

Abstract
Ret signaling is critical for formation of the enteric nervous system (ENS) because Ret activation promotes ENS precursor survival, proliferation, and migration and provides trophic support for mature enteric neurons. Although these roles are well established, we now provide evidence that increasing levels of the Ret ligand glial cell line-derived neurotrophic factor (GDNF) in mice causes alterations in ENS structure and function that are critically dependent on the time and location of increased GDNF availability. This is demonstrated using two different strains of transgenic mice and by injecting newborn mice with GDNF. Furthermore, because different subclasses of ENS precursors withdraw from the cell cycle at different times during development, increases in GDNF at specific times alter the ratio of neuronal subclasses in the mature ENS. In addition, we confirm that esophageal neurons are GDNF responsive and demonstrate that the location of GDNF production influences neuronal process projection for NADPH diaphorase-expressing, but not acetylcholinesterase-, choline acetyltransferase-, or tryptophan hydroxylase-expressing, small bowel myenteric neurons. We further demonstrate that changes in GDNF availability influence intestinal function in vitro and in vivo. Thus, changes in GDNF expression can create a wide variety of alterations in ENS structure and function and may in part contribute to human motility disorders.
Introduction
The enteric nervous system (ENS) controls intestinal motility, regulates mucosal secretion, and integrates intestinal sensory stimuli. To perform these activities, there are many enteric neuron subtypes with distinct transmitter phenotypes, neurite extension patterns, electrophysiology, and function (Furness, 2000). Although some mechanisms controlling ENS development are known (Gershon, 1999; Gariepy, 2001, 2004; Newgreen and Young, 2002a,b; Heanue and Pachnis, 2007; Amiel et al., 2008), mechanisms controlling the number of neurons in each subclass and directing neurite growth are poorly understood.
The ENS forms from a population of proliferating multipotent neural crest-derived cells that migrate into the bowel (Yntema and Hammond, 1954; Le Douarin and Teillet, 1973). As in the CNS, precursors for distinct enteric neuron subtypes exit the cell cycle at different times during development (Pham et al., 1991). In mice, for example, most myenteric plexus precursors stop dividing by birth, but some submucosal neuron precursors proliferate until postnatal day 15 (P15) (Pham et al., 1991). There are also subtle differences between myenteric neuron subtypes in the timing of their last mitosis before differentiation. For example, serotonergic and choline acetyltransferase (ChAT)-positive (ChAT+) neuronal precursors become postmitotic much earlier in development than cells destined to become vasoactive intestinal peptide (VIP)/nitric oxide (NO)-expressing neurons. These temporal differences in the timing of the last mitosis for precursors that give rise to specific neuron classes suggest that the timing and intensity of trophic factor expression may critically determine the ratio of neuron subtypes within the ENS by altering the proliferation of precursors determined to become specific types of enteric neurons.
Many aspects of ENS development and function depend on Ret signaling. Ret is a transmembrane tyrosine kinase expressed in the developing and mature ENS that acts as the receptor for glial cell line-derived neurotrophic factor (GDNF), neurturin, artemin, and persephin. These factors activate Ret by binding their respective coreceptors GFRα1, GFRα2, GFRα3, and GFRα4 (Baloh et al., 2000; Airaksinen and Saarma, 2002). Because Ret activation by GDNF/GFRα1 is required for ENS precursor survival, proliferation, and migration (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996; Cacalano et al., 1998; Chalazonitis et al., 1998; Enomoto et al., 1998; Hearn et al., 1998; Heuckeroth et al., 1998; Young et al., 2001; Natarajan et al., 2002; Iwashita et al., 2003), Ret deficiency causes intestinal aganglionosis (Hirschsprung disease) in mice (Schuchardt et al., 1994) and humans (Gabriel et al., 2002; Passarge, 2002).
The current studies were designed to test the hypothesis that ENS development is influenced not only by the presence of GDNF but also by the timing, location, and intensity of GDNF expression. More specifically, we hypothesized that proliferating ENS precursors that give rise to the submucosal plexus could be induced to proliferate more vigorously by excess GDNF administered at a time when myenteric neuron precursors have stopped dividing. This should then result in an increase in submucosal but not myenteric neurons. Furthermore, we hypothesized that the final location of neuronal processes for some but not all subclasses of enteric neurons might be influenced by the location of GDNF expression. To test these hypotheses, we used transgenic mice expressing GDNF in skeletal muscle from the myogenin promoter (Myo–Gdnf) (Nguyen et al., 1998) or in enteric glia from the glial fibrillary acidic protein promoter (GFAP–Gdnf) (Zhao et al., 2004). We also injected newborn mice with GDNF to provide GDNF systemically (Keller-Peck et al., 2001). These analyses demonstrate that alterations in the temporal and spatial expression patterns for GDNF create a wide variety of defects in ENS structure and function.
Materials and Methods
Animals.
Myo–Gdnf (Nguyen et al., 1998), GFAP–Gdnf (Zhao et al., 2004), and Gfrα1 (Enomoto et al., 1998) mice have been described previously. Myo–Gdnf mice produce excess GDNF in developing and mature skeletal muscle. GFAP–Gdnf mice produce excess GDNF in CNS and enteric glia. Gdnf+/− mice were a generous gift from Genentech (Moore et al., 1996). Gdnf+/− mice were C57BL/6 genetic background. Because of difficulty breeding GFAP–Gdnf mice into a C56BL/6 genetic background and high mortality when newborn C57BL/6 mice were injected with GDNF, GFAP–Gdnf- and GDNF-injected mice were CF-1 background. Myo–Gdnf mice were B6C3F1 genetic background. Because of differing genetic backgrounds and ages, all anatomic and functional comparisons were performed with wild-type (WT) littermates. The use and care of mice were accredited and approved by the Washington University Animal Care Committee.
GDNF treatment.
Newborn CF-1 mice were injected with either recombinant histidine (His)-tagged GDNF (Creedon et al., 1997) (2 μg GDNF/g mouse; 1.25 mg/ml, s.c., on the back) or PBS twice a day for 30 d and analyzed at P30. For bromodeoxyuridine (BrdU) studies, mice were injected with the same dose of GDNF from P0 until the day of analysis. For studies of His-tagged GDNF distribution in GDNF-injected mice, animals were injected twice a day from P14 to P19.
Quantitative ENS analysis.
Structural analyses for transgenic mice were performed at 8–12 weeks. The gut was opened along the mesenteric border, pinned flat, and fixed in 4% paraformaldehyde (1 h) before muscle layers were dissected from the submucosa. Sequential 4 cm segments starting at the pylorus were analyzed after staining for NADPH diaphorase (NADPH-d) (Neuhuber et al., 1994), Cuprolinic blue, acetylcholinesterase, and tryptophan hydroxylase (TH) (1:500; Millipore Bioscience Research Reagents) (Karaosmanoglu et al., 1996; Enomoto et al., 1998; Heuckeroth et al., 1999). For the colon, 2 cm segments were analyzed in the same proximal to distal sequence. For ChAT/NADPH-d staining, 2 cm of proximal duodenum were first stained using the NADPH-d method and then by ChAT immunohistochemistry (1:10; AB144P; Millipore Bioscience Research Reagents). For GDNF-injected mice, 3 cm small bowel and 1.5 cm colon segments were analyzed. Cell number was determined by counting neurons in 20 random 0.5 × 0.5 mm areas per mouse. For submucosal whole-mount acetylcholinesterase staining, blood vessels were visualized by phase contrast microscopy. Cell size was determined using Zeiss Axiovision software (n = 40 cells per mouse). Neuronal fiber density was determined by counting the number of neuronal fiber bundles or single fibers that crossed the left and top borders of a 0.5 × 0.5 mm grid (20× objective lens). In some cases in which fiber density was high, counting was performed at 40× and data are presented after multiplying by two, so that all fiber data are based on the same size region of the bowel. “Fiber bundles” were defined as containing at least two closely adherent fibers but in most cases contain many closely adherent fibers. An attempt was made to stretch all segments evenly. Data for GFAP–Gdnf mice were reproduced by two investigators working independently. All analyses used three to six animals, and counts were done without knowledge of mouse genotype. Because mice analyzed were different ages and from different mouse strains, all comparisons are between age- and strain-matched littermate WT and transgenic or GDNF-injected animals. For comparing single parameters in WT mice to the same parameter in mice with altered GDNF levels, Student's t tests or Mann–Whitney rank sum tests were used.
Immunohistochemistry.
Additional primary antibodies used were goat anti-human GDNF (1:10,000; AF-212-NA; R & D Systems), mouse anti-polyhistidine tag (1:200; MAB050; R & D Systems), guinea pig anti-GFAP (1:100; catalog #31223-200; Advanced Immnunochem), and guinea pig anti-PGP9.5 (1:100; catalog #GP14104; Neuromics). Secondary antibodies used were Alexa Fluor 488 or 594 conjugated (1:400; Invitrogen). Activated caspase3 antibody staining (1:100; D175; Cell Signaling Technology) was visualized using a tyramide signal amplification–cyanine 3 (Cy3) amplification kit (PerkinElmer Life and Analytical Sciences) as described previously (Gianino et al., 2003).
Bromodeoxyuridine analysis.
Mice were analyzed 3 h after BrdU injection as described previously (Gianino et al., 2003), except that denaturation used 4 m HCl (10 min) and the sodium tetraborate step was omitted. PGP9.5 and BrdU were detected with Cy3 secondary (1:500; Jackson ImmunoResearch) and Alexa Fluor 488-conjugated anti-BrdU antibodies (1:50; Caltag), respectively.
Real-time PCR for GDNF.
RNA isolated with TRIZOL was purified by RNeasy Mini kit (Qiagen), DNase digested on the column, and reverse transcribed with Powerscript (Clontech). Quantitative real-time PCR (qRT-PCR) was performed in triplicate using individual cDNA samples, SYBR green PCR Master mix (Applied Biosystems), and the iCycler iQ (Bio-Rad). Control reactions omitted the reverse transcriptase. GDNF mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for each sample. Threshold cycles (CT value) in which increased PCR products were first detected were used to calculate ΔCT [CT (GDNF) − CT (GAPDH)]. A standard curve of serially diluted gut cDNA was used to correlate changes in ΔCT with fold changes in GDNF mRNA. Three mice were analyzed for each data point. The following primers were used: GDNF, cttgggtttgggctatgaaa and acaggaaccgctgcaatatc; GAPDH, aactttggcattgtggaagg and gtcttctgggtggcagtgat.
Functional motility studies.
Intestinal and colonic contraction strength and neurotransmitter release were measured in an oxygenated organ bath as described previously (Heuckeroth et al., 1999). Intestinal transit in vivo was performed using fluorescein-labeled dextran (FITC-dextran) (5 mg/ml; molecular weight, 70,000) as described previously (Moore et al., 2003) except that mice were fasted 18 h before testing and killed 60 min after feeding with dextran. Geometric center = Σ (percentage of total fluorescent signal in each segment × segment number) was determined as described previously (Miller et al., 1981).
Results
Increased GDNF availability starting after most myenteric neuron precursors have stopped proliferating significantly increases submucosal neuron number with minimal effect on myenteric plexus neuron density
To test the hypothesis that the timing of GDNF expression influences the ratio of submucosal to myenteric neurons within the ENS, we used two model systems. First, we examined the ENS in mice expressing GDNF from the glial fibrillary acidic protein promoter (GFAP–Gdnf transgenic mice). GFAP expression in enteric glia is first detectable at embryonic day 17 (E17) (Rothman et al., 1986) and continues into adult life. We therefore anticipated that GDNF production from the GFAP promoter would also begin at about this time. We have confirmed that Gdnf mRNA levels are elevated in the bowel of GFAP–Gdnf mice as early as E18 by qRT-PCR and remain elevated compared with adult WT mice (GFAP–Gdnf vs WT mRNA levels: E18 small bowel, 2-fold increase; E18 colon, 1.22-fold increase; P0 small bowel, 5.1-fold increase; P0 colon, 3.4-fold increase; P35 small bowel, 6.8-fold increase; P35 colon, 5.5-fold increase; n = 3–4 mice at each age; p < 0.05 for each parameter) (Fig. 1K). As reported previously, Gdnf mRNA levels are higher during development than in the adult mouse bowel (Golden et al., 1999). In addition, we demonstrated by immunohistochemistry that elevated levels of GDNF protein are found in the region of the ENS at E18 in GFAP–Gdnf mice (Fig. 1A–C,F–H) and near both submucosal and myenteric ganglion cells in adult animals (Fig. 1D,I). These findings are similar to the elevations noted in the brain and spinal cord (Zhao et al., 2004) of these animals.
Figure 1.
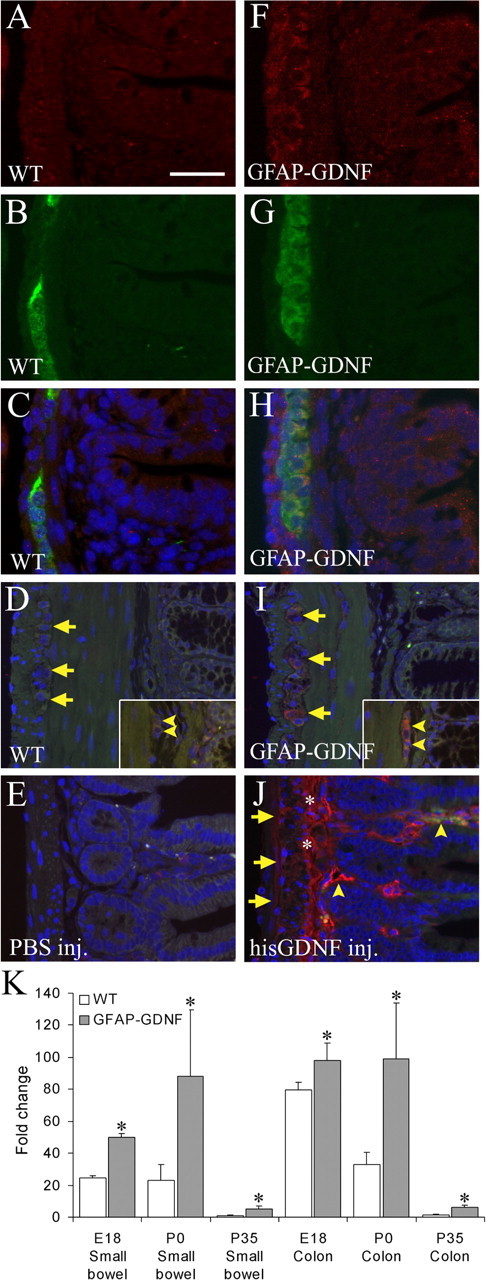
GDNF abundance is elevated near the ENS in GFAP–Gdnf and GDNF-injected mice. Immunohistochemistry for GDNF was performed on WT (A–D) and GFAP–Gdnf mouse small bowel (F–I) at E18 (A–C, F–H) and on adult colon (D, I). A, C, D, F, H, I, GDNF immunohistochemistry. B, C, G, H, PGP9.5 immunohistochemistry. C, H, Merged E18 images. C–E, H–J, Images have DAPI counterstaining to show nuclei. D, E, I, J, Autofluorescence in the green channel was used to highlight the anatomy in adult mice. E, J, Immunohistochemistry for the poly-His tag in the small bowel of mice injected with PBS (E) or His-tagged GDNF (J). D, I, Arrows indicate myenteric ganglia. Arrowheads indicate submucosal ganglia. J, Arrows show the myenteric region. * indicates the submucosal region. Arrowheads identify blood vessels. Scale bar: (in A) A–C, F–H, 25 μm; D, E, I, J, 50 μm. K, Gdnf mRNA levels are elevated in GFAP–Gdnf mice compared with WT littermates in both the small bowel and colon at E18, P0, and P35. Relative gut Gdnf mRNA levels were determined by quantitative real-time PCR after reverse transcription. The level of Gdnf mRNA in WT P35 small bowel was set to 1. *p < 0.05 versus WT for each time point.
