

Abstract
During replication, RNA viruses rapidly generate diverse mutant progeny which differ in their ability to kill host cells. We report that the progeny of a single RNA viral genome diversified during hundreds of passages in cell culture and self-organized into two genetically distinct subpopulations that exhibited the competition-colonization dynamics previously recognized in many classical ecological systems. Viral colonizers alone were more efficient in killing cells than competitors in culture. In cells coinfected with both competitors and colonizers, viral interference resulted in reduced cell killing, and competitors replaced colonizers. Mathematical modeling of this coinfection dynamics predicted selection to be density dependent, which was confirmed experimentally. Thus, as is known for other ecological systems, biodiversity and even cell killing of virus populations can be shaped by a tradeoff between competition and colonization. Our results suggest a model for the evolution of virulence in viruses based on internal interactions within mutant spectra of viral quasispecies.
Keywords: evolution, quasispecies, self-organization, virulence
RNA viruses replicate as complex mutant distributions termed viral quasispecies (1 –4). Viral clones diversify upon replication because of mutation rates in the range of 10−3 to 10−5 substitutions per nucleotide copied, due to absence (or low efficiency) of proofreading-repair functions in viral RNA dependent RNA polymerases (5 –7). Little is known of the evolution of virulence when a viral clone diversifies to produce a broad mutant distribution. Large population passages result in fitness gain (8, 9). However, fitness and virulence are not necessarily correlated traits, as shown with clones of the important picornaviral pathogen foot-and-mouth disease virus (FMDV) using cell killing as a marker for virulence (10), or using Tobacco Etch virus and reduction in seed production in planta as a marker for virulence (11).
FMDV has been used as a model system to study quasispecies dynamics (12). A biological clone of FMDV of serotype C termed C-S8c1 was extensively passaged in BHK-21, resulting in genetic diversification and fitness increase (13, 14). At passage 143, a monoclonal antibody (MAb) SD6-resistant mutant termed MARLS was isolated (14). This mutant differed in 32 mutations from its parental C-S8c1 virus and displayed high fitness and a 103-fold greater ability to kill cells than C-S8c1 (10, 15). In the present study we have examined the mutant composition of the population that resulted after 240 serial passages of FMDV C-S8c1 (Fig. 1). The clonal population diversified into two genotypically and phenotypically distinct classes of FMDV genomes that correspond to competitors and colonizers, as previously recognized in ecological systems (16, 17). We provide evidence that cell killing is the result of a compromise between the two phenotypes that coexist in the populations and develop a mathematical model for the competition-colonization dynamics.
Fig. 1.

Genetic characterization of p200 and MARLS viruses. (A) Maximum likelihood reconstruction of consensus nucleotide sequences (nucleotides 1033 through 1154 and 1570 through 3853, see Materials and Methods) of populations before and after three sequential low-MOI infections (indicated by “p3d”). (B) Maximum likelihood reconstruction [nucleotides 1033 through 3853; residue numbering is as described in (10)] of biological clones. Clones are identified by passage number and clone number (e.g., 225c6). Consensus reference sequences at passages 0, 200 (p0, p200), and of MARLS virus are included. Confidence values higher than 80% are indicated.
Results
High Fitness FMDV Clones Were Suppressed by the Dominant Population.
