

Abstract
Mice in which the genes encoding the parathyroid hormone (PTH)-related peptide (PTHrP) or the PTH/PTHrP receptor have been ablated by homologous recombination show skeletal dysplasia due to accelerated endochondral bone formation, and die at birth or in utero, respectively. Skeletal abnormalities due to decelerated chondrocyte maturation are observed in transgenic mice where PTHrP expression is targeted to the growth plate, and in patients with Jansen metaphyseal chondrodysplasia, a rare genetic disorder caused by constitutively active PTH/PTHrP receptors. These and other findings thus indicate that PTHrP and its receptor are essential for chondrocyte differentiation. To further explore the role of the PTH/PTHrP receptor in this process, we generated transgenic mice in which expression of a constitutively active receptor, HKrk-H223R, was targeted to the growth plate by the rat α1 (II) collagen promoter. Two major goals were pursued: (i) to investigate how constitutively active PTH/PTHrP receptors affect the program of chondrocyte maturation; and (ii) to determine whether expression of the mutant receptor would correct the severe growth plate abnormalities of PTHrP-ablated mice (PTHrP−/−). The targeted expression of constitutively active PTH/PTHrP receptors led to delayed mineralization, decelerated conversion of proliferative chondrocytes into hypertrophic cells in skeletal segments that are formed by the endochondral process, and prolonged presence of hypertrophic chondrocytes with delay of vascular invasion. Furthermore, it corrected at birth the growth plate abnormalities of PTHrP−/− mice and allowed their prolonged survival. “Rescued” animals lacked tooth eruption and showed premature epiphyseal closure, indicating that both processes involve PTHrP. These findings suggest that rescued PTHrP−/− mice may gain considerable importance for studying the diverse, possibly tissue-specific role(s) of PTHrP in postnatal development.
Skeletal development depends on two mechanisms, intramembranous and endochondral bone formation. The former, in which mesenchymal cells develop directly into osteoblasts, is involved in the formation of the flat skull bones; the latter, accounting for the development of most other bones, involves a two stage mechanism, whereby chondrocytes form a matrix template in which osteoblasts differentiate and initiate the ossification process (1). An understanding of this process at the molecular level is beginning to emerge. Both in vitro and in vivo findings implicate parathyroid hormone-related peptide (PTHrP) and its receptor, the PTH/PTHrP receptor, in the developmental program that controls epiphyseal growth and cartilaginous mineralization. PTHrP and the related hormone, parathyroid hormone (PTH), both bind to and activate the PTH/PTHrP receptor, a G protein-coupled receptor with seven transmembrane domains (2). PTH, through its actions on bone and kidney, is one of the major regulators of mineral ion homeostasis in mammals (3). PTHrP was first discovered as the cause of the syndrome of the humoral hypercalcemia of malignancy; however, like the PTH/PTHrP receptor (2), PTHrP is produced in many diverse normal adult and fetal tissues (4), suggesting that it plays a broad role as an auto/paracrine factor.
Dramatic developmental abnormalities occur in each of several different transgenic mouse models in which PTHrP overexpression was targeted to different tissues, or in which the genes encoding PTHrP or the PTH/PTHrP receptor were disrupted through homologous recombination (5–9). The essential role of PTHrP in growth plate development is the best documented paracrine effect of this peptide. Mice in which the PTHrP gene was ablated by homologous recombination (PTHrP−/−) die immediately after birth, and show skeletal dysplasia with short limbs due to accelerated chondrocyte maturation (8, 10–11). Interestingly, transgenic mice that overexpress PTHrP in proliferating chondrocytes have also shorter and thicker limbs, but as the result of decelerated, rather than accelerated, terminal chondrocyte differentiation and delayed mineralization (12, 13). Abundant evidence suggests that PTHrP mediates its actions on chondrocyte differentiation through the PTH/PTHrP receptor. This receptor is highly expressed in prehypertrophic chondrocytes (14), and ablation of its gene (PTH/PTHrP receptor−/−) causes growth plate abnormalities that are very similar to the ones observed in PTHrP−/− mice (9). The cellular and molecular pathways that account for the striking phenotypes of the PTHrP−/− and PTH/PTHrP receptor−/− mice, and the ones that overexpress PTHrP in the growth plate, are only partially elucidated (9, 15).
Recently, we identified in patients with Jansen metaphyseal chondrodysplasia two different PTH/PTHrP receptor mutations, H223R and T410P, that lead to ligand-independent constitutive cAMP accumulation (16, 17). This rare autosomal dominant disorder is characterized by short limbed dwarfism, due to impaired growth plate development, and by hypercalcemia and hypophosphatemia, despite low or normal blood PTH and PTHrP levels (18). Based on the current knowledge of the biological functions mediated by the PTH/PTHrP receptor, ligand-independent activity of mutant receptors provides a plausible explanation for the multiple abnormalities identified in Jansen metaphyseal chondrodysplasia (19).