By E18 when Gdnf mRNA levels are clearly elevated in the bowel of GFAP–Gdnf mice, most small bowel myenteric neuron precursors have stopped proliferating. In contrast, small bowel submucosal neuron precursors proliferate until 2 weeks after birth (Pham et al., 1991). We therefore hypothesized that GDNF expression from the GFAP promoter might increase the proliferation of submucosal but not myenteric neuron precursors in the small bowel. To test this hypothesis, we first determined submucosal and myenteric neuron density in GFAP–Gdnf mice using acetylcholinesterase (Enomoto et al., 1998) and Cuprolinic blue (Karaosmanoglu et al., 1996) staining, respectively (Table 1A) (supplemental Table 1, available at www.jneurosci.org as supplemental material). These methods are excellent for total enteric neuron counts. Control experiments demonstrate that TuJ1 (neuronal class III β tubulin) and acetylcholinesterase staining completely overlap in the submucosal plexus, and Cuprolinic blue staining has been validated against other staining methods for myenteric neurons. These analyses demonstrated a 19% increase in small bowel submucosal neuron density (WT, 115.2 ± 4.7 cells/mm2; GFAP–Gdnf, 137.3 ± 5.8 cells/mm2; p = 0.004; n = 6 mice of each genotype) but no change in the density of small bowel myenteric neurons of GFAP–Gdnf versus WT mice (WT, 104.8 ± 6.1 cells/mm2; GFAP–Gdnf, 110.7 ± 8.7 cells/mm2; p = 0.56; n = 6 mice of each genotype). Similarly, there was a 70% increase in colon submucosal neuron density (WT, 24.3 ± 3.3 cells/mm2; GFAP–Gdnf, 41.9 ± 4.7 cells/mm2; p = 0.007; n = 6 mice of each genotype) and a smaller change (28%) in colon myenteric neuron density (WT, 185.2 ± 8.0 cells/mm2; GFAP–Gdnf, 236.6 ± 9.4 cells/mm2; p < 0.001; n = 6 mice of each genotype). These observations suggest that increased GDNF availability starting in late fetal development could change the ratio of neurons within different regions of the ENS based on the proliferative capacity of their precursor cells because submucosal neuron numbers increase much more than myenteric neuron numbers in these GFAP–Gdnf mice.
Table 1.
Quantitative neuron density data
| Cell density (cells/mm2) | Fold change | p value | ||
|---|---|---|---|---|
| WT |
GFAP–Gdnf mice |
|||
| A, GDNF transgene in glia significantly increases submucosal neuron density with minimal effect on myenteric neurons | ||||
| Small bowel submucosal acetylcholinesterase | 115.2 ± 4.7 | 137.3 ± 5.8 | 1.19 | 0.004 |
| Colon submucosal acetylcholinesterase | 24.3 ± 3.3 | 41.9 ± 4.7 | 1.7 | 0.007 |
| Small bowel myenteric Cuprolinic | 104.8 ± 6.1 | 110.7 ± 8.7 | 0.56 | |
| Colon myenteric Cuprolinic | 185.2 ± 8.0 | 236.6 ± 9.4 | 1.28 | <0.001 |
| PBS-injected mice |
GDNF-injected mice |
|||
| B, Systemic GDNF injection dramatically increases submucosal neuron density but not myenteric neuron density | ||||
| Small bowel submucosal acetylcholinesterase | 209.6 ± 5.7 | 513.5 ± 20.7 | 2.44 | <0.001 |
| Colon submucosal acetylcholinesterase | 42.3 ± 5.6 | 131.6 ± 18.2 | 3.1 | 0.003 |
| Small bowel myenteric Cuprolinic | 214.8 ± 41 | 216.7 ± 49 | 0.978 | |
| Colon myenteric Cuprolinic | 592 ± 83 | 501 ± 25 | 0.356 | |
| WT |
GFAP–Gdnf mice |
|||
| C, Excess GDNF increases NADPH-d+ neuron density but not TH+ or ChAT+ neuron density | ||||
| Small bowel myenteric NADPH-d | 39.7 ± 3.4 | 51.7 ± 3.3 | 1.3 | 0.017 |
| Small bowel myenteric TH | 0.12 ± 0.05 | 0.20 ± 0.07 | 0.336 | |
| Small bowel myenteric ChAT | 59.2 ± 4.4 | 46.0 ± 6.4 | 0.148 | |
| Colon myenteric NADPH-d | 87.4 ± 7.1 | 128.4 ± 6.8 | 1.47 | <0.001 |
| Colon myenteric ChAT | 94.4 ± 17.6 | 104.4 ± 9.6 | 0.63 | |
| PBS-injected mice |
GDNF-injected mice |
|||
| Small bowel myenteric NADPH-d | 53.7 ± 1.2 | 84.5 ± 7.2 | 1.57 | 0.006 |
| Colon myenteric NADPH-d | 313.5 ± 24 | 266.8 ± 12 | 0.133 | |
| WT |
GDNF+/− |
|||
| D, Decreased GDNF reduces myenteric neuron density | ||||
| Small bowel myenteric NADPH-d | 37.1 ± 4.9 | 20.1 ± 2.9 | 0.54 | 0.043 |
| Small bowel myenteric ChAT | 72.5 ± 4.2 | 34.5 ± 5.2 | 0.48 | 0.003 |
| Esophagus NADPH-d | 14.3 ± 1.0 | 3.6 ± 1.0 | 0.25 | <0.001 |
| WT |
Myo–Gdnf mice |
|||
| E, GDNF transgene in skeletal muscle does not affect small bowel or colon neuron density | ||||
| Small bowel myenteric Cuprolinic | 125.3 ± 7.3 | 155.7 ± 18.2 | 0.197 | |
| Small bowel myenteric NADPH-d | 32.3 ± 1.5 | 39.4 ± 1.5 | 0.325 | |
| Small bowel submucosal acetylcholinesterase | 146.1 ± 7.8 | 135.3 ± 5.4 | 0.319 | |
| Colon myenteric Cuprolinic | 289.9 ± 14.0 | 306.1 ± 43.1 | 1.000 | |
| Colon submucosal acetylcholinesterase | 27.9 ± 3.6 | 34.6 ± 1.8 | 0.166 | |
| WT |
Myo–Gdnf mice |
|||
| F, Increased GDNF dramatically increases esophageal neuron density | ||||
| Esophagus myenteric NADPH-d | 7.8 ± 1.0 | 14.5 ± 0.5 | 1.85 | 0.004 |
| PBS-injected mice |
GDNF-injected mice |
|||
| Esophagus myenteric NADPH-d | 19.5 ± 1.4 | 30.1 ± 3.5 | 1.54 | 0.031 |
| WT |
GFAP–Gdnf mice |
|||
| Esophagus myenteric NADPH-d | 6.8 ± 1.0 | 17.9 ± 2.4 | 2.63 | <0.001 |
Neurons per millimeter squared (±SEM) are shown for GFAP–Gdnf, Myo-Gdnf, GDNF-injected, and Gdnf+/− mice. Because these mice are different ages and different strains, all comparisons are between animals with altered GDNF abundance and WT littermates. Fold change is the ratio of mutant or GDNF-injected mouse to WT mouse. This table is organized to follow the text. The same data are also provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material), but data there are organized by mouse genotype or treatment regimen. n = 6 GFAP–Gdnf mice, 4 GDNF-injected mice, 6 Myo–Gdnf mice, and 3 Gdnf+/− mice as well as strain- and age-matched WT animals.
Systemic GDNF administration from P0 to P30 dramatically increases submucosal neuron number
Because determining the precise time that GDNF levels become elevated in GFAP–Gdnf mice was challenging, we decided to establish a model in which the time of increased GDNF availability and source of excess GDNF were unambiguous. We therefore injected newborn mice with recombinant His-tagged GDNF or PBS twice a day for 30 d and analyzed ENS structure at P30. Mice were injected with large amounts of GDNF subcutaneously on their back to ensure that GDNF reached ENS precursors via the systemic circulation. A similar protocol had been used previously to examine the effects of systemically administered GDNF on motor neuron innervation of neuromuscular junctions (Keller-Peck et al., 2001). To verify that this protocol increased GDNF levels near the postnatal ENS, we also injected P14 mice with His-tagged GDNF or PBS for 5 d and then performed immunohistochemistry using an antibody that recognizes the His tag. These studies demonstrated that recombinant GDNF levels were elevated in the bowel in the region of the submucosal and myenteric plexus (Fig. 1E,J). Particularly high levels are detected near blood vessels. Using this model, we observed no statistically significant difference in the density of Cuprolinic blue-stained myenteric neurons in the small bowel or colon of GDNF-injected mice compared with PBS-injected littermates (small bowel: WT, 214.8 ± 41 cells/mm2 and GDNF-injected, 216.7 ± 49 cells/mm2, p = 0.978; colon: WT, 592 ± 83 cells/mm2 and GDNF-injected, 501 ± 25 cells/mm2, p = 0.356; n = 4 mice of each group) (Table 1B) (supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, submucosal neuron density increased 244% in the small bowel (WT, 209.6 ± 5.7 cells/mm2; GDNF-injected, 513.5 ± 20.7 cells/mm2; p < 0.001; n = 4 mice of each group) and 310% in the colon (WT, 42.3 ± 5.6 cells/mm2; GDNF-injected, 131.6 ± 18.2 cells/mm2; p = 0.003; n = 4 mice of each group) of GDNF-injected mice compared with WT littermates (supplemental Table 1, available at www.jneurosci.org as supplemental material) (Table 1B; Fig. 2). Thus, increased GDNF availability starting at P0 can dramatically increase the density of some enteric neurons but not others.
Figure 2.
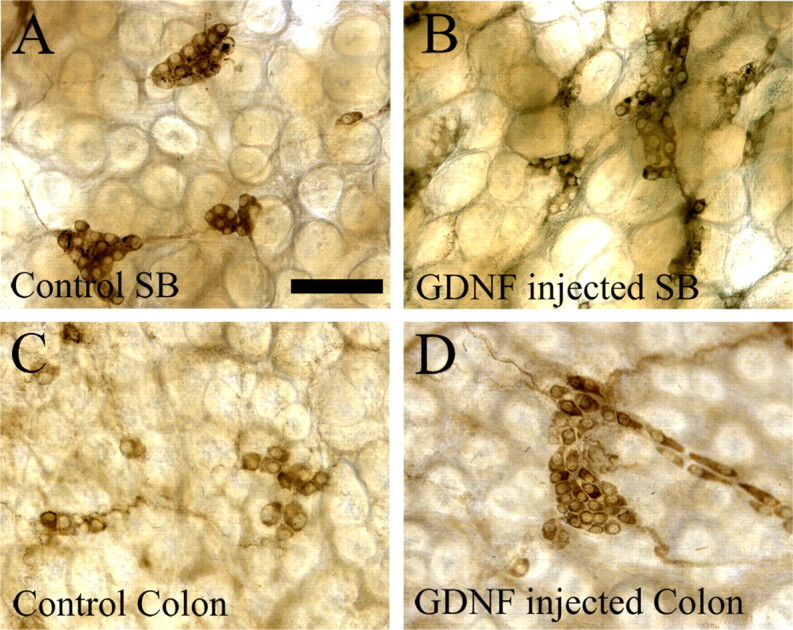
Submucosal neuron number is increased in GDNF-injected mice. A–D, Acetylcholinesterase-expressing submucosal neurons are more abundant in GDNF-injected mouse small bowel (B) and colon (D) than in PBS-injected WT littermates (A, C). Quantitative data are in Table 1 and supplemental Table 1 (available at www.jneurosci.org as supplemental material). Scale bar (in A), 100 μm.
Because His-tagged GDNF levels were highest around submucosal blood vessels (Fig. 1J), we hypothesized that injected GDNF could cause aggregation or enhanced proliferation of submucosal neurons adjacent to vessels if GDNF levels were limiting in injected mice. Quantitative analysis of submucosal neuron distribution, however, demonstrated that neuron density was similar adjacent to and farther from blood vessels, and the distribution of neurons was similar in PBS- and GDNF-injected mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (PBS-injected mice: colon submucosal neurons in fields with blood vessels, 48.3 ± 6.5 cells/mm2 or without blood vessels, 48.8 ± 4.8 cells/mm2, n = 3 mice, p = 0.95; GDNF-injected mice in fields with blood vessels, 109.5 ± 5.3 cells/mm2 or without blood vessels, 127.2 ± 6.9 cells/mm2, n = 3 mice, p = 0.11). Thus, GDNF injection may provide saturating levels of GDNF, at least in the submucosa.