A biological clone of FMDV was passaged in BHK-21 cells at a multiplicity of infection (MOI) of 1–20 plaque-forming-units per cell (PFU/cell) for a total of 240 passages (see SI Appendix).The populations at passages 200, 219, 225 and 240 were subjected to three low-MOI passages (0.006–0.02 PFU/cell) to obtain the corresponding populations termed 200p3d, 219p3d, 225p3d, and 240p3d, respectively, as detailed in Materials and Methods. The four populations and their 3d derivatives were analyzed phylogenetically (Fig. 1A). The results show that the sequence of the initially dominant populations is similar to the consensus sequence of passage 200 (p200), although the sequence of the p3d derivatives is similar to MARLS, a MAb-escape mutant isolated from the same viral lineage (14), and previously characterized as a high-fitness and high-virulence variant (10, 15). To ascertain that the viral population included individual infectious particles that were either p200 or MARLS, a total of 16 biological clones derived from the populations at passages 219, 225, and 240 were isolated and their genomic RNA analyzed by nucleotide sequencing. The phylogenetic analysis showed that indeed two subclasses of genomes were present and that they segregated into p200 and MARLS sequences (Fig. 1B). These phylogenetic comparisons suggest that MARLS clones might be suppressed by p200 variants replicating in the same population and that the MARLS variants could become dominant after the three low-MOI passages (Fig. 1). Therefore, limiting coinfection of cells by MARLS and p200 genomes permitted the former to attain dominance in the viral population.
Competitor and Colonizer FMDV Subpopulations.
To further analyze the interaction between p200 and MARLS, the two viral subpopulations were characterized phenotypically. The MARLS population and its derived clones killed BHK-21 cells faster than the p200 population and its derived clones in a cell killing assay that compared the time needed to kill 104 BHK-21 cells as a function of the initial number of PFU (10) (Fig. 2 A and B). To further evaluate this difference, viruses were tested in cell killing-interference assays (see Materials and Methods). Cells coinfected by fast-killing MARLS and slow-killing p200 viruses died at a similar rate as cells infected only by slow p200 viruses, either using whole populations or individual clones (Fig. 2 C–D). A possible dose effect was excluded because no alteration of the cell killing time was observed when cells were infected by twice the amount of fast viruses. The delay of cell killing could be attributed specifically to slow (p200) viruses because cells infected by two fast (MARLS) viruses experienced no delay (Fig. 2E). A tradeoff between higher cell killing and lower progeny production was also rejected because MARLS clone 240c2 production was statistically indistinguishable from p200 clones 240c1 and 240c13 (KS-test, P > 0.05, SI Appendix). MARLS clone 240c12 production was slightly higher than that of both p200 clones (KS-test, P < 0.05). These results demonstrate that slow p200 viruses directly interfere with the replication of fast MARLS viruses when coinfecting the same cell. We refer to p200 viruses as competitors because of their intracellular competitive advantage and to MARLS viruses as colonizers because of their higher cell killing capacity, which entails a faster dispersal upon completion of the infectious cycle.
Fig. 2.

Cell killing and cell killing-interference assays. (A and B) Cell killing capacity of viral populations and clones, measured as the time needed to kill 104 BHK-21 cells as a function of the initial number of PFU. (A) MARLS clones: 240c2 (○) and 240c12 (Δ); p200 clones: 240c1 (▲) and 240c13 (×). (B) Population p200 (▪), population p200p5d, MARLS population (□). For each point, the average and standard deviation from three independent determinations are indicated. (C–E) Cell killing-interference assays. Number of infectious centers required by p200, MARLS, or the mixture of both viruses to kill 104 BHK-21 cells in 9.5h; 2×, double dose of MARLS clone or population. Each bar represents the mean and standard deviation from triplicate assays. (C) p200 clone and MARLS clone correspond to 240c1 and 240c12, respectively; *The mean of the “Clone mix” is higher than the mean of the “MARLS clone” with marginal significance (P < 0.08, T-Student). (D) population mix indicates a mixture of MARLS population and p200 population. *The mean of the “population mix” is significantly higher (P < 0.05, T-Student) than the mean of the MARLS population. (E) C-S8p260p3d is the virus recovered by passage of C-S8c1 after 260 transfers in cell culture and 3 passages at low-MOI; this virus has a virulence similar to MARLS (10).
Mathematical Model of Viral Coinfection Dynamics.