In the present study, we generated transgenic mice that express, under the control of the rat α1(II) collagen promoter a constitutively active human PTH/PTHrP receptor, HKrk-H223R, in the growth plate. The goal of the study was two-fold: (i) to investigate how constitutively active PTH/PTHrP receptors modify the program of chondrocyte maturation and differentiation; and (ii) to determine whether expression of the transgene in PTHrP−/− mice would correct their growth plate abnormalities, thus, allowing their prolonged survival.
MATERIALS AND METHODS
Generation and Identification of Transgenic Mice.
A 2,922-bp DNA fragment containing 1,880-bp encoding the human mutant PTH/PTHrP receptor (HKrk-H223R) (16) and 1,042-bp from the pcDNAI vector, which provides the consensus polyadenylation signal absent in HKrk-H223R (20), was excised by BamHI/AvrII digestion. The ends of this fragment were made blunt with Klenow polymerase and ligated to a blunt-ended BamHI site in the p1757 plasmid containing the rat α1(II) collagen promoter, kindly provided by Y. Yamada (National Institute of Dental Research, National Institutes of Health, Bethesda, MD) (21). The BamHI site at the 5′ end of the construct was regenerated in the resulting p1757-HKrk-H223R plasmid (Fig. 1A). The cDNA encoding the mutant human PTH/PTHrP receptor was thus placed down-stream of the rat α1 (II) collagen promoter element (−977 to +110) (22), between a 640-bp fragment containing a rabbit β-globin intronic sequence (23) and an enhancer element specific for chondrocytes (24) (Fig. 1A). Nucleotide sequence analysis confirmed the correct orientation of the cDNA, the presence of an “in frame” stop codon and of the native Kozak consensus sequence upstream of the translation initiation codon. The construct insert was released from the vector by digestion with AflIII and AgeI (Fig. 1A), and was microinjected into fertilized eggs from C57BL/6 and FVB/N females. Microinjections were performed under contract by the Beth Israel Transgenic Facility, Boston. The injected eggs were then transferred to pseudopregnant female mice. Genomic DNA extracted from tail biopsies (8) was digested with PvuII, and the subsequent Southern blot was probed with a 32P-labeled 640-bp fragment from the cDNA encoding HKrk (+470 to +840, probe A) (Fig. 1A) (20). The PvuII digestion of mouse genomic DNA containing the human transgene yields one hybridizing DNA species of ≈1.2 kb corresponding to a fragment of the human PTH/PTHrP receptor (+490 to +1,640 of the cDNA encoding HKrk-H223R), and one hybridizing DNA species of ≈1.8 kb from the equivalent region of the mouse PTH/PTHrP receptor gene. F1 progeny were bred to generate homozygous F2 offspring. Homozygosity was confirmed by outcrossing the transgenic mice into wild-type animals, and by demonstrating through Southern blot analysis that the transgene was present in all progeny. To determine the number of insertion sites of the transgene, Southern blots of PvuII-digested genomic DNA were also hybridized with the pcDNAI vector fragment that provided the polyadenylation sequence (probe B) (Fig. 1A). Since the injected construct does not contain additional PvuII sites downstream of the ones present in the cDNA encoding the human PTH/PTHrP receptor, the presence of multiple insertion sites would be expected to generate multiple hybridizing DNA species.
Figure 1.
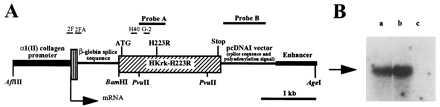
Construction and expression of the transgene. (A) Schematic representation of the transgene construct showing the location of probes A and B for Southern blot analysis of tail genomic DNA, and of oligonucleotides 2F and G-2, and 2FA and H40 that were used for RT-PCR of total RNA and for Southern blot analysis of RT-PCR products, respectively; locations of the translation initiation codon (ATG), the receptor mutation H223R and the stop codon (Stop) in the cDNA encoding a human PTH/PTHrP receptor mutant, HKrk-H223R, are shown. The positions of restriction sites for the enzymes AflIII, BamHI, PvuII, and AgeI are also indicated. (B) Southern blot analysis of the RT-PCR products using total RNA from sternum of newborn Tg-A (a) and Tg-B (b) animals, respectively. (c) Control reaction performed in absence of total RNA. The blot was hybridized with the 32P-radiolabeled oligonucleotide H40. The arrow indicates the location of the 600-bp DNA marker.
To rescue PTHrP−/− mice (C57BL/6/SV129J background), transgenic animals (Tg) were mated with PTHrP+/− animals (8), and offspring heterozygous for the transgene and for the PTHrP gene ablation were bred to generate mice that expressed the transgene and were homozygous for the PTHrP gene ablation (Tg/PTHrP−/−). The PTHrP gene ablation was confirmed by Southern blot analysis as described (8).