Excess GDNF alters the ratio of myenteric neuron subclasses
One possible explanation for the increase in submucosal but not myenteric neurons in GDNF-injected mice is that injection results in higher levels of GDNF in the submucosa than in the myenteric region of the bowel. To determine more definitively if differential effects of excess GDNF on enteric neuron subclasses could result from differing proliferative capacity of precursor cells and not simply from different levels of GDNF exposure, we decided to evaluate the density of selected myenteric neuron subclasses. Because different classes of myenteric neuron precursors become postmitotic at different stages of development (Pham et al., 1991), one potential way to differentiate between the mechanisms outlined above was to examine the effect of increased GDNF on subpopulations of myenteric neurons that presumably will be exposed to nearly identical amounts of GDNF because these neurons are directly adjacent to each other within single ganglia. We hypothesized therefore that, if proliferative capacity of ENS precursors in response to GDNF underlies the differential effects of excess GDNF on submucosal versus myenteric neurons, then GFAP–Gdnf and GDNF-injected mice might also have changes in the ratio of neuron subclasses within the myenteric plexus based on the time that these precursors exit the cell cycle. More specifically, precursors for myenteric neurons producing VIP are among the last to exit the cell cycle with some precursors still proliferating at P5 (Pham et al., 1991). For technical reasons, we analyzed NO-producing neurons (NADPH-d) in GFAP–Gdnf transgenic and GDNF-injected mice because almost all NO-producing neurons also express VIP (Sang and Young, 1996). These analyses demonstrated a 30% increase in small bowel and 47% increase in colon NADPH-d neuron density in the myenteric plexus of GFAP–Gdnf mice (small bowel: WT, 39.7 ± 3.4 cells/mm2 and GFAP–Gdnf, 51.7 ± 3.3 cells/mm2, p = 0.017; colon: WT, 87.4 ± 7.1 cells/mm2 and GFAP–Gdnf, 128.4 ± 6.8 cells/mm2, p < 0.001; n = 6 mice of each genotype) and a 57% increase NADPH-d small bowel myenteric neurons in GDNF-injected mice (WT, 53.7 ± 1.2 cells/mm2; GDNF-injected, 84.5 ± 7.2 cells/mm2; p = 0.006; n = 6 mice of each genotype) (Fig. 3A,B,E,F; Table 1C) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Interestingly, this increase in the density of NADPH-d neuron occurred without a statistically significant increase in total Cuprolinic blue-stained neurons in the small bowel of GFAP–Gdnf or GDNF-injected mice. We also examined the density of serotonergic neurons (TH-expressing cells) in GFAP–Gdnf mice because the precursors for these cells are among the earliest to exit the cell cycle (Pham et al., 1991). Although this analysis is difficult because of the small number of these cells within the myenteric plexus, no statistically significant difference in serotonergic neuron density was found in GFAP–Gdnf mice versus WT littermates (WT, 0.12 ± 0.05 cells/mm2; GFAP–Gdnf, 0.20 ± 0.07 cells/mm2; p = 0.336; n = 6 mice of each genotype) (Fig. 3C,G; Table 1C). To be more convinced that different subpopulations of myenteric neurons are differentially affected by GDNF overexpression late in gestation, we also evaluated the abundance of ChAT-positive cells in the small bowel and colon of GFAP–Gdnf and WT mice because ChAT precursors exit the cell cycle at E15, 2–3 d earlier than Gfap is expressed in the mouse ENS. To make these results as unambiguous as possible, NADPH-d and ChAT staining were performed on the same tissue specimens, and we counted neurons per ganglion of each subtype (Fig. 4). These studies confirmed that the number of NADPH-d neurons in the myenteric plexus is increased in GFAP–Gdnf versus WT mice, but there was no difference in the abundance of ChAT neurons in the same whole-mount myenteric plexus preparations (small bowel: WT, 59.2 ± 4.4 cells/mm2 and GFAP–Gdnf, 46.0 ± 6.4 cells/mm2, p = 0.148; colon: WT, 94.4 ± 17.6 cells/mm2 and GFAP–Gdnf, 104.4 ± 9.6 cells/mm2, p = 0.63; n = 4 mice of each genotype) (Fig. 4G,H; Table 1C) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Overall, these data demonstrate that changes in GDNF abundance can significantly alter the ratio of neurons within the ENS even when these cells are directly adjacent to each other, making differential exposure to GDNF an unlikely explanation for these findings.
Figure 3.
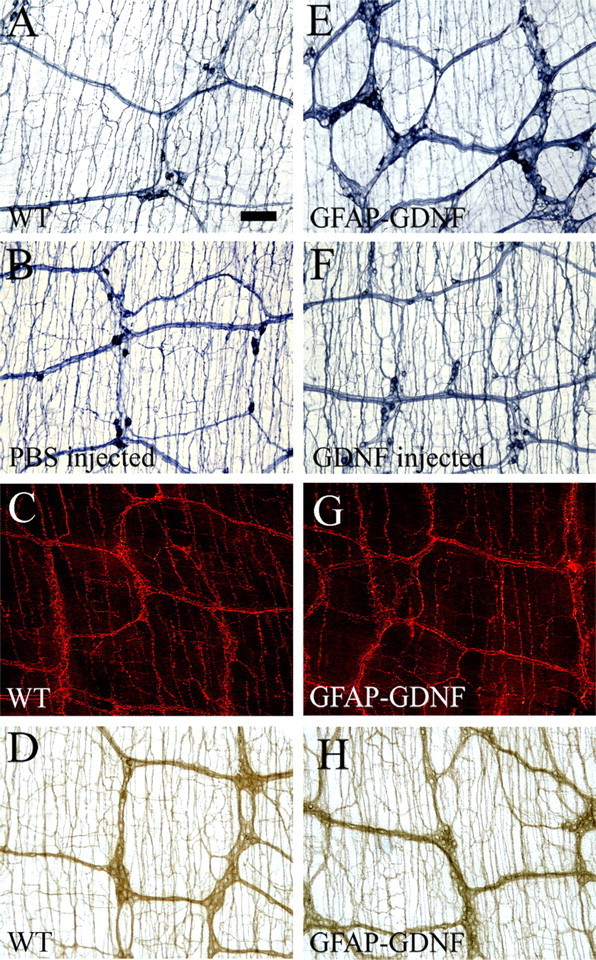
Increased GDNF influences some aspects of myenteric plexus morphogenesis. NADPH-d myenteric neuron fibers have a higher density around enteric ganglia in GFAP–Gdnf mice (E) than WT animals (A), but mice injected with GDNF (F) do not have an increase in NADPH-d-stained neuronal fibers around enteric ganglia compared with controls (B). Tryptophan hydroxylase (C, G) and acetylcholinesterase (D, H) stained myenteric plexus in WT (C, D) and GFAP–Gdnf transgenic mice (G, H) appear similar. Scale bar (in A), 100 μm.
Figure 4.
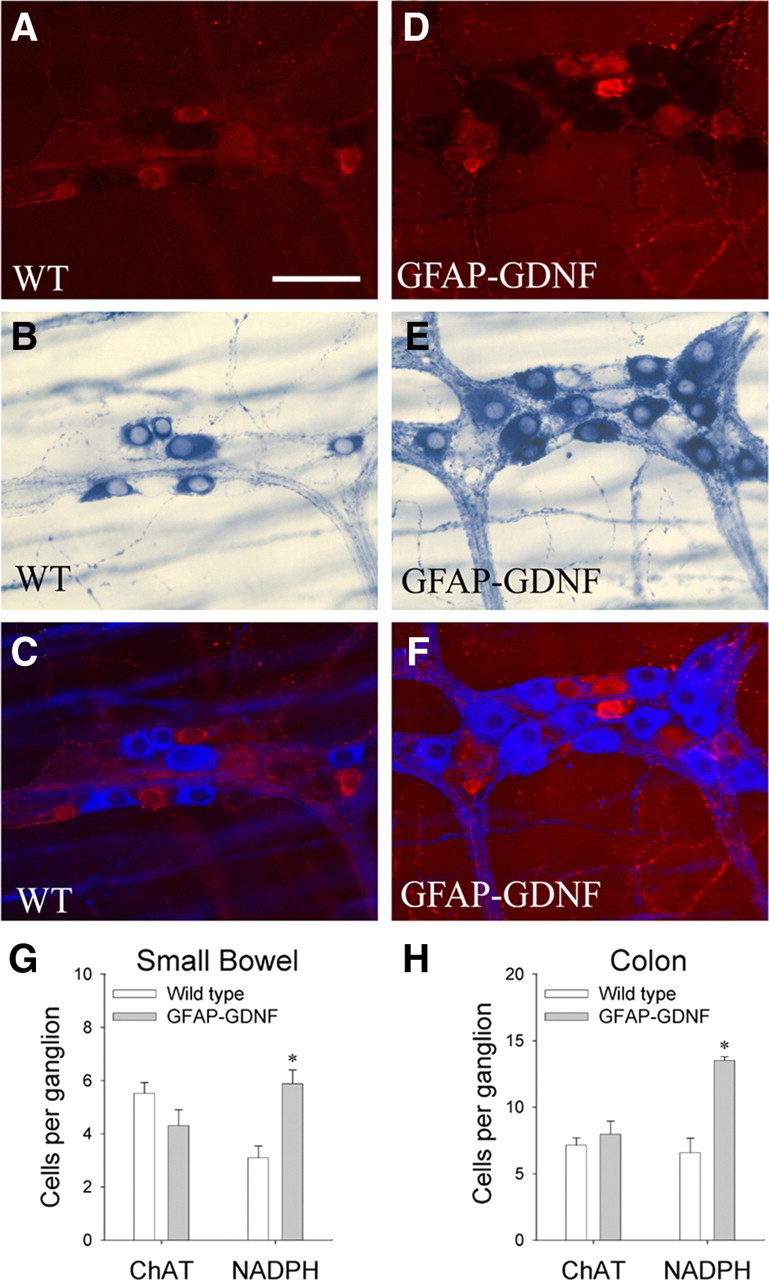
NADPH-d-positive myenteric neurons are increased in GFAP–Gdnf mice, but ChAT neurons are comparable in abundance to WT animals. To determine whether GDNF overexpression differentially affects the abundance of specific neuronal subpopulations, colon myenteric plexus was simultaneously labeled by ChAT immunohistochemistry (A, C, D, F) and NADPH-d histochemistry (B, C, E, F). C, F, For these images, NADPH-d neurons and fibers are shown using pseudo-color to facilitate combining fluorescent (A, D) and bright-field (B, E) images and to make details in both images visible. G, H, Quantitative analysis of the number of ChAT or NADPH-d neurons per ganglia in the small bowel (G) and colon (H). n = 4 mice of each genotype and 20 fields (0.25 mm2) per mouse. Error bars represent ± SEM. *p < 0.05 versus WT. Scale bar, 50 μm.
Because we hypothesized that the timing of GDNF expression is a critical determinant of the abundance of enteric neuron subtypes, a corollary to this hypothesis is that increasing or reducing GDNF abundance throughout development should affect all neuronal subpopulations equivalently. Because we do not have an available model of increased GDNF abundance throughout development, we examined Gdnf+/− mice that we and others (Shen et al., 2002; Gianino et al., 2003) had demonstrated previously have substantially fewer myenteric and submucosal neurons than WT animals. In Gdnf+/− mice, we found approximately equivalent reductions in ChAT-positive (48% of WT; WT small bowel, 72.5 ± 4.2 cells/mm2; Gdnf+/−, 34.5 ± 5.2 cells/mm2; p = 0.003; n = 3 of each genotype) and NADPH-d-positive (54% of WT; WT small bowel, 37.1 ± 4.9 cells/mm2; Gdnf+/−, 20.1 ± 2.9 cells/mm2; p = 0.043; n = 3 mice of each genotype) (supplemental Table 1D, available at www.jneurosci.org as supplemental material) neurons in the small bowel myenteric plexus, a reduction that is not statistically different from the reduction in total Cuprolinic blue-stained myenteric neurons [Gdnf+/− neuron density is 57% of WT (Gianino et al., 2003)] in the small bowel. Although it is possible that increased cell death could account for these findings rather than decreased proliferation, our previous studies make this less likely (Gianino et al., 2003). Furthermore, we found no evidence of apoptotic cell death in the ENS of Gdnf+/− mice by activated caspase-3 immunohistochemistry at E14, E16, P0, P14, or in adult animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that differences between WT and Gdnf+/− mice are not attributable to increased cell death in the mutant animals but instead to reduced cell proliferation (>300 PGP9.5-expressing cells examined at each age). Collectively, these data demonstrate that the timing and abundance of GDNF critically influences the abundance of neuronal subpopulations in the ENS.
Endogenous GDNF is retained close to its site of synthesis
The observation that GFAP–Gdnf mice have a relatively little change in small bowel submucosal neuron density compared with that observed in GDNF-injected mice suggested that GDNF might be retained close to its site of synthesis. This could explain why small bowel NADPH-d myenteric neuron density in GFAP–Gdnf and GDNF-injected mice increases by a similar amount (30 vs 57%), but the effect in the submucosal plexus of increasing GDNF via these two mechanisms is remarkably different (19 vs 244%). This could occur, for example, if production of GDNF from the GFAP promoter was minimal in the region adjacent to developing submucosal neuron precursors. As an additional test of whether local GDNF synthesis is important for effects on ENS development, we examined ENS structure in Myo–Gdnf transgenic mice. Myo–Gdnf mice express high levels of GDNF in skeletal muscle starting during fetal development and continuing into adult life, resulting in hyperinnervation of neuromuscular junctions (Cheng et al., 1992; Merlie et al., 1994; Nguyen et al., 1998). Quantitative analyses demonstrated that neuron number, neuron size, and neuronal fiber density were normal in Myo–Gdnf small bowel and colon (Tables 1E, 2C, 3C) (supplemental Table 1, available at www.jneurosci.org as supplemental material). This suggested that GDNF produced in skeletal muscle was not available to ENS precursors even when overexpressed. Enzyme linked immunoassay confirmed that GDNF levels are very low in the blood of these animals (Zhao et al., 2004). Thus, it seems likely that GDNF is retained close to its site of synthesis.
Table 2.