To pinpoint the coevolutionary dynamics of competitors and colonizers, and to generate testable hypotheses, we developed a mathematical model of virus dynamics (18, 19) in cell culture that accounts for intracellular interactions. Two different viruses, whose abundances are denoted by v 1 and v 2, compete for an uninfected cell pool of size x and give rise to singly infected and coinfected cell populations of size y 1, y 2, and y 12, respectively. The dynamics are given by the ordinary differential equations (ODE)
 |
This ODE system describes uninfected cells being infected with efficiency β, infected cells dying and releasing viral offspring at rate a, and free virus being produced at rate k and inactivated at rate u. Because cell monolayers are confluent, no additional parameters describing any external supply of cells or cell division are required. The model parameters a and k are indexed by virus type, and the intracellular competition parameter c denotes the probability that viral offspring of a coinfected cell is of type 1. Virus 1 is the competitor and its parameters have been measured from p200 viruses, whereas virus 2 is the colonizer, characterized by parameters obtained from MARLS viruses (Table 1). Specifically, competitor viruses are more likely to be produced by coinfected cells (c > 1/2) as determined by high-MOI infection experiments (see SI Appendix). Interference is reflected by the condition a
12 = a
1, which expresses the delay of colonizers in cells coinfected by competitors and which was demonstrated in cell killing-interference assays (Fig. 2 C–E). As shown in the cell killing experiments (Fig. 2 A and B), colonizers are more efficient in cell killing (a
2 > a
1), and because the burst sizes ( ) of the two viruses are equal, colonizers also replicate faster (k
2 > k
1). By introducing a linear change of coordinates the parameter space of the ODE system can be seen to be 2D (see SI Appendix). It is given by the rescaled parameters
) of the two viruses are equal, colonizers also replicate faster (k
2 > k
1). By introducing a linear change of coordinates the parameter space of the ODE system can be seen to be 2D (see SI Appendix). It is given by the rescaled parameters 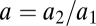 , the ratio of cell death rates, and c, the intracellular competition parameter. The competitors p200 and the colonizers MARLS differ only in precisely these two parameters (Table 1).
, the ratio of cell death rates, and c, the intracellular competition parameter. The competitors p200 and the colonizers MARLS differ only in precisely these two parameters (Table 1).
Table 1.
Parameters and estimated values of the model of virus competition in cell culture
| Parameter∗ | Value | Description | Method of measurement† |
| a1 | 0.14 h−1 | Cell killing rate of virus 1 | Counting of live cells |
| a2 | 0.25 h−1 | Cell killing rate of virus 2 | Calculated from the basic reproductive ratio |
| u | 0.28 h−1 | Viral inactivation rate | Slope of exponential decay of infectivity |
| β | 7.8·10−8mlh−1 | Infectivity rate‡ | Calculated from the basic reproductive ratio |
| c | 0.62 | Probability of a coinfected cell to produce virus 1 | Specific RNA quantification in high-MOI infections |
| K | 250 1 | Burst size | Titration of viral progeny after complete cell lysis |
∗ See SI Appendix for details.
†Virus 1 and virus 2 correspond to p200 (competitor) and MARLS (colonizer) viruses, respectively.
‡The initial number of susceptible cells was x 0 = 2 × 106 cells in a volume of 2 ml.
We have solved the ODE system numerically for different initial viral densities to assess the winner of the competition as the virus type produced with the highest total abundance. For the competitor-colonizer region of the parameter space, i.e., for c > 1/2 and a > 1, the winner of a competition can be either the competitor or the colonizer, depending on the initial MOI (see SI Appendix). The advantage of the colonizer decreases with the initial density of viruses, to the point where the competitor can outcompete the colonizer under high-density conditions. In the limit of high MOI, we have also found an analytical solution of the ODE system which confirms this observation. Competitors outcompete colonizers in total numbers if their intracellular advantage c is larger than the critical value
 |
In the competitor-colonizer regime (a > 1), this threshold is always greater than 1/2. Increasing initial viral densities v
0 shift the fitness advantage from colonizers to competitors because  . Thus, the model predicts a density-dependent outcome of the coinfection.
. Thus, the model predicts a density-dependent outcome of the coinfection.
Experimental Test of Model Prediction.