Assessment of Transgene Expression.
To assess expression of the mutant PTH/PTHrP receptor transgene, total RNA was extracted from sternal cartilage of newborn transgenic mice, and was purified by CsCl gradient centrifugation as described (20). Total RNA was then reverse transcribed into single-stranded DNA using the HKrk-specific reverse primer G2 (5′-TAGTTGGCCCACGTCCTGT-3′) (Fig. 1A) and the First Strand cDNA synthesis kit (Pharmacia). PCR was then performed using the same reverse primer G2 and the forward primer 2F (5′-TCCGAGAGCCTCCTGCATGAG-3′) (Fig. 1A). The PCR conditions were as follows: 95°C for 1 min, 58°C for 45 sec, 72°C for 1 min, and additional 10 min at 72°C at the end of the 45 cycles. The PCR products were analyzed by Southern blot using the 32P-labeled oligonucleotides H40 (5′- GTCATAAATGTAGTCCGGA-3′) and 2FA (5′-TAGAGACCCGGACCCGCTCCG-3′), respectively (Fig. 1 A, B). Their identity was also confirmed by the presence of a unique restriction site for BamHI (Fig. 1A).
Skeletal Morphology and Histology.
For staining and visualization of whole skeleton, cleared skeletons of 18.5 days postcoitum or newborn mice were stained with Alizarin red S as described (25). For light microscopy, tissues from 17.5 days postcoitum, newborn, and 2- to 3-week-old mice were fixed in 10% formalin/PBS (pH 7.4), and stored in fixative at 4 C. In selective cases, hindlimbs and/or sternebrae were decalcified in 10% formalin/PBS (pH 7.4) containing 20% EDTA. Paraffin blocks were prepared by standard histological procedures. Sections (5–6 μm thickness) were cut at several levels, and stained with hematoxylin and eosin, or with Alcian blue. Selected samples were stained by the method of von Kossa (35) to identify mineralized tissues.
In Situ Hybridization.
In situ hybridizations were performed as described (11) using complementary 35S-labeled riboprobes (complementary RNAs, cRNAs), that were transcribed from the plasmids encoding the pro-α1(II) chain of rat type II collagen (nucleotides 1–521, GenBank accession no. K02804) (26), mouse osteopontin (nucleotides 158-1144, GenBank accession no. X14882) (27), and mouse type X collagen (nucleotides 903-2215, GenBank accession no. X65121) (28) (kindly provided by M. Young, National Institute of Health, Bethesda, MD; S. Nomura, Osaka Univerisity, Osaka; and B. Olsen, Harvard Medical School, Boston, respectively).
RESULTS
Generation and Identification of Transgenic Mice.
Two transgenic founders expressing a constitutively active human PTH/PTHrP receptor, HKrk-H223R (16) under the control of the rat α1(II) collagen promoter were established in the C57BL/6 (Tg-A) and in the FVB/N (Tg-B) strain, respectively. Other founders, obtained with either strain, were either sterile or did not transmit the transgene. Founder Tg-A was shown by Southern blot analysis to carry only one copy of the transgene, while founder Tg-B carried several concatemeric copies with two closely linked integration sites (not shown).
PCR analysis of reverse transcribed total RNA, extracted from newborn sternal cartilage, was performed to detect the transgene transcript. Primers 2F and G-2 (Fig. 1A) were selected to amplify specifically the transgene; PCR products were analyzed by Southern blot using primers H40 and (Fig. 1 A, B) and 2FA (data not shown). Specificity of the PCR products was furthermore confirmed by digestion with BamHI (data not shown). Expression of the transgene in sternal cartilage was detected in both transgenic lines (Fig. 1B).
Gross and Histological Phenotype of Transgenic Mice.
The two transgenic mouse lines, Tg-A and Tg-B, showed a similar phenotype, characterized by delayed endochondral bone formation. The phenotype was more pronounced in Tg-B than in Tg-A, and in both founder lines homozygous animals were more affected than heterozygous littermates. Tg-A mice were grossly indistinguishable from their normal littermates, while Tg-B animals had foreshortening of the limbs and the tail (data not shown).
In comparison to normal littermates, both transgenic lines showed mild but definite skeletal changes at different stages of pre- and postnatal development. The intact skeleton of Tg-A mice, stained with Alizarin red S, showed at birth reduced or absent mineralization of many elements that develop by endochondral bone formation, i.e. supraoccipital bone, sternum, ischium and pubic bone (Fig. 2A). Also, the ossification centers of the vertebrae were reduced in size (Fig. 2A), and their appearance in the tail was delayed (data not shown). All findings were more pronounced in the homozygous transgenic animals. In addition to these abnormalities, homozygous Tg-B mice had a severe mineralization defect of radius and metatarsal bones, and short and deformed hindlimbs with widening of the metaphysis (Fig. 2B). Consistent with the α1 (II) collagen promoter-dependent expression of constitutively active PTH/PTHrP receptors, no abnormalities were noted in those parts of the cranio-facial skeleton that develop by intramembranous ossification (Fig. 2A).