Quantitative cell size data
| Cell size (μm2) | Fold change | p value | ||
|---|---|---|---|---|
| WT |
GFAP–Gdnf mice |
|||
| A, Excess GDNF increases cell size of some mature submucosal and myenteric neurons in small bowel and colon> | ||||
| Small bowel myenteric Cuprolinic | 206.5 ± 9.3 | 260.8 ± 9.6 | 1.26 | <0.001 |
| Colon myenteric Cuprolinic | 213.1 ± 8.7 | 229.3 ± 7.1 | 1.07 | 0.028 |
| Small bowel myenteric NADPH-d | 170.5 ± 4.4 | 202.7 ± 6.2 | 1.18 | <0.001 |
| Colon myenteric NADPH-d | 192.9 ± 5.6 | 245.4 ± 7.2 | 1.27 | <0.001 |
| Small bowel myenteric TH | 1099.7 ± 158.7 | 1107 ± 132.1 | 0.261 | |
| Small bowel submucosal acetylcholinesterase | 254.5 ± 6.9 | 269.7 ± 7.9 | 0.181 | |
| Colon submucosal acetylcholinesterase | 451.1 ± 17.0 | 453.9 ± 16.1 | 0.909 | |
| PBS-injected mice |
GDNF-injected mice |
|||
| Small bowel myenteric Cuprolinic | 203.7 ± 6 | 241.5 ± 7 | 1.19 | <0.001 |
| Colon myenteric Cuprolinic | 214.3 ± 6 | 245.6 ± 7 | 1.14 | 0.002 |
| Small bowel myenteric NADPH-d | 183.4 ± 6 | 229.8 ± 6 | 1.25 | <0.001 |
| Colon myenteric NADPH-d | 203.1 ± 4 | 233.7 ± 5 | 1.15 | <0.001 |
| Small bowel submucosal acetylcholinesterase | 243.5 ± 6 | 265.7 ± 6 | 1.09 | 0.002 |
| Colon submucosal acetylcholinesterase | 396.7 ± 14 | 420.4 ± 14 | 0.142 | |
| WT |
GFAP–Gdnf mice |
|||
| B, Excess GDNF increases cell size of esophageal neurons | ||||
| Esophagus myenteric NADPH-d | 214.0 ± 9.8 | 318.3 ± 9.9 | 1.49 | <0.001 |
| PBS-injected mice |
GDNF-injected mice |
|||
| Esophagus myenteric NADPH-d | 181.5 ± 6 | 311.8 ± 7 | 1.72 | <0.001 |
| WT |
Myo–Gdnf mice |
|||
| C, GDNF trangene in skeletal muscle does not increase cell size of mature enteric neurons | ||||
| Esophagus myenteric NADPH-d | 238.9 ± 24 | 296.1 ± 29 | 0.199 | |
| Small bowel myenteric Cuprolinic | 182.1 ± 12 | 163.2 ± 25 | 0.531 | |
| Small bowel myenteric NADPH-d | 212.0 ± 15 | 237.2 ± 45 | 0.626 | |
| Small bowel submucosal acetylcholinesterase | 218.2 ± 9 | 214.0 ± 13 | 0.801 | |
| Colon myenteric Cuprolinic | 189.2 ± 24 | 176.4 ± 31 | 0.762 | |
| Colon submucosal acetylcholinesterase | 369.4 ± 32 | 324.9 ± 11 | 0.263 | |
The sizes of enteric neurons (±SEM) are shown for GFAP–Gdnf, Myo–Gdnf, and GDNF-injected mice. Because these mice are different ages and different strains, all comparisons are between animals with altered GDNF abundance and WT littermates. This table is organized to follow the text. The same data are also provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material), organized by mouse genotype or treatment regimen. Animal numbers are the same as in Table 1.
Table 3.
Quantitative neuronal fiber density data
| Fiber density | Fold change | p value | ||
|---|---|---|---|---|
| WT |
GFAP–Gdnf mice |
|||
| A, GFAP–Gdnf transgenic mice versus WT | ||||
| Esophagus myenteric NADPH-d fiber bundles | 0.0 ± 0.0 | 1.96 ± 0.2 | ** | <0.001 |
| Esophagus myenteric NADPH-d single fibers | 1.94 ± 0.2 | 4.86 ± 0.6 | 2.52 | <0.001 |
| Esophagus submucosal NADPH-d single fibers | 35.8 ± 7.0 | 84.6 ± 14.8 | 2.36 | 0.018 |
| Small bowel myenteric NADPH-d fiber bundles | 3.2 ± 0.2 | 4.2 ± 0.2 | 1.31 | <0.001 |
| Small bowel myenteric NADPH-d single fibers | 38.6 ± 1.6 | 41.4 ± 1.4 | 0.162 | |
| Small bowel myenteric TH fiber bundles | 4.4 ± 0.1 | 4.4 ± 0.1 | 0.823 | |
| Small bowel myenteric TH single fibers | 12.2 ± 0.3 | 11.2 ± 0.4 | 0.92 | 0.035 |
| Small bowel myenteric ChAT fiber bundles | 4.4 ± 0.4 | 5.4 ± 0.2 | 1.23 | 0.02 |
| Small bowel myenteric ChAT single fibers | 10.6 ± 1.0 | 8.2 ± 1.2 | 0.07 | |
| Small bowel myenteric acetylcholinesterase fiber bundles | 4.0 ± 0.2 | 4.5 ± 0.2 | 1.13 | 0.04 |
| Small bowel myenteric acetylcholinesterase single fibers | 44.6 ± 1.2 | 46.4 ± 1.4 | 0.292 | |
| Colon myenteric NADPH-d fiber bundles | 4.0 ± 0.2 | 5.5 ± 0.2 | 1.37 | <0.001 |
| Colon myenteric NADPH-d single fibers | 51.0 ± 2.0 | 46.6 ± 1.2 | 0.059 | |
| Colon myenteric acetylcholinesterase fiber bundles | 5.2 ± 0.2 | 5.7 ± 0.2 | 0.108 | |
| Colon myenteric acetylcholinesterase single fibers | 45.4 ± 1.4 | 44.0 ± 1.0 | 0.448 | |
| PBS-injected mice |
GDNF-injected mice |
|||
| B, GDNF-injected mice versus PBS-injected controls | ||||
| Esophagus myenteric NADPH-d fiber bundles | 0.0 ± 0.0 | 4.8 ± 0.26 | ** | <0.001 |
| Esophagus myenteric NADPH-d single fibers | 4.0 ± 0.4 | 8.4 ± 0.6 | 2.07 | <0.001 |
| Small bowel myenteric NADPH-d fiber bundles | 3.6 ± 0.1 | 4.3 ± 0.1 | 1.19 | 0.002 |
| Small bowel myenteric NADPH-d single fibers | 47.8 ± 1.0 | 46.6 ± 0.6 | 0.228 | |
| Small bowel myenteric TH fiber bundles | 4.2 ± 0.1 | 4.1 ± 0.1 | 0.809 | |
| Small bowel myenteric TH single fibers | 14.1 ± 0.3 | 14.5 ± 0.4 | 0.431 | |
| Small bowel myenteric acetylcholinesterase fiber bundles | 4.7 ± 0.1 | 5.2 ± 0.2 | 1.11 | 0.018 |
| Small bowel myenteric acetylcholinesterase single fibers | 43.6 ± 0.8 | 45.4 ± 1.0 | 0.257 | |
| Colon myenteric NADPH-d fiber bundles | 6.7 ± 0.1 | 6.3 ± 0.1 | 0.94 | 0.031 |
| Colon myenteric NADPH-d single fibers | 41.2 ± 0.6 | 44.8 ± 0.8 | 1.09 | <0.001 |
| Colon myenteric acetylcholinesterase fiber bundles | 5.8 ± 0.2 | 6.1 ± 0.1 | 0.242 | |
| Colon myenteric acetylcholinesterase single fibers | 44.8 ± 0.8 | 45.8 ± 0.8 | 0.317 | |
| Colon myenteric TH fiber bundles | 6.0 ± 0.2 | 5.8 ± 0.2 | 0.337 | |
| Colon myenteric TH single fibers | 12.6 ± 0.2 | 12.0 ± 0.2 | 0.147 | |
| WT |
Myo–Gdnf mice |
|||
| C, Myo–Gdnf mice versus WT | ||||
| Esophagus myenteric NADPH-d single fibers | 7.8 ± 2.4 | 37.4 ± 4.5 | 4.79 | <0.001 |
| Esophagus submucosal NADPH-d single fibers | 22.6 ± 1.3 | 21.6 ± 1.3 | 0.598 | |
| Small bowel myenteric NADPH-d fiber bundles | 6.9 ± 0.5 | 7.5 ± 1.0 | 0.573 | |
| Small bowel myenteric NADPH-d single fibers | 45.4 ± 2.6 | 48.8 ± 2.6 | 0.385 | |
| Small bowel myenteric acetylcholinesterase fiber bundles | 9.9 ± 0.8 | 9.6 ± 1.0 | 0.801 | |
| Small bowel myenteric acetylcholinesterase single fibers | 45.0 ± 5.0 | 41.0 ± 4.2 | 0.584 | |
| Colon myenteric acetylcholinesterase fiber bundles | 12.8 ± 1.4 | 12.1 ± 1.7 | 0.779 | |
| Colon myenteric acetylcholinesterase single fibers | 37.6 ± 4.6 | 43.0 ± 3.2 | 0.387 | |
| WT |
Gdnf+/− |
|||
| D, Gdnf+/− mice versus WT | ||||
| Esophagus myenteric NADPH-d single fibers | 7.1 ± 1.4 | 2.6 ± 0.6 | 0.37 | 0.047 |
| Small bowel myenteric NADPH-d single fibers | 52.0 ± 2.0 | 42.4 ± 1.2 | 0.82 | 0.026 |
The number of neuronal fibers (±SEM) crossing the left or upper border of a 0.5 × 0.5 mm counting grid are shown. Fiber bundles and single fibers were counted separately. This table is organized to follow the text. The same data are also provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material), organized by mouse genotype or treatment regimen. Animal numbers are the same as in Table 1.
**Cannot be calculated because of division by zero.
Increased GDNF dramatically affects esophageal innervation
Esophageal neurons are also influenced by Ret signaling, but, unlike the small bowel and colon, some esophageal neurons are present in Ret−/− mice (Schuchardt et al., 1994; Durbec et al., 1996; Gershon, 1997; Yan et al., 2004). To determine the influence of excess GDNF on esophageal neurons, we evaluated Myo–Gdnf, GFAP–Gdnf, and GDNF-injected mice after NADPH-d staining (Fig. 5). The Myo–Gdnf transgene in this case might be expected to produce GDNF near esophageal neuron precursors because, in the adult mouse, the entire esophagus has skeletal muscle, not smooth muscle, and transgenes driven by the myogenin promoter are detected in the esophagus as early as E15.5 (Kablar et al., 2000). These analyses demonstrated a 1.85-fold increase in NADPH-d esophageal neurons in Myo–Gdnf mice (WT, 7.8 ± 1.0 cells/mm2; Myo–Gdnf, 14.5 ± 0.5 cells/mm2; p = 0.004; n = 6 mice of each genotype), 1.54-fold increase in GDNF-injected mice (WT, 19.5 ± 1.4 cells/mm2; GDNF-injected 30.1 ± 3.5 cells/mm2; p = 0.031; n = 4 mice of each group), and a 2.63-fold increase in GFAP–Gdnf mice (WT, 6.8 ± 1.0 cells/mm2; GFAP–Gdnf, 17.9 ± 2.4 cells/mm2; p < 0.001; n = 6 mice of each group) (Table 1F). Additional support for the normal role of GDNF in determining the density of esophageal neurons is provided by the 75% reduced density of NADPH-d neurons in the esophagus of Gdnf+/− mice compared with WT littermates (WT, 14.3 ± 1 cells/mm2; Gdnf+/−, 3.6 ± 1 cells/mm2; p < 0.001; n = 4 WT and 3 Gdnf+/− mice). Thus, several lines of evidence confirm that esophageal neuron number depends on GDNF availability and that GDNF may affect esophageal neuron density even when administered after P0 (Table 1D,F; Fig. 5).
Figure 5.
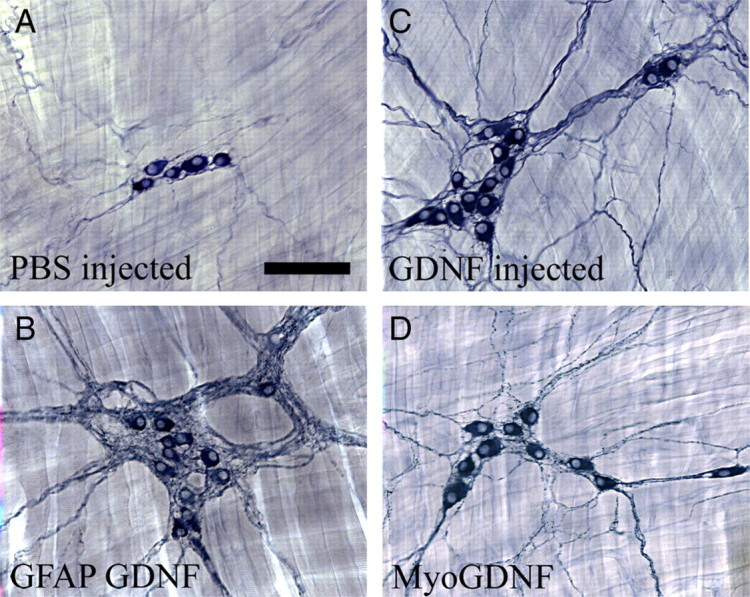
Esophageal innervation is influenced by increased GDNF. A, NADPH-d myenteric neurons in WT esophagus or from PBS-injected mice are typically found in small clusters with a few small neuronal fibers between ganglia. B, GFAP–Gdnf mice had more esophageal neurons and a dramatic accumulation of neuronal fibers near enteric ganglia. GDNF-injected mice (C) and Myo–Gdnf (D) had more abundant neurons, as well as a higher density of neuronal fibers. Scale bar, 100 μm.
