl esters were more active than the original hit compound (SID 14735057), which has an isopropyl group (Fig. 1C). However, the t-butyl derivative was more toxic than the other ester analogs (compare 1 to 2). The ester could be replaced by a ketone with approximately 2-fold loss in allosteric effect (compare 1 to 6), but a bulkier ester (7) or the acid (5 and 8) eliminated all activity. In addition, only a furan group or a similar sized thiophene (12) or phenyl group (13) maintained activity; whereas replacing the furan or thiophene ring with another cyclohetero ring, for instance pyridine or piperidine (Fig. S3), caused a complete loss of activity. Overall, the best compounds that emerged so far have a 6- or 7-membered aliphatic ring, a methyl, ethyl, or t-butyl ester, and a furan ring. These compounds showed greater than 3-fold potentiation of EP2 with high selectivity and no cellular toxicity up to at least 100 μM in vitro (SI Text, Fig. 2B, and Table 2).
Cell-Based Label-Free Assay.
We used the Epic biosensor cell-based assay as an alternative assay to evaluate the effect of the compounds on PGE2 induced cAMP accumulation in living cells. EP2 cells were grown on 384-well Epic plates coated with the optical biosensor. Upon activation of EP2 receptors by PGE2, the cell mass near the bottom of the biosensor is redistributed, which leads to a change in the refractive index at the sensor surface. The reflected wavelength difference (∆pm) before and after ligand stimulation is defined as the dynamic mass redistribution (DMR) signal (21), which reveals an integrated cellular response to cAMP elevation.
Our results demonstrated that activation of EP2 receptors by PGE2 induces a DMR signal in both C6G-EP2 and HCT-15-EP2 cells (Fig. 4A) that exhibited a Gαs-signaling signature (21) characterized by a decline in DMR at low agonist concentration transitioning to a rise in DMR as agonist concentration is raised (compare 0.15 nM PGE2 and 111 nM PGE2). The addition of 10-μM compound 3 by itself caused little effect on the DMR but greatly potentiated the effect of PGE2 (Fig. 4A). Indeed, 10-μM compound 3 pretreatment caused a 36-fold shift to the left in the PGE2 concentration-response curve in the Epic assay, from 3.26 nM to 0.09 nM (Fig. 4B). This shift was substantially larger than we had observed in the TR-FRET assay of global cAMP accumulation after EP2 receptor activation (Table 1), suggestive of a striking nonlinear response of the cell’s mass redistribution to elevation in cAMP level.
Fig. 4.

Consequences of allosteric potentiation of EP2 receptors. (A) The PGE2-induced real time DMR response in EP2 cells is significantly increased with compound pretreatment for 30 min (0.15 nM PGE2 + 10 μM 3) compared to 0.15 nM PGE2 alone. The compound (10 μM 3) pretreatment brings the smaller DMR signal induced by lower concentration of PGE2 (0.15 nM) to the maximum DMR response similar to that induced by 111 nM PGE2. Compound alone (10 μM 3) has no effect on the DMR signal. (B) The dose-response curve of the PGE2-induced DMR signal is shifted to the left with compound 3 (10 μM) pretreatment. The maximum DMR signal was measured and Δresponse was calculated for increasing concentrations of PGE2 with or without compound 3. (Control, EC50 = 3.26 nM; 10 μM 3, EC50 = 0.09 nM). (C) NMDA induces LDH release in the cultured hippocampal neurons (DIV14), which can be attenuated by pretreatment with butaprost (5–20 μM), EP2 allosteric compounds 1 (5–20 μM), or 3 (20 μM), but not by compound 5 (20 μM), which is inactive as an EP2 potentiator (Table 2). Data are mean ± SEM with n = 4 to 9 independent experiments each done in quadruplicate. *, P < 0.01; **, P < 0.001 by ANOVA with posthoc Bonferroni. (D) The neuroprotection by the EP2 allosteric potentiators is dependent on the fold shift in PGE2 EC50. A 50% increase in PGE2 potency (∼1.5-fold shift to the left in the PGE2 concentration-response curve) is required to produce half-maximal neuroprotection by the EP2 allosteric potentiators.