Figure 2.
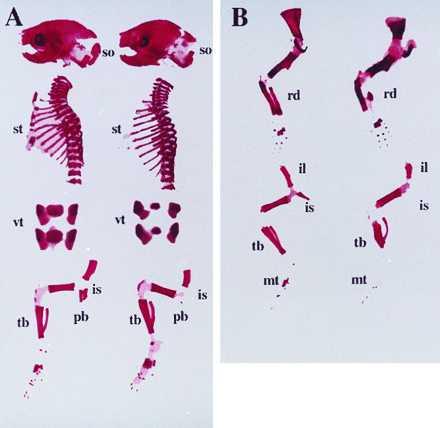
Alizarin red S staining of the skeletons of newborn wild-type and transgenic mice. (A) Lateral view of skull, thoracic cage, and hindlimb; and frontal view of lumbar vertebra of a wild-type mouse (left), and a homozygous Tg-A (right) littermate; (so) supraoccipital bone, (st) sternum, (vt) lumbar vertebrae, (is) ischium, (pb) pubic bone, (tb) tibia. (B) Lateral view of forelimb (upper) and hindlimb (lower) of a wild-type mouse (left), and a homozygous Tg-B (right) littermate; (rd) radius, (il) ilium, (is) ischium, (pb) pubic bone, (mt) metatarsal bones, (tb) tibia.
The histological analysis of the skeletons of Tg-A and Tg-B animals confirmed the delay of endochondral bone formation. For example, sections of the sternebrae of normal newborn mice showed hypertrophic differentiation of chondrocytes, blood vessel invasion, primary spongiosa and bone marrow cavity formation (Fig. 3A). In contrast, sternebrae of newborn heterozygous Tg-A animals had no blood vessel invasion, although hypertrophic differentiation of chondrocytes was evident in the middle of the cartilage (Fig. 3B). The delay in homozygous Tg-A mice was more dramatic, their sternebrae having almost no hypertrophic chondrocytes (Fig. 3C). The lack of hypertrophic chondrocytes in sterna of homozygous Tg-A mice was confirmed by in situ hybridization analysis, which revealed, in comparison to the wild-type animals, a virtual absence of collagen type X mRNA expression (Fig. 4 A–D), yet abundant type II collagen mRNA expression, a marker of proliferative chondrocytes (Fig. 4 E–H). Findings were equivalent in the Tg-B line (data not shown).
Figure 3.
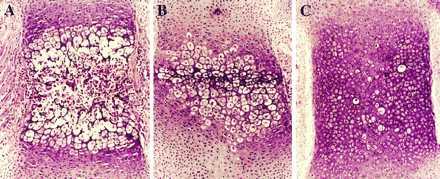
Histological sections, stained with hematoxylin and eosin, of decalcified sternum from a newborn wild-type mouse (A) and Tg-A littermates that were heterozygous (B) or homozygous (C) for the transgene.
Figure 4.
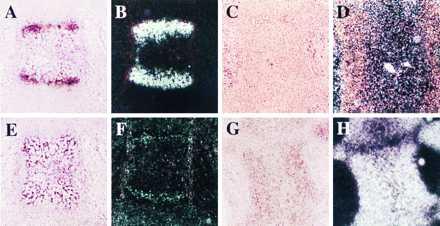
In situ hybridization with 35S-labeled type X collagen (A–D) or type II collagen (E–H) cRNAs of serial sections of decalcified sternum from newborn wild-type mouse (A, B, E, F) and homozygous Tg-A (C, D, G, H) littermate. The sections were counter-stained with hematoxylin and eosin; bright field (A, C, E, G) and dark field views (B, D, F, H) are shown.
An abnormal and delayed process of endochondral bone formation was also evident by analysis of the hindlimbs. Tibiae of 17.5-day-old wild-type fetuses consisted of cartilaginous epiphysis at the proximal and distal end, and a bony diaphysis in the middle (Fig. 5 A, B). Although the mutant and the wild-type bones did not differ in total length, the middle part of the tibiae of heterozygous and, particularly, of homozygous Tg-A fetuses was still occupied by cartilage with only the beginning of vascular invasion (Fig. 5 C–F). This cartilaginous mold consisted of hypertrophic chondrocytes, surrounded by a cartilaginous mineralized matrix, as shown by Alcian blue (data not shown) and von Kossa stainings (Fig. 5I), respectively. Furthermore, these cells expressed low levels of type II and X collagen mRNAs, and large amounts of osteopontin mRNA (data not shown). As suggested by Alizarin red staining (see above), the histological findings in the tibiae of Tg-B mice, showed, in comparison to Tg-A animals, a much more dramatic delay in endochondral bone formation. Tibiae of newborn homozygous Tg-B mice were shorter and thicker than those of control littermates, and consisted mainly of proliferative and hypertrophic chondrocytes (Fig. 5 G, H). Furthermore, contrary to the regular pattern of differentiation, hypertrophy and subsequent blood vessel invasion began at the periphery of the diaphysis, and not in the center (Fig. 5 H, J). By two weeks of age the long bones from either transgenic line appeared to be histologically normal, despite being shorter and thicker in Tg-B animals in comparison to the control littermates (data not shown).