Proliferating precursors respond to changes in GDNF levels
The preceding analyses demonstrated dramatic effects on submucosal and esophageal neurons with minimal effect on the myenteric plexus when GDNF levels increase starting at P0. One hypothesis that could explain these observations would be increased proliferation of submucosal and esophageal neuron precursors in response to excess GDNF. If this is the correct explanation, one might expect a higher postnatal proliferation rate for the precursors that give rise to these cells than for ENS precursors in the myenteric plexus. Furthermore, there should be increased proliferation demonstrable in the submucosal plexus and esophagus of GDNF-injected mice. To evaluate this possibility, proliferation rates of ENS precursors were determined by BrdU/PGP9.5 double labeling at E17, E18, P0, P3, P5, and P8 (Fig. 6A,C,D). These proliferation data were then compared with the effects of GDNF injection (P0–P30) on enteric neuron number. There were several specific effects of injected GDNF that we wanted to correlate with proliferation rates for ENS precursors in WT mice. First, small bowel submucosal neuron number increased 244% in GDNF-injected mice, but colon submucosal neuron number increased 310%. This suggested that submucosal neuron precursors continue to proliferate longer in the colon than in the small bowel of WT mice. Although there were inadequate numbers of submucosal PGP9.5-expressing cells for quantitative analysis of proliferation at P0, proliferation rates in the WT colon were 57% (p < 0.005) and 43% (p < 0.001) higher at P3 and P8, respectively, than in the small bowel. Second, although submucosal neurons increased dramatically in GDNF-injected mice, Cuprolinic blue-stained myenteric neuron numbers did not. This correlates with the observation that, at P3, P5, and P8, colon submucosal neuron precursor proliferation rates are 400% (p < 0.001), 180% (p < 0.074), and 240% (p < 0.001) higher, respectively, than in the colon myenteric plexus. Similarly, in the small bowel, submucosal neuron precursor proliferation rates are 230% (p = 0.004), 175% (p = 0.002), and 238% (p = 0.001) higher in the submucosal plexus at P3, P5, and P8, respectively, than in the myenteric plexus. Finally, in the esophagus, there was a 54% increase in neuron number in the GDNF-injected mice. This correlates with the observation that, in WT esophagus, the rate of proliferation for ENS precursors increases from E17 to P5. Furthermore, compared with the small bowel myenteric plexus, for example, proliferation rates for PGP9.5+ cells in the esophagus are 198% (p = 0.001) and 235% (p = 0.009) higher at P5 and P8, respectively. Overall, these data support the hypothesis that changes in GDNF availability differentially affect specific enteric neuron populations based on the normal timing of withdrawal from the cell cycle for their precursor cells.
Figure 6.
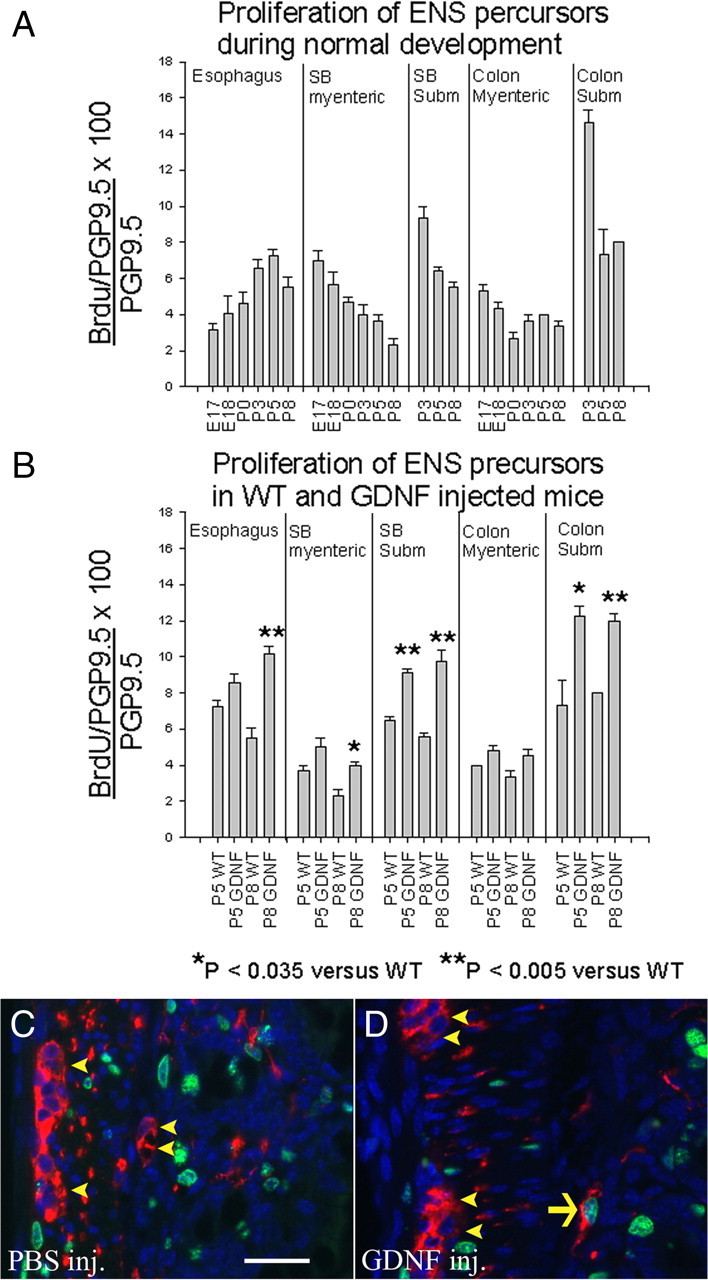
Because ENS precursors have regional and temporal differences in proliferation rates that change during development, increased GDNF leads to increased precursor proliferation in subsets of cells that have not yet exited the cell cycle. A, The percentage of PGP9.5-expressing cells that labeled with BrdU was determined in different regions of the bowel at E17, E18, P0, P3, P5, and P8. B, Proliferation rates for PGP9.5-expressing enteric neurons were also determined in mice injected with GDNF daily from P0 to P5 or P0 to P8. C, D, PGP9.5/BrdU double-labeled immunohistochemistry in mice injected with PBS (C) or GDNF (D) from P0 to P5. Mice were analyzed at P5, 3 h after BrdU injection. Arrow indicates a PGP9.5/BrdU double-positive cell. Arrowheads indicate PGP9.5+ but BrdU-negative cells. n = 3 mice at each age. Error bars represent ± SEM. *p < 0.035 versus WT; **p < 0.005 versus WT. Scale bar, 25 μm.
To more conclusively demonstrate that the changes in cell density observed in subsets of enteric neurons result from increased proliferation of their precursors, we directly measured precursor proliferation rates in mice injected with GDNF from P0 to P5 or from P0 to P8 (Fig. 6B). These studies confirmed that GDNF injection led to substantial increases in the proliferation rates for both submucosal and esophageal neuron precursors, the two populations most dramatically affected by our GDNF injection paradigm. In contrast, proliferation rates for neuronal precursors within the myenteric plexus did not significantly increase as a result of GDNF injections with the exception of a small increase in P8 small bowel myenteric neuron precursors. This could correlate with the increase in NADPH-d myenteric neurons detected in the small bowel of GDNF-injected mice. In conjunction with the other data provided above, these BrdU data demonstrate that postnatal increases in GDNF can increase proliferation rates for ENS precursors that have not yet exited the cell cycle in WT mice and thus alter the ratio of neuron subtypes within the ENS.
GDNF provides trophic support for some mature submucosal and myenteric neurons in the small bowel and colon
Although an essential role for GDNF during ENS development is already established, GDNF effects on the mature ENS are less well known. To determine whether excess GDNF could support mature enteric neurons, we measured neuronal cell size in WT, GFAP–Gdnf, and GDNF-injected mice (Table 2A). GFAP–Gdnf and GDNF-injected mice had 7–27% increases in small bowel and colon Cuprolinic blue-stained or NADPH-d+ myenteric neuron cell size (small bowel: Cuprolinic, WT, 206.5 ± 9.3 μm2 and GFAP–Gdnf, 260.8 ± 9.6 μm2, p < 0.001; colon: Cuprolinic, WT, 213.1 ± 8.7 μm2 and GFAP–Gdnf, 229.3 ± 7.1 μm2, p = 0.028; small bowel: NADPH-d+, WT, 170.5 ± 4.4 μm2 and GFAP–Gdnf, 202.7 ± 6.2 μm2, p < 0.001; colon: NADPH-d+, WT, 192.9 ± 5.6 μm2 and GFAP–Gdnf, 245.4 ± 7.2 μm2, p < 0.001; for GDNF-injected mice, small bowel: Cuprolinic, WT, 203.7 ± 6 μm2 and GDNF-injected, 241.5 ± 7 μm2, p < 0.001; colon: Cuprolinic, WT, 214.3 ± 6 μm2 and GDNF-injected, 245 ± 7 μm2, p = 0.002; small bowel: NADPH-d+, WT, 183.4 ± 6 μm2 and GDNF-injected, 229.8 ± 6 μm2, p < 0.001; colon: NADPH-d+, WT, 203.1 ± 4 μm2 and GDNF-injected, 233.7 ± 5 μm2, p < 0.001), and GDNF-injected mice had a 9% increase in small bowel submucosal neuron size compared with control littermates (WT, 243.5 ± 6 μm2; GDNF-injected, 265.7 ± 6 μm2; p = 0.002). In addition, there was a 9% increase in the density of small NADPH-d neuronal fibers in the colon myenteric plexus (Table 3B) (WT, 41.2 ± 0.6; GDNF-injected, 44.8 ± 0.8; p < 0.001; n = 4 mice each) and a dramatic increase in the thickness of fiber bundles innervating the villi of GDNF-injected mice (Fig. 7A,B). Finally, there was an 18% decrease in the density of small NADPH-d neuronal fibers in the small bowel of GDNF heterozygous mice (WT, 52.0 ± 2.0; Gdnf+/−, 42.4 ± 1.2; p = 0.026; n = 3 mice each), supporting the hypothesis that GDNF normally provides trophic support to at least some mature enteric neurons including NADPH-d cells.
Figure 7.
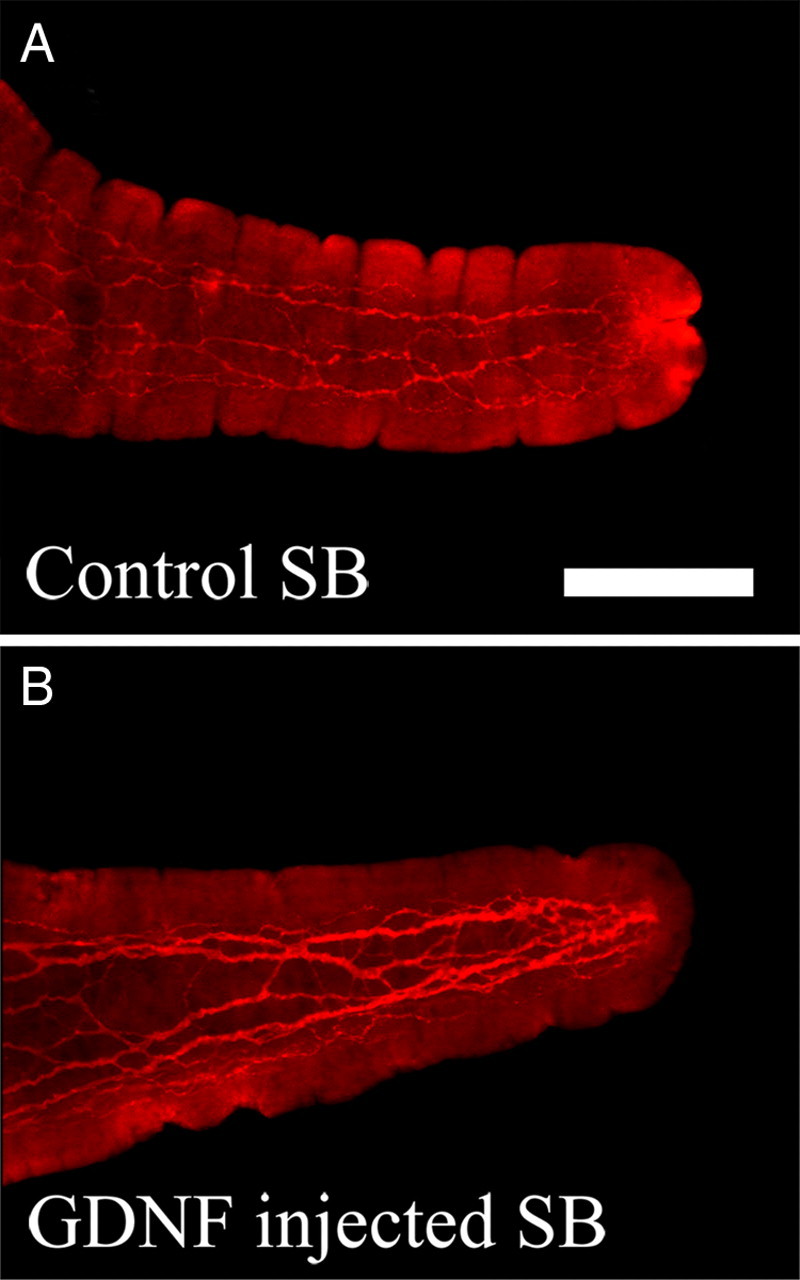
Nerve fiber density in the villus of GDNF-injected mice is increased compared with control littermates. A, PGP9.5 immunoreactive neuronal fibers in a villus from a control mouse. B, PGP9.5-immunoreactive neuronal fibers in a villus from a GDNF-injected mouse. Scale bar, 100 μm.
In contrast to these findings, there was no change in the size of submucosal neurons in the small bowel or colon of GFAP–Gdnf mice (Table 2A) (small bowel: WT, 254.5 ± 6.9 μm2 and GFAP–Gdnf, 269.7 ± 7.9 μm2, p = 0.181; colon: WT, 451.1 ± 17.0 μm2 and GFAP–Gdnf, 453.9 ± 16.1 μm2; p = 0.909; n = 6 mice of each genotype) or in the colon of GDNF-injected mice (Table 2A) (WT, 396.7 ± 14 μm2; GDNF-injected, 420.4 ± 14 μm2; p = 0.142; n = 4 mice each). There was also no change in the size of TH-expressing cells in the small bowel myenteric plexus of GFAP–Gdnf mice (WT, 1099.7 ± 158.7 μm2; GFAP–Gdnf, 1107 ± 132.1 μm2; p = 0.261). Furthermore, with the exception of colonic NADPH-d-stained small neuronal fibers in GDNF-injected mice (WT, 41.2 ± 0.6; GDNF-injected, 44.8 ± 0.8; p < 0.001), there was no change in the density of small neuronal fibers in the colon or small bowel myenteric plexus of GFAP–Gdnf (Fig. 3A,E) or GDNF-injected mice detected by NADPH-d (Fig. 3B,F) or acetylcholinesterase (Fig. 3D,H; Table 3A,B) staining. Overall, these data suggest that, although GDNF is essential for proliferation and survival of developing enteric neurons (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996), GDNF provides trophic support to only a subset of mature enteric neurons.