Neuroprotection by EP2 Allosteric Potentiators.
Excitotoxicity caused by NMDA receptor activation is well recognized to induce neuronal apoptosis both in vitro and in vivo (22, 23). NMDA receptor activation can, via COX2, cause prostaglandin production, especially PGE2 (24, 25). Activation of the EP2 receptor by PGE2 can be neuroprotective following cerebral ischemia (3, 4). We therefore examined the effect of EP2-directed compounds on NMDA-induced excitotoxicity. Hippocampal cultures (DIV14) were preincubated with butaprost or the EP2 allosteric potentiator compounds at various concentrations for 20 to 30 min, followed by 30-μM NMDA treatment overnight in the presence of butaprost or the EP2 compound. Lactate dehydrogenase (LDH) release was used as the indicator of neuronal injury. NMDA treatment induced a significant increase of LDH release in the cultured hippocampal neurons, which was attenuated in a concentration-dependent manner by preincubation with either butaprost or EP2 compound 1 (Fig. 4C). Two active EP2 allosteric potentiators (1 and 3) (20 μM) produced significant neuroprotection (P < 0.001), whereas the inactive acid analog 5 (20 μM) did not (Table 2 and Fig. 4C). Fig. 4D shows that the neuroprotection by EP2 allosteric potentiators is dependent on the fold shift in PGE2 EC50. Surprisingly, only a 50% increase in PGE2 potency is required to produce half-maximal neuroprotection by the EP2 allosteric potentiators.
Discussion
Activation of COX2 signaling pathways mediates neuronal injury and neuroinflammation (2, 5–8), but can also be neuroprotective (3, 4), which suggests that the COX2 downstream mechanisms promoting and opposing brain injury are complicated. Over the past decade, allosteric modulators have been identified for ligand-gated ion channels and a small number of GPCRs (15, 16); however, no allosteric modulator of prostaglandin receptors has yet been reported. We now report a series of small molecules that act as allosteric potentiators of the human EP2 receptor by enhancing the potency of its natural agonist, PGE2. These compounds share a thiophene carboxylate scaffold. The most active compounds increased the potency of PGE2 on EP2 receptors 4- to 5-fold at 10 to 20 μM concentration. These highly EP2 selective compounds have no detectable activity on the Gαs-coupled EP4 and β2-adrenergic receptors. Interestingly, these EP2 modulators form nanoparticles in aqueous solution that appear to act as active reservoirs for bioactive monomer. Finally, these EP2 allosteric potentiators attenuated excitotoxicity in cultured hippocampal neurons and did not show significant cellular toxicity up to at least 100 μM in vitro.
Unlike PGE2 and the selective EP2 agonist butaprost, the thiophene carboxylates do not have a prostaglandin-like structure, therefore are unlikely to target the agonist binding pocket or show conventional agonist-like effects on the receptor (SI Text and Figs. S6 and S7). Allosteric potentiation could be a result of increased affinity of PGE2 to the orthosteric (agonist binding) site via conformational change of the receptor (type I allosterism), improved coupling between the receptor and its associated Gαs protein (type II), or both (type III) (16). Our data do not distinguish among these mechanisms, but do rule out possible actions of the thiophene carboxylates up- or downstream of the EP2 receptor itself. Their lack of effect on EP4 and β2-adrenergic receptors (Figs. 1C and 2D and Table 1) rules out a direct action on the Gαs protein, adenylate cyclase, phosphodiesterase, or the TR-FRET assay, and their inactivity on the parent C6G cell line (Fig. 1C) rules out action on a distinct GPCR. The lack of effect on the PGE2-activated EP4 receptor (Fig. 2D) argues against formation of a bioactive complex with PGE2 itself. These compounds potentiate EP2 activation in two different cell lines (C6G and HCT-15) by approximately the same degree, further supporting the conclusion that the observed modulation is on the EP2 receptor itself.