Figure 5.

Light microscopy of tibiae from wild-type mice, and Tg-A or Tg-B littermates. (A–F) Histological sections of undecalcified tibiae stained with hematoxylin and eosin from wild-type animals (A, B), heterozygous (C, D), and homozygous (E, F) Tg-A littermates at day 17.5 of fetal development. Panels B, D, F are higher magnifications of the boxed area in A, C, and E, respectively. (G and H) Histological sections of undecalcified tibiae from a newborn wild-type mouse (G) and a homozygous Tg-B (H) littermate stained with hematoxylin and eosin. (I and J) Histological sections of undecalcified tibiae from a 17.5-day-old homozygous Tg-A fetus (I) and a homozygous newborn Tg-B mouse (J) stained by the method of von Kossa (35).
Gross and Histological Phenotype of PTHrP−/− Mice Expressing Constitutively Active PTH/PTHrP Receptors.
PTHrP−/− animals die around birth and show an acceleration of the endochondral bone formation process, with markedly diminished circumference of the rib cage (8), which may lead to impaired lung development and gas exchange. In an attempt to reverse the neonatal lethality of PTHrP−/− mice by correcting their growth plate abnormalities, Tg-A animals were mated with heterozygous PTHrP+/− mice (C57BL/6 and C57BL/6/SV129J background, respectively), to generate animals that express constitutively active PTH/PTHrP receptors in their growth plate but lack PTHrP (Tg-A/PTHrP−/−). Transgene expression resulted in prolonged postnatal survival of PTHrP−/− animals by up to two months. At birth, or immediately before, the rescued mice were of normal appearance (data not shown) and their skeletons were indistinguishable from Tg-A/PTHrP+/− (Fig. 6) and Tg-A/PTHrP+/+ animals (data not shown). In particular, bones formed by an endochondral process, such as the supraoccipital bone, the tympanic bulla, and the rib cage (Fig. 6) exhibited no premature and/or abnormal mineralization. Furthermore, in the rescued mice the mandible was grossly normal in size, and the doming of the skull and the untimely ossification of the synchondrosis between the basoccipital bone and the basisphenoid bone were absent (Fig. 6). Finally, the shortening and the deformities of long bones and the markedly diminished height of growth plate cartilage in PTHrP−/− mice were almost completely corrected by the targeted expression of constitutively active PTH/PTHrP receptors (Fig. 6 and Fig. 7 A–C). After birth, however, rescued but “malnourished” animals grew less than control littermates, and they showed a premature disappearance of the growth plates and their secondary ossification centers (Fig. 8). Interestingly, tooth eruption was lacking although tooth buds had developed. However, the number of ameloblasts was reduced and the odontoblast layer was disorganized (data not shown). The cause of death of the rescued mice at about 1–2 months of age is still unknown and requires further investigations.
Figure 6.
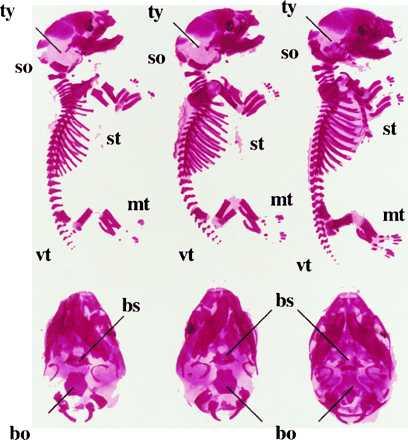
Alizarin red S staining of total cleared skeleton and base of the skull of 18.5 d old fetal Tg-A/PTHrP+/− mouse (Left), a Tg-A/PTHrP−/− (Center), and a PTHrP−/− littermate lacking the transgene (Right). Tg-A/PTHrP+/− and Tg-A/PTHrP−/− animals were heterozygous for the transgene; (so) supraoccipital bone, (ty) tympanic bulla, (st) sternum, (vt) vertebrae, (mt) metatarsal bones, (bo) basoccipital bone, (bs) basisphenoid bone.
Figure 7.