GDNF can provide trophic support to esophageal neurons
Esophageal neuron size is increased 49 and 72%, respectively, in GFAP–Gdnf and GDNF-injected mice compared with WT littermates (Table 2B) (WT, 214.0 ± 9.8 μm2 and GFAP–Gdnf, 318 ± 9.9 μm2, p < 0.001, n = 6 mice of each genotype; for GDNF-injected mice: WT, 181.5 ± 6 μm2 and GDNF-injected, 311.8 ± 7 μm2, p < 0.001, n = 4 mice each). In contrast, esophageal neuron size was normal in Myo–Gdnf transgenic mice (WT, 238.9 ± 24 μm2; Myo–Gdnf, 296.1 ± 29 μm2; p = 0.199; n = 6 mice of each genotype). This does not, however, mean that GDNF was unavailable to esophageal neurons in Myo–Gdnf mice. In fact, there is a 479% increase in the density of neuronal fibers in the esophagus of adult Myo–Gdnf compared with WT mice (Table 3C) (WT, 7.8 ± 2.4; Myo–Gdnf, 37.4 ± 4.5; p < 0.001) and a 7.7-fold increase in Gdnf mRNA in the P8 esophagus (p < 0.001). Similarly, there were 252 and 207% increases in the neuronal fiber density in the esophagus of GFAP–Gdnf (Table 3A) (WT, 1.94 ± 0.2; GFAP–Gdnf, 4.86 ± 0.6; p < 0.001; n = 6 mice of each genotype) and GDNF-injected (Table 3B) (WT, 4.0 ± 0.4; GDNF-injected, 8.4 ± 0.6; p < 0.001; n = 4 mice of each genotype) mice, respectively, and a 236% increase in esophageal submucosal neuron fiber density in GFAP–Gdnf mice (Table 3A) (WT, 35.8 ± 7; GFAP–Gdnf, 84.6 ± 14.8; p = 0.018; n = 6 mice of each genotype). Furthermore, the density of NADPH-d neuronal fibers in the esophagus of Gdnf+/− mice is also reduced by 63% (Table 3D) (p = 0.047). Thus, GDNF provides trophic support for mature esophageal neurons, but the site of synthesis may influence whether there are effects on neuronal projections or the size of the cell body.
The location of GDNF production determines the pattern of myenteric plexus NADPH-d neuronal projections
The preceding analysis defined a number of changes in ENS structure as a result of increased GDNF availability, but the quantitative analysis missed one of the most striking changes in ENS structure that occurs in GFAP–Gdnf mice. This is the observation that GFAP–Gdnf animals have a remarkable increase in neuronal fibers surrounding myenteric ganglia and extending in thick fiber bundles between ganglia (Figs. 3, 5) compared with WT mice. This accumulation of NADPH-d neuronal filaments matches the distribution of GFAP-expressing enteric glia (Fig. 8) and suggests that the final distribution of NADPH-d-expressing myenteric neuronal processes is influenced by the site of GDNF production within the bowel wall. This pattern of innervation is particularly striking in the esophagus (Fig. 5B), especially compared with Myo–Gdnf (Fig. 5D) mice in which there are very few NADPH-d fibers around the ganglia, but a dramatic increase in the number of small neuronal fibers innervating the myogenin producing skeletal muscle (Table 3C). The importance of the location of GDNF production is further supported by the observation that, in Myo–Gdnf mice, the NADPH-d neuronal fibers in the esophageal submucosa are normal (WT, 22.6 ± 1.3; Myo–Gdnf, 21.6 ± 1.3; p = 0.598; n = 6 mice of each genotype). In contrast to each of these transgenic models, systemic administration of GDNF causes both more small neuronal fibers extending into esophageal skeletal muscle and thicker bundles of fibers extending between ganglia (Fig. 5C).
Figure 8.
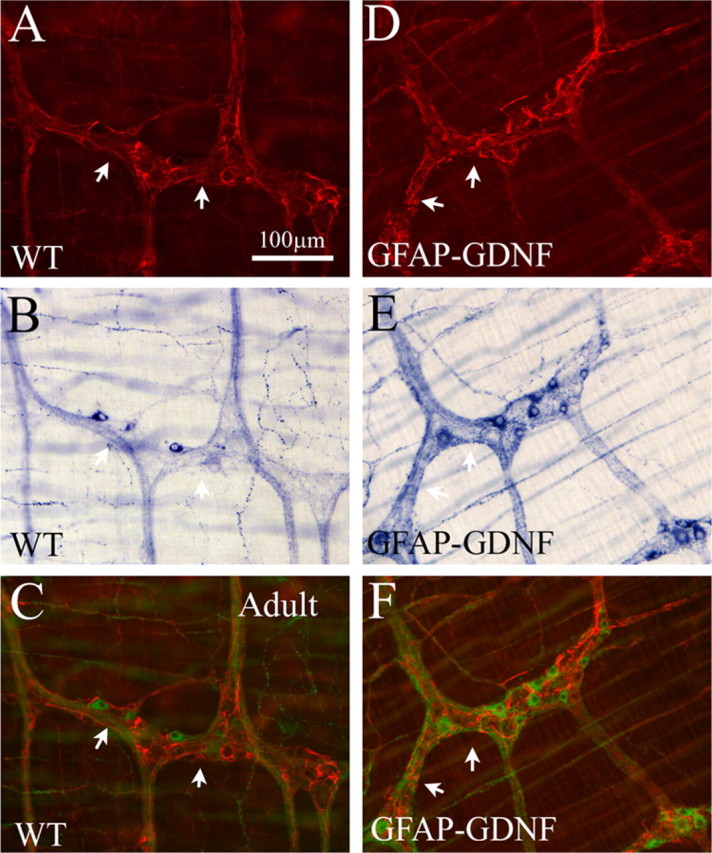
GFAP–Gdnf mice have increased neuronal fiber density near enteric glia. A, D, GFAP immunohistochemistry in WT and GFAP–Gdnf mice. B, E, The same region of the bowel was simultaneously stained using NADPH-d histochemistry. C, F, For these merged images, NADPH-d cells and fibers are pseudo-colored green to facilitate combining fluorescent (A, D) and bright-field (B, E) images and to provide increased contrast to the GFAP immunohistochemistry. Thick nerve fiber bundles accumulate near GFAP-expressing cells in the transgenic mice. Arrows highlight corresponding areas of GFAP and NADPH-d in images. Scale bar, 100 μm.
Similar increases in the density of NADPH-d neuronal fibers surrounding myenteric ganglia occur in the small bowel (Fig. 3A,E) and colon (data not shown) of GFAP–Gdnf mice. Interestingly, however, the density of small NADPH-d neuronal fibers between ganglia in GFAP–Gdnf mice is normal (Table 3A) (small bowel: WT, 38.6 ± 1.6 and GFAP–Gdnf, 41.4 ± 1.4, p = 0.162; colon: WT, 51.0 ± 2.0 and GFAP–Gdnf, 46.6 ± 1.2, p = 0.059; n = 6 mice of each genotype), suggesting once again that these fibers are attracted to GDNF produced in enteric glia. This redistribution of NADPH-d neuronal fibers to enteric ganglia does not occur in GDNF-injected mice even in the small bowel in which the increase in NADPH-d neuron number and cell size is similar to the increase that occurs in GFAP–Gdnf animals (Fig. 3B,F; Tables 1, 2). In addition, although redistribution of NADPH-d neuronal fibers toward enteric ganglia in GFAP–Gdnf mice is striking, we did not see similar changes in the myenteric plexus of GFAP–Gdnf mice after staining with acetylcholinesterase (Fig. 3D,H), tryptophan hydroxylase (Fig. 3C,G), or ChAT (data not shown). Thus, only a subset of neuronal fibers have their distribution influenced by the location of GDNF production within the bowel wall.
Increased GDNF availability alters intestinal contractility
Although excess GDNF clearly influences ENS structure, we wanted to determine whether increased GDNF also influenced ENS function in adult mice. We therefore measured intestinal contractility and neurotransmitter release in response to electric field stimulation for small bowel and colon segments of GFAP–Gdnf and WT littermates in an oxygenated organ bath (Fig. 9). For both circular and longitudinal muscle, the force of contraction was 25–70% greater for the GFAP–Gdnf mice than for WT animals at the higher intensities of electric field stimulation studied. We also examined levels of VIP and substance P release because these are major inhibitory and excitatory neurotransmitters in the bowel and coexpressed in NO- and ChAT-expressing neurons, respectively. These studies demonstrated a 25–75% increase in both VIP and substance P release from the small bowel and colon of GFAP–Gdnf compared with WT mice despite more dramatic effects of the transgene on NADPH-d than on ChAT neuron numbers. These functional changes in vitro, however, correlated well with the markedly reduced contractility and transmitter release that we observed previously in Gdnf+/−, Gfrα1+/−, and Ret+/− mice (Gianino et al., 2003) even when the animals lacked striking demonstrable changes in ENS anatomy. To determine whether these in vitro changes reflect alterations in intestinal motility in vivo, intestinal transit was evaluated by feeding WT, GFAP–Gdnf, and Gfrα1+/− mice FITC-dextran. For this comparison, Gfrα1+/− mice were selected because they have only subtle changes in ENS anatomy but had significantly reduced gut contractility in organ bath experiments (Gianino et al., 2003). Analyses of FITC concentrations along the gastrointestinal tract 1 h after feeding demonstrated delayed transit in the Gfrα1+/− mice (geometric center: WT, 10.76 ± 0.12 and GFRα1+/−, 9.33 ± 0.16; p < 0.001). In contrast, GFAP–Gdnf mice had significantly more FITC in the distal colon than WT controls (p < 0.001) but an equivalent distribution of FITC along the small bowel (Fig. 10). Note that there are also differences in the distribution of FITC between the two WT control groups used that presumably reflect strain differences between the mouse lines (C57BL/6 vs CF-1). Thus, both increases and decreases in GDNF/GFRα1/Ret signaling significantly influence ENS structure and function.
Figure 9.
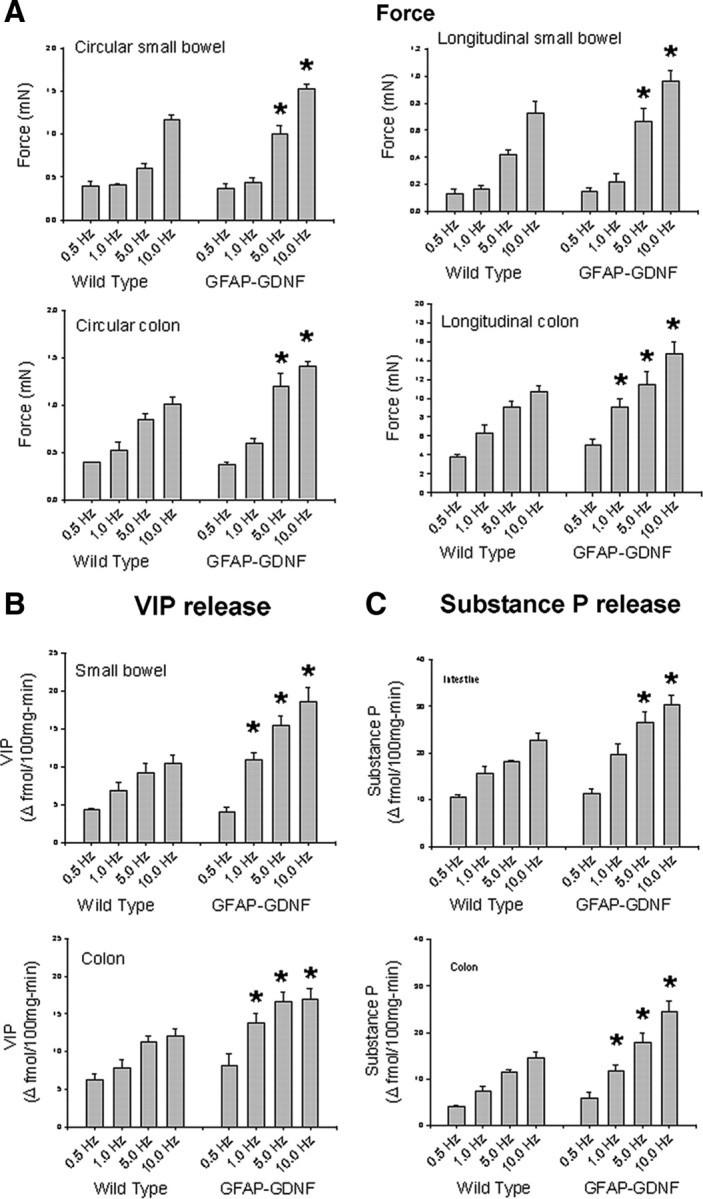
Intestinal contractility and neurotransmitter release are increased in GFAP–Gdnf transgenic mice. A, Circular and longitudinal muscle contractile force was determined after various intensities of electric field stimulation (0.5, 1, 5, and 10 Hz). In all regions evaluated, contractility was stronger in GFAP–Gdnf than in WT mice at 5 or 10 Hz stimulation. B, C, Electric field stimulation results in increased neurotransmitter release for VIP (B) and substance P (C) in both WT and GFAP–Gdnf mice. The increase in transmitter release is greater in GFAP–Gdnf than in WT animals. n = 4 mice of each genotype. Error bars represent ±SEM. *p < 0.05 for GFAP–Gdnf versus WT mice at each electric field strength.
Figure 10.

Intestinal transit is reduced in Gfrα1+/− and accelerated in GFAP–Gdnf mice. The distribution of FITC-dextran along the length of the bowel was analyzed 1 h after ingestion. A, Gfrα1+/− mice had less FITC in the distal bowel than WT mice. B, GFAP–Gdnf mice had more FITC in the distal bowel than WT mice. WT controls for each mutant mouse line were matched for strain background of the mutant, and transit is different for the two WT mouse strains analyzed. n = 4 mice of each mutant genotype and 4 matched controls.
Discussion
For normal ENS function, a complex network of interacting neurons must be established within the bowel. This process requires regulated ENS precursor proliferation, cell fate determination, precursor migration, and neuronal process extension. In addition, the development of neuron subtypes must be controlled independently to generate appropriate numbers of neurons within each class. Finally, subtype-specific mechanisms must direct axon pathfinding and maintain specific cell–cell interactions.