To test the density-dependent outcome of the infection predicted by the model, we determined the final abundances of viruses for different initial viral densities both computationally, using the parameters measured from p200 and MARLS (Table 1; an estimate of the values of the parameters is included in the SI Appendix), and experimentally. The p200 and MARLS clones and populations were subjected to standard virus competition assays (see Materials and Methods) at different initial MOI (see SI Appendix). The average fitness of MARLS relative to p200 was found to be dependent on the initial density: the higher the MOI, the lower the relative fitness of MARLS viruses (Fig. 3). The observed fitness values are in good agreement with the predicted fitness values and they confirm the predicted power law dependence of fitness on MOI, although the effect of the space (2D nature of cell monolayers) may be responsible for the lack of exact match. In particular, the experimental data validate the main prediction of the model, namely that fitness is density-dependent and that both competitors and colonizers can be winners of this coevolutionary process.
Fig. 3.

Model predictions and experimental results of virus competition experiments. The basic model of virus competition in cell culture was implemented and solved for the parameters listed in Table 1, to obtain relative fitness of MARLS compared to p200 which are plotted as a function of MOI ( ). Experimental fitness values were obtained from virus competition experiments. Two representative clones each of MARLS and p200 subpopulations were mixed at equal PFU, and challenged in competitions with variable initial MOI. Reference p200 and MARLS populations (p200p5d) were also mixed in direct competition at variable initial MOI. Relative fitness values obtained in each competition for both clones and populations are plotted against MOI (▲). Experimental measurements with standard errors (solid line) and model predictions (dashed line) were fitted by linear regression. Procedures for fitness value measurements and competition experiments are detailed in Materials and Methods and the SI Appendix.
). Experimental fitness values were obtained from virus competition experiments. Two representative clones each of MARLS and p200 subpopulations were mixed at equal PFU, and challenged in competitions with variable initial MOI. Reference p200 and MARLS populations (p200p5d) were also mixed in direct competition at variable initial MOI. Relative fitness values obtained in each competition for both clones and populations are plotted against MOI (▲). Experimental measurements with standard errors (solid line) and model predictions (dashed line) were fitted by linear regression. Procedures for fitness value measurements and competition experiments are detailed in Materials and Methods and the SI Appendix.
Discussion
We have described rapid self-organization in the progeny mutant spectrum of a single viral clone into two different ecological niche specialists. The coexistence of the two strategies relies on intracellular reproductive success due to interference (competition) and on intercellular spread due to increased virulence (colonization) (17, 20). The rapid quasispecies rearrangement was probably facilitated by a high mutation rate (21 –23) and the fact that cell killing in RNA viruses can be modulated by a small subset of mutations at potentially many different genomic sites (10, 11). The suppression of colonizers by competitors at high viral density can lead to the attenuation of a viral population. This mechanism could also explain the previous observation of mutant suppression or density-dependent selection in dissimilar viral systems such as vesicular stomatitis virus (24, 25), FMDV (26), and bacteriophages ϕ6 and ϕX174 (27, 28). In these studies, a subset of clones or subpopulations that showed high fitness in low-MOI infections was outcompeted under high-MOI conditions. The faster replicators under low-MOI conditions probably acted as colonizers, whereas the suppressors at high-MOI acted as competitors. Defective interfering particles maintenance in high-MOI infections strongly attenuates viral infections (29), whereas serial bottleneck (low-density) transfers maintained the cell killing capacity of an FMDV clone despite its reduced replicative fitness (10). Fitness determinants of FMDV are scattered along the viral genome (9, 10) whereas BHK-21 cell-killing determinants mapped mainly in the nonstructural protein-coding region (10).