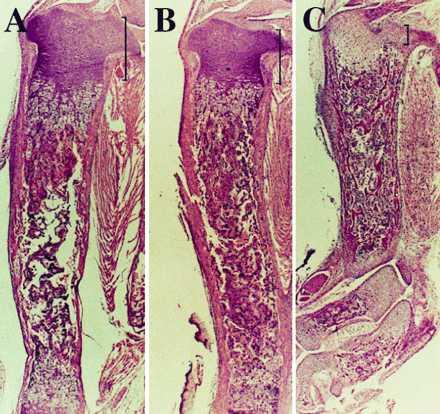
Histological sections of decalcified tibiae from a newborn Tg-A/PTHrP+/+ mouse (A), a Tg-A/PTHrP−/− (B) littermate (both animals were homozygous for the transgene), and a representative newborn PTHrP−/− (C) mouse. The sections were stained with hematoxylin and eosin. The height of the proliferative chondrocyte layer is indicated by brackets. A–C show pictures at the same magnification.
Figure 8.

In situ hybridization with 35S-labeled type X collagen cRNA of decalcified tibiae from a 3-week-old Tg-A/PTHrP+/+ mouse (A) and a Tg-A/PTHrP−/− (B) littermate; both animals were homozygous for the transgene. The sections were stained with hematoxylin and eosin; the bright field view of the proximal epiphysis is shown.
DISCUSSION
The mutant PTH/PTHrP receptors identified as the cause of Jansen metaphyseal chondrodysplasia induce constitutive cAMP accumulation in vitro (17), and previous studies had shown that cAMP, as well as PTHrP, suppress terminal differentiation of chondrocytes (9, 15, 30). Taken together, these data suggest that the PTH/PTHrP receptor modulates the program of chondrocytes differentiation, at least partly, through activation of the cAMP signaling pathway. To gain further insights into the mechanisms through which the PTH/PTHrP receptor affects growth plate development, we generated transgenic mice that express a constitutively active PTH/PTHrP receptor, HKrk-H223R, under the control of the collagen α1 (II) promoter to target transgenic expression to chondrocytes (12, 21, 29); collagen type II has an expression pattern in the growth plate that is similar to that of the PTH/PTHrP receptor (10, 11). The HKrk-H223R mutant was chosen instead of the HKrk-T410P mutant because of its higher levels of ligand-independent cAMP accumulation, and the lack of constitutive and agonist-dependent inositol phosphate accumulation (17).
Transgenic animals with PTHrP expression driven by the α1 (II) collagen promoter show a decrease in mineralization in the endochondral skeleton at birth and a severe delay in chondrocyte differentiation (12). On the contrary, mice that lack the PTHrP or the PTH/PTHrP receptor gene present a mirror image phenotype that is characterized by an increased mineralization and an accelerated program of chondrocyte differentiation (8, 9). Interestingly, in all three animal models, and in patients affected by Jansen metaphyseal chondrodysplasia, limbs are shorter and deformed, but very likely as result of different mechanisms.
Consistent with these observations, targeted expression of constitutively active PTH/PTHrP receptors delayed mineralization and decelerated the chondrocyte maturation program in skeletal segments that are formed by the endochondral process. This delay was dependent on the expression levels of the mutant PTH/PTHrP receptor, i.e., it was more accentuated in homozygous than in heterozygous animals. Tg-B mice showed the most severe phenotype, which led to shortened and deformed limbs, reminiscent of the findings in patients with Jansen disease (18) and in mice with transgenic expression of PTHrP in the growth plate (12).
Interestingly, tibiae of 17.5 d-old Tg-A showed a delay in blood vessel invasion and an accumulation and persistence of “late hypertrophic” chondrocytes, which were defined on the basis of surrounding mineralized matrix, significantly reduced collagen type X expression (31), and high concentrations of osteopontin mRNA (14). During endochondral bone formation, vascularization of hypertrophic cartilage is a key step that is required for bone deposition; however the biochemical mechanisms involved in this process are not yet known. Since reduced but detectable levels of type II collagen mRNA were present even in these late hypertrophic cells, it seems likely that constitutively active PTH/PTHrP receptors continued to be expressed, and that their presence delayed blood vessel invasion. Thus, expression of constitutively active PTH/PTHrP receptors in chondrocytes affected two key transition points in the differentiation pathway: 1) the conversion of proliferating chondrocytes into postmitotic chondrocytes that synthesize type X collagen, and 2) the replacement of hypertrophic chondrocytes by bone cells that is associated with vascular invasion.
At 2 weeks of age, long bones of mutant Tg-A and Tg-B mice were histologically similar to those of wild-type animals, but especially those of Tg-B animals were distinctly shorter. The “normalization” of the histological findings was likely due to a progressively decreasing number of cells that express constitutively active PTH/PTHrP receptors, as growth plates become physiologically smaller with age. The same phenomenon was described in mutant mice overexpressing PTHrP in chondrocytes (12), and in patients affected by Jansen metaphyseal chondrodysplasia, whose growth plate abnormalities subside radiologically later in life without treatment (19).