The downstream consequences of cAMP accumulation include cellular shape change and redistribution of intracellular organelles (21). We used the Epic biosensor cell-based assay to evaluate the effects of the thiophene carboxylates on PGE2-induced cellular shape changes. PGE2 had approximately the same potency in the TR-FRET and Epic assays (∼1–4 nM), but 10-μM compound 3 pretreatment caused a substantially larger leftward shift in the PGE2 concentration response curve in the Epic assay (Fig. 4 A and B) compared to that for PGE2-induced global cAMP accumulation. This suggests that the intracellular transduction of cAMP accumulation to shape change is strikingly nonlinear.
We used DLS and electron microscopy to show that the thiophene carboxylates form nanoparticles of uniform diameter in aqueous solution, when the bulk concentration is in the bioactive range (Table 2 and Fig. 3). Over the past decade, major attention has been directed to the aggregates formed by many small molecules. Those nano- to submicron colloid-like particles often nonspecifically inhibit target enzymes and are a major source of false-positive hits in high-throughput screening (19). Interestingly, even some marketed drugs can aggregate in vitro (19). How compound aggregates promiscuously inhibit their target enzymes is not fully understood, but enzyme molecules and other proteins are considered to be adsorbed onto the surface of aggregates and undergo surface denaturation (19). However, in contrast to the typical properties of aggregates, the thiophene carboxylates are (i) highly EP2-selective rather than promiscuous, (ii) EP2 potentiators rather than inhibitors, and (iii) active in a cell-based assay rather than cell-free assay (Table 1 and Fig. 2D), all unexpected features of known aggregates (19, 20). Indeed, rather than being promiscuous, these compounds are inactive in most other cell-free and cell-based PubChem bioassays. For example, the hit compound (SID 14735057) has been tested in 227 assays and found active only at EP2. Similarly, compound 1 has been tested in 295 bioassays and found positive in only six, three of which are our assays of EP2; the other three showed weak potentiation at 38 μM in a phenotypic assay of cAMP response element-driven luciferase activity, weak (IC50 = 32 μM) in vitro inhibition of hydroxysteroid (17-beta) dehydrogenase 4, and in vitro inhibition of CYP2C9 at 5 μM. Thus, the allosteric activity of these compounds is highly specific compared to the nonspecific promiscuous inhibition by aggregates described previously. Spinning down the nanoparticles eliminates biologic activity in the supernatant (Fig. 3C), demonstrating their involvement in EP2 potentiation. An alternative explanation for the mode of action of these nanoparticles is suggested by two important differences between the behavior of the EP2 modulators and the previously described nonspecific aggregates. First, the effects of DMSO, BSA, and three well-known nanoparticle-forming molecules do not support the hypothesis that the nanoparticles themselves are responsible for EP2 potentiation (Fig. 3). Second, compound 7, which forms nanoparticles of similar size, is inactive at EP2 receptors (Table 2), confirming that nanoparticle formation is not sufficient to impart pharmacologic activity. We propose that nanoparticles are the active forms of these EP2 allosteric modulators but potentiate receptor activation through release of monomers locally around or within the hydrodynamic layers of the particle cores (Fig. 5). Raising bulk compound concentration is expected to increase the number of nanoparticles surrounding the cells. Recognizing that local monomer concentration near the nanoparticle should be much higher than that of free monomer in bulk solution; this could account for the observed concentration-dependent potentiation above the critical aggregation concentration in bulk solution (Fig. 2B).
Fig. 5.

Nanoparticles are the active forms of the EP2 allosteric potentiators. Nanoparticles formed by thiophene carboxylate compounds in aqueous solution potentiate receptor activation through release of monomers locally around or within the hydrodynamic layers of the particle cores. The local compound monomers with a much higher concentration than free monomers in the bulk solution, directly mediate the allosteric potentiation of EP2 receptors.
Five key adsorption, distribution, metabolism, and excretion characteristics of these EP2 allosteric compounds were estimated by QikProp (26). The predicted properties of all active EP2 modulators fall into the projected range of 95% of known drugs (Table S1).