Mutants with a high cell-killing efficiency that replicate in independent cells may have a selective advantage because they spread faster (30 –32). However, viruses share gene products inside coinfected cells allowing the progression of dominant-negative mutants (24, 33 –35) during processes such as pathogen-derived resistance (36), lethal defection (34, 37), or pseudotype formation (38). Therefore, unlike in bacteria or protozoa (39, 40), coinfection would tend to attenuate virus populations. Our model might also explain the attenuation of a clone of FMDV after mice-to-mice transfers because attenuated strains were only isolated in organs in which the virus spreads locally thereby increasing the probability of coinfection (41). Coinfection of cells by two or more viral particles must be abundant in vivo, as judged by the frequency of viral recombinants that are described as epidemiologically relevant for many viral systems (42 –44). Recombination could be enhanced by the fact that double infections of cells might be more frequent than expected from dual hits occurring at random as documented for some virus-host systems (45, 46). Coinfection of cells has been described in HIV and Dengue infections (47, 48). The analyses with FMDV reported here document that mutants within a viral quasispecies behave as ecological entities and follow competition-colonization dynamics. This compromise, which ensues from intrapopulation self-organization, can modulate the cell killing capacity of the entire viral population. In addition to implications for virulence as a trait that can be modulated in complex viral populations, the results reinforce the concept that mutant spectra can act as a unit of selection (4, 22, 33, 34).
Materials and Methods
Cells, Viruses, and Infections.
Infections of BHK-21 cell monolayers with FMDV have been previously described (10, 15, 26). The FMDVs used in the present study are the initial clone C-S8c1 (p0, GenBank NC 002554) (49) and viral populations derived by passage of p0 at high MOI (1-10 PFU/cell). They are labeled by “p” followed by the passage number (e.g., p200, GenBank FJ824812). MARLS (GenBank AF274010) is a monoclonal antibody-escape mutant of FMDV C-S8c1, selected as a minority component from p213 (15). Biological clones were obtained by dilution and plating in semisolid agar medium (50). Clones are labeled with the number of the population, followed by “c” and a clone number (e.g., p240c1). Specifically, p240c1 and p240c13 have a MARLS sequence, and p240c2 and p240c12 have a p200 sequence, as shown by phylogenetic analysis (see Fig. 1). Serial low-MOI passages (MOI = 0.006–0.02 PFU/cell) were carried with p200, p219, p225, and p240 to obtain populations p3d after three low-MOI passages, and p5d after five low-MOI passages (seeSI Appendix). Population p200p5d was employed throughout the study as the reference MARLS population. C-S8p260p3d derives from C-S8c1 after 263 passages at high-MOI (1-10PFU/cell), and its cell killing capacity is similar to that of MARLS (10).
Competition Between Viruses.
Competition between viruses was carried out as described previously (10). Briefly, known numbers of PFU of the two viruses were adsorbed onto BHK-21 cell monolayers for 1 h at 37 °C; then the monolayers were washed to remove unadsorbed virions, and the infection was allowed to proceed until complete cytopathology. Viral RNA specific for each virus, present in the cell culture supernatants was quantitated by real-time RT-PCR. The primers that specifically amplify MARLS and p200 sequences are CACGTACTATTTTTCTGATTTG and CACGTACTACTTTTCTGATCTG, respectively. Relative fitness values were calculated as described in the SI Appendix.
Cell-Killing Assay.
The capacity of FMDV to kill BHK-21 cells was measured as previously described (10). The assay consists in determining the minimum number of PFU required to kill 104 BHK-21 cells after variable times of infection. The experiments were performed using multiwell M96 plates seeded with 104 BHK-21 cells per well and then infected by serial dilutions of the virus to be tested, following the standard infection protocol. At different times postinfection the cells were fixed with 2% formaldehyde. The results are expressed as the logarithm of the number of PFU (PFU/mL of the virus used for the infection) as a function of the time needed to kill the 104 BHK-21 cells. Control viruses with previously measured cell killing capacities were included in all of the experiments. Cell viability was measured by cell counting after trypan blue staining (37).
Cell Killing-Interference Assay.