The targeted expression of constitutively active PTH/PTHrP receptors was able to reverse, almost completely, the severe skeletal abnormalities of newborn homozygous PTHrP−/− mice. The correction of the growth plate defects, and, consequently, of the rib cage deformities is a plausible explanation for their survival.
After birth, however, the rescued animals grew less rapidly than wild-type or transgenic mice, and premature epiphyseal closure was observed. This striking finding is likely to be caused by the absence of PTHrP and not by the expression of the “Jansen” receptors, since comparable findings were not observed in mice with the transgene alone. The failure of constitutively active PTH/PTHrP receptors to prevent the growth plate closure in the rescued animals could be related to an age-dependent reduction in the number of cells that express the transgene, to insufficient constitutive activity, or to a lack of inositol phosphate accumulation by the mutant receptor (17; 3). Alternatively, PTHrP may normally preserve growth plate chondrocytes through mechanisms that do not involve the PTH/PTHrP receptor (e.g., through its nuclear actions; ref. 32).
Rescued mice lacked tooth eruption even though enamel organs developed. It is not surprising, therefore, that they became malnourished after weaning, which probably contributed to their premature death. Interestingly, PTHrP and the PTH/PTHrP receptor have been shown to be expressed in the tooth buds and in the surrounding osseous tissue (33, 34), respectively, but due to the specificity of the promoter used in this study the transgene was not targeted to either one of these structures.
In summary: (i) targeted expression of constitutively active PTH/PTHrP receptors led to slowed conversion of proliferative chondrocytes into hypertrophic chondrocytes, and to prolonged presence of hypertrophic chondrocytes with delayed vascular invasion; (ii) correction of the growth plate abnormalities by targeted expression of constitutively active PTH/PTHrP receptors allowed postnatal survival of PTHrP−/− mice; the perinatal death of PTHrP−/− mice is therefore very likely due to skeletal abnormalities; (iii) absence of PTHrP caused the premature closure of growth plates and the lack of tooth eruption. Our findings suggest that rescued PTHrP−/− mice may represent an attractive model for studying the post-natal role(s) of PTHrP in different tissues and organs.
Acknowledgments
We thank Eleonore Samson for her excellent technical support. This work was supported by National Institutes of Health Grants DK-50708–01 (to H.J.) and DK 47308 (to H.M.K.). C.S.K. is recipient of a grant from the Medical Research Council of Canada.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: PTHrP, parathyroid hormone-related peptide; PTH, parathyroid hormone; Tg, transgenic.
References
- 1.Erlebacher A, Filvaroff E H, Gitelman S E, Derynck R. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 2.Jüppner H. Curr Opin Nephrol Hypertens. 1994;3:371–378. [PubMed] [Google Scholar]
- 3.Kronenberg H M, Bringhurst F R, Nussbaum S, Jüppner H, Abou-Samra A B, Segre G V, Potts J T., Jr . In: Handbook of Experimental Pharmacology: Physiology and Pharmacology of Bone. Mundy G R, Martin T J, editors. Heidelberg: Springer; 1993. pp. 185–201. [Google Scholar]
- 4.Broadus A E, Stewart A F. In: The Parathyroids. Basic and Clinical Concepts. Bilzikian J P, Levine M A, Marcus R, editors. New York: Raven; 1994. pp. 259–294. [Google Scholar]
- 5.Wysolmerski J J, McCaughern-Carucci J F, Daifotis A G, Broadus A E, Philbrick W M. Development (Cambridge, UK) 1995;121:3539–3547. doi: 10.1242/dev.121.11.3539. [DOI] [PubMed] [Google Scholar]
- 6.Wysolmerski J J, Broadus A E, Zhou J, Fuchs E, Milstone L M, Philbrick W M. Proc Natl Acad Sci USA. 1994;91:1133–1137. doi: 10.1073/pnas.91.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasavada R C, Cavaliere C, D’Ercole A J, Dann P, Burtis W J, Madlener A L, Zawalich K, Zawalich W, Philbrick W M, Stewart A F. J Biol Chem. 1996;271:1200–1208. doi: 10.1074/jbc.271.2.1200. [DOI] [PubMed] [Google Scholar]