Direct activation of the EP2 receptor by butaprost has been reported to promote or oppose neuronal injury (27, 28). We confirmed that direct activation of EP2 by butaprost opposes excitotoxicity in primary hippocampal cultures, and that compounds 1 and 3, but not the inactive analog 5, are also neuroprotective (Fig. 4C). NMDA causes the release of PGE2 from in vitro mouse brain cultures (25), and EP2 receptor expression is widespread in hippocampal cultures (Fig. S8), providing the substrate for a neuroprotective effect of allosteric EP2 receptor potentiators. These EP2 allosteric potentiators provide a starting point for development of alternative therapeutic strategies for brain injury caused by status epilepticus, ischemia, or other neuronal disorders, although the role of the EP2 receptor in promoting neuroprotection or neurotoxicity is controversial (13, 14). In addition, the discovery of these previously undescribed EP2 allosteric potentiators suggests that development of allosteric modulators for other prostaglandin receptors is feasible. Considering that prostaglandin signaling plays central roles in many pathological pathways (29, 30), allosteric modulators of prostaglandin receptors could lead to novel selective and efficacious therapies for central nervous system disorders and cancers, and serve as probes to elucidate related biochemical pathways. Future studies are directed toward improving the potency and solubility of the thiophene carboxylates as EP2 allosteric potentiators, but these compounds are already proving valuable as tools for more detailed investigation of the cellular functions of EP2 receptor activation.
Materials and Methods
cAMP TR-FRET Assay Development in 384-Well Format.
cAMP was measured with a homogeneous TR-FRET method (Cisbio Bioassay). For the suspension assay, cells were grown overnight in flasks, pretreated with the cAMP-selective phosphodiesterase inhibitor rolipram (20 μM) for 2 h, then dissociated with EDTA and seeded in 384-well plates at various cell densities in 10 μl Hank’s Buffered Salt Solution (HyClone) plus 20μM rolipram. The cells were treated with or without tested EP2 compounds or drugs for 5 min before addition of PGE2 (0–1 μM), butaprost (0–1 μM), or forskolin (10 μM) for 40 min. The cells were lysed in 10-μL lysis buffer containing the FRET acceptor cAMP-d2 and 1 min later 10-μL lysis buffer with anti-cAMP-Cryptate was added. After 60 to 90 min incubation at room temperature, the TR-FRET signal was detected by an Analyst HT Multimode Reader (Molecular Devices). Cryptate excitation was at 330/80 nm, and emission from cryptate donor and d2 acceptor was measured at 620/7.5 and 665/7.5 nm, respectively, with the dichroic mirror set at 380 nm. All FRET signals were expressed as F665/F620 × 104. For the adherent assay, cells were seeded in 384-well plates with 4,000 cells per well in 40-μL medium and grown overnight. Medium was withdrawn and 10-μL HBSS plus 20-μM rolipram added for 2 h, then the suspension assay procedure was followed. The C6G and HEK cell assays were conducted in suspension, while the HCT-15 assays were conducted with adherent cells.
In Vitro Analysis of Neuroprotection.
Hippocampal neurons were isolated from embryonic-day 16 to 18 (E16–18) embryos of timed-pregnant Sprague-Dawley rats. Cells were plated onto poly-D-lysine coated 24-well plates at a density of 150,000 cells per well in Neurobasal medium plus B27 and 5% FBS (FBS) (Invitrogen). Cultures were incubated at 37 °C in 95% air/5% CO2 and half of the culture medium was replaced every 3 to 4 days with serum-free medium. After 14 days in culture, cells were pretreated with vehicle (0.2% DMSO), various concentrations of butaprost, or compounds 1, 3, or 5 for 20 to 30 min. Excitotoxicity was induced by treating neurons with NMDA (30 μM) plus glycine (10 μM) overnight in the presence of test drugs. Excitotoxic damage was assessed by measuring the fraction of LDH released into the culture medium (Roche Applied Science). Released LDH % was calculated as 100 × LDH released/(LDH released + LDH in the cells), where the LDH in the cells was determined in the cell lysate. Relative LDH release % was defined as 100 × (LDH drug − LDH control)/(LDH NMDA − LDH control), where the LDH drug was the released LDH % from wells treated with variable concentration of drugs plus NMDA; LDH control was the released LDH % without any treatment; LDH NMDA was the released LDH % treated by NMDA only. Neuroprotection % is defined as 100 − relative LDH release percent. Cultures in which the LDH control was greater than 5% or the (LDH NMDA − LDH control) was less than 5% were discarded.