For many aspects of ENS development, Ret activation is critical because Ret supports ENS precursor survival, proliferation, migration, and axon extension. For this reason, Ret, GFRα1, or GDNF deficiency cause extensive aganglionosis. Although these roles are well established, we now demonstrate several novel aspects of ENS development affected by Ret signaling. (1) Increased GDNF increases enteric neuron number and cell size in the small bowel, colon, and esophagus. (2) Temporal regulation of GDNF production influences the ratio of specific ENS neuronal subtypes. (3) The location of GDNF synthesis influences neuronal projections for some but not all enteric neuron subtypes. (4) Intestinal contractility, neurotransmitter release (at least for VIP and substance P), and intestinal transit are enhanced by increased GDNF, but effects of increased or decreased GDNF (Gianino et al., 2003) on ENS function are not readily predicted based simply on anatomy. Thus, this analysis reveals several new aspects of Ret signaling and highlights how changes in GDNF expression that occur during normal development (Golden et al., 1999; Natarajan et al., 2002) may influence ENS morphogenesis and function. These findings may have clinical significance because many transcription factors, neurotransmitters, injury paradigms, proinflammatory cytokines, hormones, medications, and lifestyle decisions (i.e., calorie restriction and exercise) influence GDNF expression (Saavedra et al., 2008). In particular, altered GDNF abundance induced by bowel inflammation or infection (Steinkamp et al., 2003; von Boyen et al., 2006; Starke-Buzetti and Oaks, 2008) could account for some symptoms of postinfectious irritable bowel syndrome (Spiller and Garsed, 2009). In addition, a report of intestinal ganglioneuromatosis in a 38-year-old woman whose colon adenocarcinoma produced excess GDNF and neurturin (Qiao et al., 2009) suggests that adult human ENS structure and function is affected by Ret ligand abundance.
The timing of GDNF production critically regulates ENS morphogenesis
Controlling neuron number for specific subclasses is likely essential for normal intestinal function, but mechanisms regulating this are poorly understood. For some neuronal subpopulations, particular trophic or transcription factors are required. For example, Mash-1−/− mice are missing most enteric serotonergic and esophageal neurons (Blaugrund et al., 1996). Similarly, Trk C or neurotrophin-3 deficiency reduces myenteric neuron number and dramatically reduces calcitonin gene-related peptide-positive submucosal neurons (Chalazonitis et al., 2001). Data presented here support an additional hypothesis about how neuron numbers in specific ENS subpopulations are regulated. It is based on three observations. (1) GDNF abundance influences enteric neuron number by regulating ENS precursor proliferation (Gianino et al., 2003). (2) Caspase-3-, Bax-, and Bid-dependent cell death is rare in developing ENS of WT mice and only occurs in settings that reduce neuron number [e.g., Ret−/− mice (Taraviras et al., 1999)]. (3) Different enteric neuron precursor subpopulations exit the cell cycle at different times during development (Pham et al., 1991). We now present strong evidence that increased GDNF levels after P0 significantly increases submucosal and esophageal neurons without dramatically affecting total myenteric neuron density. We further demonstrate a correlation between these increased cell numbers and the time that their precursors normally stop dividing. We also directly demonstrate increased proliferation of PGP9.5+ cells in the esophagus and submucosal plexus of GDNF-injected mice that correlates well with changes observed in ENS structure. Although it remains possible that caspase-3-independent cell death as has been demonstrated recently in the avian ENS (Wallace et al., 2009) occurs in mice and could account for some of our observations, these data strongly support the hypothesis that the timing and intensity of GDNF signaling critically regulates enteric neuron number and can alter the ratio of myenteric to submucosal neurons by regulating cell proliferation.
For GDNF-injected mice, it is possible that the effect of GDNF on submucosal versus myenteric neuron numbers reflects the distribution of the injected protein rather than the timing of withdrawal of precursor cells from the cell cycle. Differential effects of GDNF on the abundance of myenteric neuron subclasses in GFAP–Gdnf mice, however, are more difficult to explain on the basis of protein distribution because ChAT+ and NADPH-d+ neurons are directly adjacent to each other in myenteric ganglia. Instead, increased density of NADPH-d-expressing, but not ChAT/substance P-expressing, neurons in the small bowel of these mice is consistent with the observation that NADPH-d precursors exit the cell cycle later than ChAT/substance P-expressing precursors (P5 vs E15, respectively) (Pham et al., 1991) and that GDNF levels increase in late gestation in GFAP–Gdnf mice. These data are also consistent with the idea that GDNF responsiveness of enteric neurons changes over time as neurons differentiate. Collectively, these data support the hypothesis that the timing, location, and abundance of GDNF critically control neuronal subtype ratios by regulating the proliferation of committed precursor pools.
There are several caveats that challenge this simple data interpretation. First, colon myenteric plexus NADPH-d+ neurons in GDNF-injected mice do not increase, but small bowel NADPH-d myenteric neurons increase 57% in GDNF-injected animals. We suspect that this is attributable to differences in the time that small bowel and colon NADPH-d myenteric neuron precursors exit the cell cycle. The presence of increased NADPH-d neurons in GFAP–Gdnf mouse colon myenteric plexus is also consistent with earlier cell cycle exit (i.e., before P0) for colonic NADPH-d ENS precursors than for small bowel NADPH-d+ cells, but our data neither support nor refute this hypothesis. Another potential concern is that total (Cuprolinic blue stained) myenteric neuron density in the small bowel or colon of mice injected with GDNF or in the small bowel for GFAP–Gdnf animals is the same as in WT mice. In GFAP–Gdnf mice, this may reflect the fact that the NADPH-d neurons are only a subset of the total cells (i.e., 38% in WT mice), and small increases (i.e., 30% change in NADPH-d cells) in cell number are difficult to detect because of the variability intrinsic to ENS structure (i.e., expected change in Cuprolinic blue-stained cells, 0.3 × 0.38 = 0.114 or 11.4% change in total neurons). In 30-d-old GDNF-injected mice in which NADPH-d neuron density increases by 57%, we also would only have anticipated small changes in total neuron density (i.e., ∼15–21%) if the increase in NADPH-d neurons was attributable solely to increased precursor proliferation. Thus, although we do demonstrate small statistically significant increases in proliferation rate for small bowel myenteric neuron precursors in GDNF-injected mice at P8, it is not possible to know whether this accounts for observed increases in NADPH-d-expressing neurons. We also note that the 28% increase in Cuprolinic blue-stained myenteric neurons in the colon but not in the small bowel of GFAP–Gdnf mice compared with WT animals is difficult to explain simply based on proliferation rates of precursors in WT mice because myenteric neurons in the small bowel and colon appear to exit the cell cycle at comparable times. We suspect that this is attributable to subtle differences in transgene expression between colon and small bowel but have been unable to verify this experimentally. An alternative explanation is that excess GDNF alters gene expression within some myenteric neuron precursors to increase NADPH-d cells by changing cell fate. We also cannot exclude the possibility that some effects of GDNF on the ENS are indirect and that excess GDNF triggers changes via feedback loops between neurons and glial cells or between different classes of enteric neuron that ultimately determine neuron subtype numbers. In either case, the data convincingly demonstrate that changes in GDNF abundance alter the ratio of myenteric neuron subtypes.
The location of GDNF production influences neuronal projections for some enteric neurons
GDNF has been proposed to guide the migration of neural crest derivatives along the bowel. This hypothesis is based on the ability of GDNF to attract ENS precursors in culture and production of Gdnf mRNA in the bowel in advance of migrating ENS precursors (Young et al., 2001; Natarajan et al., 2002; Iwashita et al., 2003; Sato and Heuckeroth, 2008). Our analysis also suggests that the location of GDNF production influences GDNF function. For example, Myo–Gdnf mice have a 479% increase in small neuronal fibers within the esophageal skeletal muscle but no change in neuronal fiber density in the adjacent submucosa. The altered distribution of NADPH-d+ fibers throughout the gastrointestinal tract of GFAP–Gdnf compared with GDNF-injected or WT animals also suggests that NADPH-d+ fiber location is determined at least in part by the site of GDNF production. Interestingly, alterations in neuronal fiber distribution were not observed for acetylcholinesterase-, tryptophan hydroxylase-, or ChAT-expressing myenteric neurons. Thus, GDNF expression patterns influence neuronal process guidance for only some enteric neurons subtypes. These effects, and the absence of significant serum GDNF levels in Myo–Gdnf or GFAP–Gdnf mice (Zhao et al., 2004), suggest that GDNF protein is retained close to its site of synthesis consistent with the observation that the location of GDNF administration in the CNS determines the location of axon sprouting (Rosenblad et al., 1999; Kirik et al., 2000). Two mechanisms could maintain GDNF protein close to its site of synthesis. First, GDNF may be bound by GFRα1. Second, GDNF binds 2-O-sulfate-rich heparin-related glycosaminoglycan (Rickard et al., 2003). These effects also may explain why high doses are required to observe systemic effects of injected GDNF (Keller-Peck et al., 2001). Doses used are 1000-fold higher than needed for tissue culture [i.e., 2 μg GDNF/g mouse vs 2 ng/ml culture media) (Heuckeroth et al., 1998)].
GDNF dose and location influence esophageal innervation
Quantitative evaluation of esophageal innervation in mice missing GDNF signaling receptors Ret or GFRα1 demonstrated dramatic but incomplete loss of esophageal neurons (Schuchardt et al., 1994; Durbec et al., 1996; Enomoto et al., 2004; Yan et al., 2004). Experiments with cultured E11.5 esophageal explants also confirmed that crest-derived cells in the esophagus responded to GDNF by migration and axon extension (Yan et al., 2004). Our data that Gdnf+/− mice have a reduced density of esophageal neurons is also consistent with the hypothesis that GDNF abundance determines esophageal neuron number. Given reduced responsiveness of esophageal crest-derived cells to GDNF as development proceeds (Yan et al., 2004), we were surprised to find dramatically increased esophageal neuron numbers in Myo–Gdnf, GFAP–Gdnf, and GDNF-injected mice. BrdU data, however, confirmed that esophageal crest-derived cells proliferate postnatally, providing a potential explanation for increased esophageal neuron number in GDNF-injected mice. Furthermore, we directly demonstrated that GDNF injection increases the rate of esophageal neuron precursor proliferation at P8. These data, in conjunction with the analysis of Ret−/−, Gfrα1−/−, Myo–Gdnf, GFAP–Gdnf, and GDNF-injected mice clearly demonstrate that esophageal neurons are Ret/GFRα1/GDNF responsive and thus confirm and extend previous studies.
GDNF overexpression alters intestinal function
Accompanying changes in ENS anatomy, there are increases in small bowel and colon contractility, substance P and VIP release, and intestinal transit in GFAP–Gdnf compared with WT mice. These changes are the opposite of the effects observed in Gdnf+/−, Gfrα1+/−, and Ret+/− mice (Shen et al., 2002; Gianino et al., 2003) and demonstrate that altered GDNF abundance could change intestinal motility. The relationship between the anatomical and the functional changes is complex and likely to involve multiple subsets of enteric neurons that will require additional study. However, the present data suggest a congruent relationship between the anatomical and functional findings. Thus, GFAP–Gdnf mice have increased NADPH-d neurons that coexpress VIP in nearly all regions. This is consistent with the increased release of VIP in response to electrical stimulation. These mice also have increased transit and increased release of substance P in response to electrical stimulation. Because substance P and VIP represent major contractile and relaxant neurotransmitters released from excitatory and inhibitory motor neurons, respectively, both of these classes of motor neurons would be activated as components of the propulsive motility responsible for enhanced transit. Thus, although we have identified significant changes in intestinal motility as a result of altered GDNF availability, more detailed analyses will be needed to determine whether these changes reflect developmental differences in ENS anatomy, short-term effects on function, or both. In particular, the finding that substance P release and intestinal contractility are both elevated in GFAP–Gdnf mice might not have been predicted based on anatomic studies because these animals have normal numbers of ChAT neurons. These data are consistent with our previous observation that transmitter release and intestinal contractility are dramatically reduced in several mouse lines than with decreased Ret signaling in the absence of dramatic effects on anatomy (Gianino et al., 2003) and suggest that GDNF has important effects on enteric neuron physiology that extend beyond the anatomic effects documented. These results are also consistent with several previous studies demonstrating that GDNF influences neurotransmitter synthesis and release in other neuronal populations (Feng et al., 1999; Charbel Issa et al., 2001; Wang et al., 2001; Skoff et al., 2003; Wang et al., 2003; Gomes et al., 2006). Together, these data demonstrate that a wide variety of changes in ENS structure and function are created by changing the location, timing, and intensity of GDNF signaling. These data also suggest that GDNF promoter polymorphisms, changes in transcriptional regulators, or extrinsic factors (medicines, inflammation, etc.) that alter GDNF expression may dramatically affect ENS structure and function and contribute to human intestinal motility disorders.
Footnotes
This work was supported by National Institutes of Health Grants NS042794 (A.P.), RO1 DK34153 (J.R.G.), and RO1 DK57038 and RO1 DK6459201 (R.O.H.), Children's Discovery Institute Grant CH-II-2008-123 (R.O.H.), Burroughs Wellcome Fund Grant 1008525 (R.O.H.), the Digestive Disease Research Center Core/National Institutes of Health Grant P30-DK52574 (R.O.H.), and March of Dimes Grant FY02-182 (R.O.H). We appreciate the guidance of Dr. Anthony Bauer for the design of the intestinal transit studies and Jonathan Gitlin for suggestions about this manuscript. We also appreciate the support and encouragement of Dr. Jeffrey Milbrandt in the early stages of this work.