The cell killing assay was adapted to measure the interference that FMDV variants exerted on the killing of BHK-21 by other variants, as follows. Monolayers of 5·105 cells were infected at high-MOI (> 20 PFU/cell) with either individual variants or with an equal number of PFUs of the two variants (killing and interfering) to be tested. After a 1 h adsorption period, cells were detached by trypsin-EDTA treatment, washed, serially diluted, and applied onto fresh monolayers of 104 cells. The monolayers were overlaid with semisolid agar. At 9.5 h postapplication, the minimum number of infected cells required to kill 104 BHK-21 cells was measured.
Viral Phylogenies.
Phylogenetic trees were inferred by maximum likelihood, using the Tamura-Nei substitution model with Gamma distributed heterogeneous rates (TN-8Γ) (51). The genomic regions chosen for sequence comparison included the capsid-coding region and provided a sufficient average number of mutations in the consensus sequence and among individual clones to achieve the required resolution in the phylogenetic analyses (9, 10, 15, 52, 53). Nucleotides 1154 through 1570 were not considered in the analysis presented in Fig. 1A because some populations contain a proportion of genomes harboring internal deletions (52).
Supplementary Material
Acknowledgments
We thank J.F. García-Arriaza, M. Herrera, and J.J. Holland for helpful suggestions and M. Dávila and A.I. de Ávila for technical assistance. Work at Centro de Biología Molecular “Severo Ochoa” was supported by grants BFU2006-00863 from Ministerio de Educación y Ciencia, BFU2008-02816/BMC from Ministerio de Ciencia e Innovación, 36558/06 from FIPSE, and Fundación R. Areces. Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas is funded by Instituto de Salud Carlos III. S.O. was supported by a predoctoral fellowship from the MEC. Part of this work was done while N.B. was affiliated with the Program for Evolutionary Dynamics at Harvard University and funded by a grant from the Bill & Melinda Gates Foundation through the Grand Challenges in Global Health Initiative.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession nos. NC002554, FJ824812, and AF274010).
This article contains supporting information online at www.pnas.org/cgi/content/full/0909787107/DCSupplemental.
References
- 1.Eigen M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971;58:465–523. doi: 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- 2.Eigen M, Schuster P. The Hypercycle. A Principle of Natural Self-Organization. Berlin: Springer; 1979. [DOI] [PubMed] [Google Scholar]
- 3.Domingo E, Sabo D, Taniguchi T, Weissmann C. Nucleotide sequence heterogeneity of an RNA phage population. Cell. 1978;13:735–744. doi: 10.1016/0092-8674(78)90223-4. [DOI] [PubMed] [Google Scholar]
- 4.Domingo E, et al. Viruses as quasispecies: Biological implications. Curr Top Microbiol Immunol. 2006;299:51–82. doi: 10.1007/3-540-26397-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batschelet E, Domingo E, Weissmann C. The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate. Gene. 1976;1:27–32. doi: 10.1016/0378-1119(76)90004-4. [DOI] [PubMed] [Google Scholar]
- 6.Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinhauer DA, Domingo E, Holland JJ. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene. 1992;122:281–288. doi: 10.1016/0378-1119(92)90216-c. [DOI] [PubMed] [Google Scholar]
- 8.Novella IS, et al. Exponential increases of RNA virus fitness during large population transmissions. Proc Natl Acad Sci USA. 1995;92:5841–5844. doi: 10.1073/pnas.92.13.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escarmís C, Dávila M, Domingo E. Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller’s ratchet. J Mol Biol. 1999;285:495–505. doi: 10.1006/jmbi.1998.2366. [DOI] [PubMed] [Google Scholar]