Supplementary Material
Acknowledgments
We thank Dr. Jim Snyder for discussion early in the project, and Drs. Shuming Nie and Min Kuang for access to the dynamic light scattering instrument. This work was funded by National Institutes of Health Grants 1U54 HG003918 and 1U01 NS058158 (to R.D.).
Footnotes
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909310107/DCSupplemental.
The authors declare no conflict of interest.
Data deposition: All PubChem data were deposited into the PubChem database (http://pubchem.ncbi.nlm.nih.gov/ (assay ID nos. 940, 1080, 1421, and 1422).
References
- 1.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci USA. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–24799. doi: 10.1074/jbc.271.40.24794. [DOI] [PubMed] [Google Scholar]
- 3.McCullough L, et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- 5.Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakayama M, et al. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci USA. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iadecola C, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawano T, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Zhang J, Breyer RM, Chen C. Altered hippocampal long-term synaptic plasticity in mice deficient in the PGE2 EP2 receptor. J Neurochem. 2009;108:295–304. doi: 10.1111/j.1471-4159.2008.05766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houchen CW, Sturmoski MA, Anant S, Breyer RM, Stenson WF. Prosurvival and antiapoptotic effects of PGE2 in radiation injury are mediated by EP2 receptor in intestine. Am J Physiol Gastrointest Liver Physiol. 2003;284:G490–G498. doi: 10.1152/ajpgi.00240.2002. [DOI] [PubMed] [Google Scholar]
- 11.Paralkar VM, et al. An EP2 receptor-selective prostaglandin E2 agonist induces bone healing. Proc Natl Acad Sci USA. 2003;100:6736–6740. doi: 10.1073/pnas.1037343100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vukicevic S, et al. Role of EP2 and EP4 receptor-selective agonists of prostaglandin E(2) in acute and chronic kidney failure. Kidney Int. 2006;70:1099–1106. doi: 10.1038/sj.ki.5001715. [DOI] [PubMed] [Google Scholar]
- 13.Liang X, et al. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang X, et al. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol. 2008;64:304–314. doi: 10.1002/ana.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 16.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarvis RA, Patrick EA. Clustering using a similarity measure based on shared nearest neighbors. IEEE Trans Comput. 1973;22:1025–1034. [Google Scholar]
- 18.Richon A. LeadScope: data visualization for large volumes of chemical and biological screening data. J Mol Graph Model. 2000;18:76–79. [PubMed] [Google Scholar]
- 19.Coan KE, Shoichet BK. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J Am Chem Soc. 2008;130:9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 21.Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007;55:314–322. doi: 10.1016/j.vascn.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5(Suppl):1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 23.Concannon CG, et al. NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J Neurochem. 2008;105:891–903. doi: 10.1111/j.1471-4159.2007.05187.x. [DOI] [PubMed] [Google Scholar]
- 24.Lerea LS, et al. Prostaglandin F2alpha is required for NMDA receptor-mediated induction of c-fos mRNA in dentate gyrus neurons. J Neurosci. 1997;17:117–124. doi: 10.1523/JNEUROSCI.17-01-00117.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlson NG, et al. Microglial inhibition of neuroprotection by antagonists of the EP1 prostaglandin E2 receptor. J Neuroinflammation. 2009;6:5. doi: 10.1186/1742-2094-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy EM, Jorgensen WL. Prediction of properties from simulations: free energies of solvation in hexadecane, octanol, and water. J Am Chem Soc. 2000;122:2878–2888. [Google Scholar]
- 27.Takadera T, Ohyashiki T. Prostaglandin E2 deteriorates N-methyl-D-aspartate receptor-mediated cytotoxicity possibly by activating EP2 receptors in cultured cortical neurons. Life Sci. 2006;78:1878–1883. doi: 10.1016/j.lfs.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 28.Akaike A, et al. Prostaglandin E2 protects cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;663:237–243. doi: 10.1016/0006-8993(94)91268-8. [DOI] [PubMed] [Google Scholar]
- 29.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 30.Cimino PJ, Keene CD, Breyer RM, Montine KS, Montine TJ. Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr Med Chem. 2008;15:1863–1869. doi: 10.2174/092986708785132915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.