- 8.Karaplis A C, Luz A, Glowacki J, Bronson R, Tybulewicz V, Kronenberg H M, Mulligan R C. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 9.Lanske B, Karaplis A C, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize L, Ho C, Mulligan R C, Abou-Samra A B, Jüppner H, Segre G V, Kronenberg H. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 10.Amizuka N, Henderson J E, Hoshi K, Warshawsky H, Ozawa H, Goltzman D, Karaplis A C. Endocrinology. 1996;137:5055–5067. doi: 10.1210/endo.137.11.8895380. [DOI] [PubMed] [Google Scholar]
- 11.Lee K, Lanske B, Karaplis A, Deeds J, Kohno H, Nissenson R, Kronenberg H, Segre G. Endocrinology. 1996;137:5109–5118. doi: 10.1210/endo.137.11.8895385. [DOI] [PubMed] [Google Scholar]
- 12.Weir E C, Philbrick W M, Amling M, Neff L A, Baron R, Broadus A E. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick W M, Broadus A E, Baron R. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee K, Deeds J D, Chiba S, Un-no M, Bond A T, Segre G V. Endocrinology. 1994;134:441–450. doi: 10.1210/endo.134.1.8275957. [DOI] [PubMed] [Google Scholar]
- 15.Vortkamp A, Lee K, Lanske B, Segre G V, Kronenberg H M, Tabin C. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 16.Schipani E, Kruse K, Jüppner H. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 17.Schipani E, Langman C B, Parfitt A M, Jensen G S, Kikuchi S, Kooh S W, Cole W G, Jüppner H. N Engl J Med. 1996;335:708–714. doi: 10.1056/NEJM199609053351004. [DOI] [PubMed] [Google Scholar]
- 18.Kruse K, Schütz C. Eur J Pediatr. 1993;152:912–915. doi: 10.1007/BF01957529. [DOI] [PubMed] [Google Scholar]
- 19.Jüppner H. Trends Endocrinol Metab. 1996;7:157–162. doi: 10.1016/1043-2760(96)00063-x. [DOI] [PubMed] [Google Scholar]
- 20.Schipani E, Karga H, Karaplis A C, Potts J T, Jr, Kronenberg H M, Segre G V, Abou-Samra A B, Jüppner H. Endocrinology. 1993;132:2157–2165. doi: 10.1210/endo.132.5.8386612. [DOI] [PubMed] [Google Scholar]
- 21.Yamada Y, Miyashita T, Savagner P, Horton W, Brown K S, Abramczuk J, Xie H X, Kohno K, Bolander M, Bruggeman L. Ann NY Acad Sci. 1990;580:81–87. doi: 10.1111/j.1749-6632.1990.tb17920.x. [DOI] [PubMed] [Google Scholar]
- 22.Kohno K, Sullivan M, Yamada Y. J Biol Chem. 1985;260:4441–4447. [PubMed] [Google Scholar]
- 23.O’Hare K, Benoist C, Breathnach R. Proc Natl Acad Sci USA. 1981;78:1527–1531. doi: 10.1073/pnas.78.3.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horton W, Miyashita T, Kohno K, Hassell J R, Yamada Y. Proc Natl Acad Sci USA. 1987;84:8864–8868. doi: 10.1073/pnas.84.24.8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLeod N. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 26.Kohno K, Martin G R, Yamada Y. J Biol Chem. 1984;259:13668–13673. [PubMed] [Google Scholar]
- 27.Nomura S, Wills A J, Edwards D R, Heath J K, Hogan B L M. J Cell Biol. 1988;106:441–450. doi: 10.1083/jcb.106.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apte S S, Seldin M F, Hayashi M, Olsen B R. Eur J Biochem. 1992;206:217–224. doi: 10.1111/j.1432-1033.1992.tb16919.x. [DOI] [PubMed] [Google Scholar]
- 29.Nakata K, Ono K, Miyazaki J I, Olsen B R, Muragaki Y, Adachi E, Yamamura K I, Kimura T. Proc Natl Acad Sci USA. 1993;90:2870–2874. doi: 10.1073/pnas.90.7.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jikko A, Murakami H, et al. Endocrinology. 1996;137:122–128. doi: 10.1210/endo.137.1.8536602. [DOI] [PubMed] [Google Scholar]
- 31.Hillarby M C, King K E, Brady G, Grant M E, Wallis G A, Boot-Handford R P. Ann NY Acad Sci. 1996;785:263–266. doi: 10.1111/j.1749-6632.1996.tb56279.x. [DOI] [PubMed] [Google Scholar]
- 32.Henderson J E, Amizuka N, Warshawsky H, Biasotto D, Lanske B M K, Goltzman D, Karaplis A C. Mol Cell Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beck F, Tucci J, Russell A, Senior P V, Ferguson M W J. Cell Tissue Res. 1995;280:283–290. doi: 10.1007/BF00307800. [DOI] [PubMed] [Google Scholar]
- 34.Lee K, Deeds J, Segre G V. Endocrinology. 1995;136:453–463. doi: 10.1210/endo.136.2.7835276. [DOI] [PubMed] [Google Scholar]
- 35.Stevens A, Lowe J, Bancroft J D. In: Theory and Practice of Histological Techniques. Bancroft J B, Stevens A, editors. New York: Churchill Livingstone; 1996. pp. 331–333. [Google Scholar]