- 10.Herrera M, García-Arriaza J, Pariente N, Escarmís C, Domingo E. Molecular basis for a lack of correlation between viral fitness and cell killing capacity. PLoS Pathog. 2007;3:e53. doi: 10.1371/journal.ppat.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrasco P, de la Iglesia F, Elena SF. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus. J Virol. 2007;81:12979–12984. doi: 10.1128/JVI.00524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domingo E, Ruiz-Jarabo CM, Arias A, Garcia-Arriaza JF, Escarmís C. Quasispecies dynamics and evolution of foot-and-mouth disease virus. In: Sobrino F, Domingo E, editors. Foot-and-Mouth Disease. Wymondham, England: Horizon Bioscience; 2004. [Google Scholar]
- 13.García-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmís C. Evolutionary transition toward defective RNAs that are infectious by complementation. J Virol. 2004;78:11678–11685. doi: 10.1128/JVI.78.21.11678-11685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charpentier N, Dávila M, Domingo E, Escarmís C. Long-term, large-population passage of aphthovirus can generate and amplify defective noninterfering particles deleted in the leader protease gene. Virology. 1996;223:10–18. doi: 10.1006/viro.1996.0450. [DOI] [PubMed] [Google Scholar]
- 15.Baranowski E, et al. Multiple virulence determinants of foot-and-mouth disease virus in cell culture. J Virol. 1998;72:6362–6372. doi: 10.1128/jvi.72.8.6362-6372.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tilman D. Competition and biodiversity in spatially structured habitats. Ecology. 1994;75:2–16. [Google Scholar]
- 17.Tilman D, May RM, Lehman CL, Nowak MA. Habitat destruction and the extinction debt. Nature. 1994;371:65–66. [Google Scholar]
- 18.Nowak MA, May RM. Virus Dynamics. Mathematical Principles of Immunology and Virology. New York: Oxford Univ Press Inc.; 2000. [Google Scholar]
- 19.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 20.May RM, Nowak MA. Superinfection, metapopulation dynamics, and the evolution of diversity. J Theor Biol. 1994;170:95–114. doi: 10.1006/jtbi.1994.1171. [DOI] [PubMed] [Google Scholar]
- 21.Domingo E. Virus Evolution. In: Knipe DM, Howley PM, editors. Fields Virology. 5th Ed, Vol 12. Philadelphia: Lappincott Williams & Wilkins; 2007. pp. 389–421. [Google Scholar]
- 22.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness under selective pressure in mice. PLoS Pathog. 2005;1:102–110. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Torre JC, Holland JJ. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J Virol. 1990;64:6278–6281. doi: 10.1128/jvi.64.12.6278-6281.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novella IS, Reissig DD, Wilke CO. Density-dependent selection in vesicular stomatitis virus. J Virol. 2004;78:5799–5804. doi: 10.1128/JVI.78.11.5799-5804.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sevilla N, Ruiz-Jarabo CM, Gómez-Mariano G, Baranowski E, Domingo E. An RNA virus can adapt to the multiplicity of infection. J Gen Virol. 1998;79:2971–2980. doi: 10.1099/0022-1317-79-12-2971. [DOI] [PubMed] [Google Scholar]
- 27.Turner PE, Chao L. Prisoner’s dilemma in an RNA virus. Nature. 1999;398:441–443. doi: 10.1038/18913. [DOI] [PubMed] [Google Scholar]
- 28.Bull JJ, Millstein J, Orcutt J, Wichman HA. Evolutionary feedback mediated through population density, illustrated with viruses in chemostats. Am Nat. 2006;167:E39–E51. doi: 10.1086/499374. [DOI] [PubMed] [Google Scholar]
- 29.Holland JJ. Defective viral genomes. In: Fields BM, Knipe DM, editors. Virology. New York: Raven Press; 1990. pp. 151–165. [Google Scholar]
- 30.Wu H, et al. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J Virol. 2006;80:2380–2389. doi: 10.1128/JVI.80.5.2380-2389.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.May RM, Nowak MA. Coinfection and the evolution of parasite virulence. Proc Biol Sci. 1995;261:209–215. doi: 10.1098/rspb.1995.0138. [DOI] [PubMed] [Google Scholar]