References
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors: implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Blaugrund E, Pham TD, Tennyson VM, Lo L, Sommer L, Anderson DJ, Gershon MD. Distinct subpopulations of enteric neuronal progenitors defined by time of development, sympathoadrenal lineage markers, and Mash-1-dependence. Development. 1996;122:309–320. doi: 10.1242/dev.122.1.309. [DOI] [PubMed] [Google Scholar]
- Cacalano G, Fariñas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalazonitis A, Rothman TP, Chen J, Gershon MD. Age-dependent differences in the effects of GDNF and NT-3 on the development of neurons and glia from neural crest-derived precursors immunoselected from the fetal rat gut: expression of GFRa-1 in vitro and in vivo. Dev Biol. 1998;204:385–406. doi: 10.1006/dbio.1998.9090. [DOI] [PubMed] [Google Scholar]
- Chalazonitis A, Pham TD, Rothman TP, DiStefano PS, Bothwell M, Blair-Flynn J, Tessarollo L, Gershon MD. Neurotrophin-3 is required for the survival-differentiation of subsets of developing enteric neurons. J Neurosci. 2001;21:5620–5636. doi: 10.1523/JNEUROSCI.21-15-05620.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbel Issa P, Lever IJ, Michael GJ, Bradbury EJ, Malcangio M. Intrathecally delivered glial cell line-derived neurotrophic factor produces electrically evoked release of somatostatin in the dorsal horn of the spinal cord. J Neurochem. 2001;78:221–229. doi: 10.1046/j.1471-4159.2001.00430.x. [DOI] [PubMed] [Google Scholar]
- Cheng TC, Hanley TA, Mudd J, Merlie JP, Olson EN. Mapping of myogenin transcription during embyrogenesis using transgenes linked to the myogenin control region. J Cell Biol. 1992;119:1649–1656. doi: 10.1083/jcb.119.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creedon DJ, Tansey MG, Baloh RH, Osborne PA, Lampe PA, Fahrner TJ, Heuckeroth RO, Milbrandt J, Johnson EM., Jr Neurturin shares receptors and signal transduction pathways with glial cell line-derived neurotrophic factor in sympathetic neurons. Proc Natl Acad Sci U S A. 1997;94:7018–7023. doi: 10.1073/pnas.94.13.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM, Jr, Milbrandt J. GFRα1 deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Hughes I, Golden J, Baloh RH, Yonemura S, Heuckeroth RO, Johnson EM, Jr, Milbrandt J. GFRalpha1 expression in cells lacking RET is dispensable for organogenesis and nerve regeneration. Neuron. 2004;44:623–636. doi: 10.1016/j.neuron.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Feng L, Wang CY, Jiang H, Oho C, Mizuno K, Dugich-Djordjevic M, Lu B. Differential effects of GDNF and BDNF on cultured ventral mesencephalic neurons. Brain Res Mol Brain Res. 1999;66:62–70. doi: 10.1016/s0169-328x(99)00015-7. [DOI] [PubMed] [Google Scholar]
- Furness JB. Types of neurons in the enteric nervous system. J Auton Nerv Syst. 2000;81:87–96. doi: 10.1016/s0165-1838(00)00127-2. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Salomon R, Pelet A, Angrist M, Amiel J, Fornage M, Attié-Bitach T, Olson JM, Hofstra R, Buys C, Steffann J, Munnich A, Lyonnet S, Chakravarti A. Segregation at three loci explains familial and population risk in Hirschsprung disease. Nat Genet. 2002;31:89–93. doi: 10.1038/ng868. [DOI] [PubMed] [Google Scholar]
- Gariepy CE. Intestinal motility disorders and development of the enteric nervous system. Pediatr Res. 2001;49:605–613. doi: 10.1203/00006450-200105000-00001. [DOI] [PubMed] [Google Scholar]
- Gariepy CE. Developmental disorders of the enteric nervous system: genetic and molecular bases. J Pediatr Gastroenterol Nutr. 2004;39:5–11. doi: 10.1097/00005176-200407000-00003. [DOI] [PubMed] [Google Scholar]
- Gershon MD. Genes and lineages in the formation of the enteric nervous system. Curr Opin Neurobiol. 1997;7:101–109. doi: 10.1016/s0959-4388(97)80127-4. [DOI] [PubMed] [Google Scholar]
- Gershon MD. Lessons from genetically engineered animal models. II. Disorders of enteric neuronal development: insights from transgenic mice. Am J Physiol. 1999;277:G262–G267. doi: 10.1152/ajpgi.1999.277.2.G262. [DOI] [PubMed] [Google Scholar]
- Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–2198. doi: 10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- Golden JP, DeMaro JA, Osborne PA, Milbrandt J, Johnson EM., Jr Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol. 1999;158:504–528. doi: 10.1006/exnr.1999.7127. [DOI] [PubMed] [Google Scholar]
- Gomes CA, Vaz SH, Ribeiro JA, Sebastião AM. Glial cell line-derived neurotrophic factor (GDNF) enhances dopamine release from striatal nerve endings in an adenosine A2A receptor-dependent manner. Brain Res. 2006;1113:129–136. doi: 10.1016/j.brainres.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat Rev Neurosci. 2007;8:466–479. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- Hearn CJ, Murphy M, Newgreen D. GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Dev Biol. 1998;197:93–105. doi: 10.1006/dbio.1998.8876. [DOI] [PubMed] [Google Scholar]
- Heuckeroth RO, Lampe PA, Johnson EM, Milbrandt J. Neurturin and GDNF promote proliferation and survival of enteric neuron and glial progenitors in vitro. Dev Biol. 1998;200:116–129. doi: 10.1006/dbio.1998.8955. [DOI] [PubMed] [Google Scholar]
- Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, Jackman A, Molliver DC, Bardgett ME, Snider WD, Johnson EM, Jr, Milbrandt J. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory, and parasympathetic neurons. Neuron. 1999;22:253–263. doi: 10.1016/s0896-6273(00)81087-9. [DOI] [PubMed] [Google Scholar]
- Iwashita T, Kruger GM, Pardal R, Kiel MJ, Morrison SJ. Hirschsprung disease is linked to defects in neural crest stem cell function. Science. 2003;301:972–976. doi: 10.1126/science.1085649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kablar B, Tajbakhsh S, Rudnicki MA. Transdifferentiation of esophageal smooth to skeletal muscle is myogenic bHLH factor-dependent. Development. 2000;127:1627–1639. doi: 10.1242/dev.127.8.1627. [DOI] [PubMed] [Google Scholar]
- Karaosmanoglu T, Aygun B, Wade PR, Gershon MD. Regional differences in the number of neurons in the myenteric plexus of the guinea pig small intestine and colon: an evaluation of markers used to count neurons. Anat Rec. 1996;244:470–480. doi: 10.1002/(SICI)1097-0185(199604)244:4<470::AID-AR5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD. Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci. 2001;21:6136–6146. doi: 10.1523/JNEUROSCI.21-16-06136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Björklund A. Preservation of a functional nigrostriatal dopamine pathway by GDNF in the intrastriatal 6-OHDA lesion model depends on the site of administration of the trophic factor. Eur J Neurosci. 2000;12:3871–3882. doi: 10.1046/j.1460-9568.2000.00274.x. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- Merlie JP, Mudd J, Cheng TC, Olson EN. Myogenin and acetylcholine receptor alpha gene promoters mediate transcriptional regulation in response to motor innervation. J Biol Chem. 1994;269:2461–2467. [PubMed] [Google Scholar]
- Miller MS, Galligan JJ, Burks TF. Accurate measurement of intestinal transit in the rat. J Pharmacol Methods. 1981;6:211–217. doi: 10.1016/0160-5402(81)90110-8. [DOI] [PubMed] [Google Scholar]
- Moore BA, Otterbein LE, Türler A, Choi AM, Bauer AJ. Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology. 2003;124:377–391. doi: 10.1053/gast.2003.50060. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Natarajan D, Marcos-Gutierrez C, Pachnis V, de Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–5160. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- Neuhuber WL, Wörl J, Berthoud HR, Conte B. NADPH-diaphorase-positive nerve fibers associated with motor endplates in the rat esophagus: new evidence for co-innervation of striated muscle by enteric neurons. Cell Tissue Res. 1994;276:23–30. doi: 10.1007/BF00354780. [DOI] [PubMed] [Google Scholar]
- Newgreen D, Young HM. Enteric nervous system: development and developmental disturbances. Part 1. Pediatr Dev Pathol. 2002a;5:224–247. doi: 10.1007/s10024-001-0142-y. [DOI] [PubMed] [Google Scholar]
- Newgreen D, Young HM. Enteric nervous system: development and developmental disturbances. Part 2. Pediatr Dev Pathol. 2002b;5:329–349. doi: 10.1007/s10024-002-0002-4. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- Passarge E. Dissecting Hirschsprung disease. Nat Genet. 2002;31:11–12. doi: 10.1038/ng878. [DOI] [PubMed] [Google Scholar]
- Pham TD, Gershon MD, Rothman TP. Time of origin of neurons in the murine enteric nervous system: sequence in relation to phenotype. J Comp Neurol. 1991;314:789–798. doi: 10.1002/cne.903140411. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Qiao S, Iwashita T, Ichihara M, Murakumo Y, Yamaguchi A, Isogai M, Sakata K, Takahashi M. Increased expression of glial cell line-derived neurotrophic factor and neurturin in a case of colon adenocarcinoma associated with diffuse ganglioneuromatosis. Clin Neuropathol. 2009;28:105–112. doi: 10.5414/npp28105. [DOI] [PubMed] [Google Scholar]
- Rickard SM, Mummery RS, Mulloy B, Rider CC. The binding of human glial cell line-derived neurotrophic factor to heparin and heparan sulfate: importance of 2-O-sulfate groups and effect on its interaction with its receptor, GFRalpha1. Glycobiology. 2003;13:419–426. doi: 10.1093/glycob/cwg046. [DOI] [PubMed] [Google Scholar]
- Rosenblad C, Kirik D, Devaux B, Moffat B, Phillips HS, Björklund A. Protection and regeneration of nigral dopaminergic neurons by neurturin or GDNF in a partial lesion model of Parkinson's disease after administration into the striatum or the lateral ventricle. Eur J Neurosci. 1999;11:1554–1566. doi: 10.1046/j.1460-9568.1999.00566.x. [DOI] [PubMed] [Google Scholar]
- Rothman TP, Tennyson VM, Gershon MD. Colonization of the bowel by the precursors of enteric glia: studies of normal and congenitally aganglionic mutant mice. J Comp Neurol. 1986;252:493–506. doi: 10.1002/cne.902520406. [DOI] [PubMed] [Google Scholar]
- Saavedra A, Baltazar G, Duarte EP. Driving GDNF expression: the green and the red traffic lights. Prog Neurobiol. 2008;86:186–215. doi: 10.1016/j.pneurobio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Sánchez MP, Silos-Santiago I, Frisén J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Sang Q, Young HM. Chemical coding of neurons in the myenteric plexus and external muscle of the small and large intestine of the mouse. Cell Tissue Res. 1996;284:39–53. doi: 10.1007/s004410050565. [DOI] [PubMed] [Google Scholar]
- Sato Y, Heuckeroth RO. Retinoic acid regulates murine enteric nervous system precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite growth in vitro. Dev Biol. 2008;320:185–198. doi: 10.1016/j.ydbio.2008.05.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchardt A, D'Agati V, Larsson-Blomberg L, Constantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- Shen L, Pichel JG, Mayeli T, Sariola H, Lu B, Westphal H. Gdnf haploinsufficiency causes Hirschsprung-like intestinal obstruction and early-onset lethality in mice. Am J Hum Genet. 2002;70:435–447. doi: 10.1086/338712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoff AM, Resta C, Swamydas M, Adler JE. Nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF) regulate substance P release in adult spinal sensory neurons. Neurochem Res. 2003;28:847–854. doi: 10.1023/a:1023211107073. [DOI] [PubMed] [Google Scholar]
- Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136:1979–1988. doi: 10.1053/j.gastro.2009.02.074. [DOI] [PubMed] [Google Scholar]
- Starke-Buzetti WA, Oaks JA. Increased glial-derived neurotrophic factor in the small intestine of rats infected with the tapeworm, Hymenolepis diminuta. Int J Exp Pathol. 2008;89:458–465. doi: 10.1111/j.1365-2613.2008.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkamp M, Geerling I, Seufferlein T, von Boyen G, Egger B, Grossmann J, Ludwig L, Adler G, Reinshagen M. Glial-derived neurotrophic factor regulates apoptosis in colonic epithelial cells. Gastroenterology. 2003;124:1748–1757. doi: 10.1016/s0016-5085(03)00404-9. [DOI] [PubMed] [Google Scholar]
- Taraviras S, Marcos-Gutierrez CV, Durbec P, Jani H, Grigoriou M, Sukumaran M, Wang LC, Hynes M, Raisman G, Pachnis V. Signalling by RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development. 1999;126:2785–2797. doi: 10.1242/dev.126.12.2785. [DOI] [PubMed] [Google Scholar]
- von Boyen GB, Steinkamp M, Geerling I, Reinshagen M, Schäfer KH, Adler G, Kirsch J. Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: a key to the regulation of epithelial apoptosis in Crohn's disease. Inflamm Bowel Dis. 2006;12:346–354. doi: 10.1097/01.MIB.0000219350.72483.44. [DOI] [PubMed] [Google Scholar]
- Wallace AS, Barlow AJ, Navaratne L, Delalande JM, Tauszig-Delamasure S, Corset V, Thapar N, Burns AJ. Inhibition of cell death results in hyperganglionosis: implications for enteric nervous system development. Neurogastroenterol Motil. 2009;21:768–e49. doi: 10.1111/j.1365-2982.2009.01309.x. [DOI] [PubMed] [Google Scholar]
- Wang CY, Yang F, He X, Chow A, Du J, Russell JT, Lu B. Ca2+ binding protein frequenin mediates GDNF-induced potentiation of Ca2+ channels and transmitter release. Neuron. 2001;32:99–112. doi: 10.1016/s0896-6273(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen G, Lu B, Wu CP. GDNF acutely potentiates Ca2+ channels and excitatory synaptic transmission in midbrain dopaminergic neurons. Neurosignals. 2003;12:78–88. doi: 10.1159/000071817. [DOI] [PubMed] [Google Scholar]
- Yan H, Bergner AJ, Enomoto H, Milbrandt J, Newgreen DF, Young HM. Neural cells in the esophagus respond to glial cell line-derived neurotrophic factor and neurturin, and are RET-dependent. Dev Biol. 2004;272:118–133. doi: 10.1016/j.ydbio.2004.04.025. [DOI] [PubMed] [Google Scholar]
- Yntema CL, Hammond WS. The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J Comp Neurol. 1954;101:515–541. doi: 10.1002/cne.901010212. [DOI] [PubMed] [Google Scholar]
- Young HM, Hearn CJ, Farlie PG, Canty AJ, Thomas PQ, Newgreen DF. GDNF is a chemoattractant for enteric neural cells. Dev Biol. 2001;229:503–516. doi: 10.1006/dbio.2000.0100. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Alam S, Oppenheim RW, Prevette DM, Evenson A, Parsadanian A. Overexpression of glial cell line-derived neurotrophic factor in the CNS rescues motoneurons from programmed cell death and promotes their long-term survival following axotomy. Exp Neurol. 2004;190:356–372. doi: 10.1016/j.expneurol.2004.06.015. [DOI] [PubMed] [Google Scholar]