- 32.Frank SA. Models of parasite virulence. Q Rev Biol. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- 33.Crowder S, Kirkegaard K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet. 2005;37:701–709. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- 34.Perales C, Mateo R, Mateu MG, Domingo E. Insights into RNA virus mutant spectrum and lethal mutagenesis events: Replicative interference and complementation by multiple point mutants. J Mol Biol. 2007;369:985–1000. doi: 10.1016/j.jmb.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 35.Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science. 2002;296:2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- 36.Baulcombe DC. Mechanisms of pathogen-derived resistance to viruses in transgenic plants. Plant Cell. 1996;8:1833–1844. doi: 10.1105/tpc.8.10.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grande-Pérez A, Lázaro E, Lowenstein P, Domingo E, Manrubia SC. Suppression of viral infectivity through lethal defection. Proc Natl Acad Sci USA. 2005;102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valcárcel J, Ortín J. Phenotypic hiding: The carryover of mutations in RNA viruses as shown by detection of mar mutants in influenza virus. J Virol. 1989;63:4107–4109. doi: 10.1128/jvi.63.9.4107-4109.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Ami F, Mouton L, Ebert D. The effects of multiple infections on the expression and evolution of virulence in a Daphnia-endoparasite system. Evolution. 2008;62:1700–1711. doi: 10.1111/j.1558-5646.2008.00391.x. [DOI] [PubMed] [Google Scholar]
- 40.de Roode JC, et al. Virulence and competitive ability in genetically diverse malaria infections. Proc Natl Acad Sci USA. 2005;102:7624–7628. doi: 10.1073/pnas.0500078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanz-Ramos M, Díaz-San Segundo F, Escarmís C, Domingo E, Sevilla N. Hidden virulence determinants in a viral quasispecies in vivo. J Virol. 2008;82:10465–10476. doi: 10.1128/JVI.00825-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomson MM, Pérez-Alvarez L, Nájera R. Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy. Lancet Infect Dis. 2002;2:461–471. doi: 10.1016/s1473-3099(02)00343-2. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo NS, et al. Genomic features of intertypic recombinant sabin poliovirus strains excreted by primary vaccinees. J Virol. 2001;75:5740–5751. doi: 10.1128/JVI.75.13.5740-5751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agol VI. Molecular mechanisms of poliovirus variation and evolution. Curr Top Microbiol Immunol. 2006;299:211–259. doi: 10.1007/3-540-26397-7_8. [DOI] [PubMed] [Google Scholar]
- 45.Cicin-Sain L, Podlech J, Messerle M, Reddehase MJ, Koszinowski UH. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J Virol. 2005;79:9492–9502. doi: 10.1128/JVI.79.15.9492-9502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chohan B, Lavreys L, Rainwater SM, Overbaugh J. Evidence for frequent reinfection with human immunodeficiency virus type 1 of a different subtype. J Virol. 2005;79:10701–10708. doi: 10.1128/JVI.79.16.10701-10708.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science. 2006;311:236–238. doi: 10.1126/science.1115030. [DOI] [PubMed] [Google Scholar]
- 48.Jung A, et al. Recombination: Multiply infected spleen cells in HIV patients. Nature. 2002;418:144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- 49.Sobrino F, Dávila M, Ortín J, Domingo E. Multiple genetic variants arise in the course of replication of foot-and-mouth disease virus in cell culture. Virology. 1983;128:310–318. doi: 10.1016/0042-6822(83)90258-1. [DOI] [PubMed] [Google Scholar]
- 50.Escarmís C, et al. Genetic lesions associated with Muller’s ratchet in an RNA virus. J Mol Biol. 1996;264:255–267. doi: 10.1006/jmbi.1996.0639. [DOI] [PubMed] [Google Scholar]
- 51.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 52.García-Arriaza J, Ojosnegros S, Dávila M, Domingo E, Escarmís C. Dynamics of mutation and recombination in a replicating population of complementing, defective viral genomes. J Mol Biol. 2006;360:558–572. doi: 10.1016/j.jmb.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 53.Fares MA, et al. Evidence for positive selection in the capsid protein-coding region of the foot-and-mouth disease virus (FMDV) subjected to experimental passage regimens. Mol Biol Evol. 2001;18:10–21. doi: 10.1093/oxfordjournals.molbev.a003715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.