

Abstract
It is postulated that basic residues in the regulatory region of myosin light chain kinase are important for conferring autoinhibition by binding to the catalytic core. To investigate this proposal, 10 basic amino acids within the regulatory region of rabbit smooth muscle myosin light chain kinase (Lys961–Lys979) were replaced either singularly or in combination with acidic or nonpolar residues by site-directed mutagenesis. All active mutant kinases were dependent on Ca2+/calmodulin for catalytic activity. None of the mutants was active in the absence of Ca2+/calmodulin, suggesting that the autoinhibitory region has not been defined completely. Charge reversal mutants at Arg974, Arg975, and Lys976 resulted in loss of high affinity binding of calmodulin and increased the concentration of calmodulin required for half-maximal activation (KCaM). The charge reversal mutant at Lys979 also increased KCaM but to a lesser extent. Charge reversal mutants at Lys965 and Arg967 resulted in an inactive myosin light chain kinase that could not be proteolytically activated. When these residues were mutated to Ala, the expressed kinase was dependent upon Ca2+/calmodulin for activity and exhibited a decrease in KCaM. Charge reversal mutants in Lys961 and Lys962 also had decreased KCaM values. These basic residues amino-terminal of the calmodulin binding domain may play an important role in the activation of the kinase.
Myosin light chain kinase phosphorylates the regulatory light chain of myosin in a Ca2+/calmodulin-dependent manner. Distinct skeletal and smooth muscle forms of the kinase are known to exist as products of different genes (Stull et al., 1985). Recently, Gallagher et al. (1991) demonstrated that within a given animal species, non-muscle and smooth muscle myosin light chain kinases exhibit indistinguishable molecular masses and may be the same protein. In smooth muscle, regulatory light chain phosphorylation is a crucial step for the initiation of smooth muscle contraction (Kamm and Stull, 1985). Although the biochemical significance of regulatory light chain phosphorylation is becoming increasingly understood, the underlying biochemical mechanism for the Ca2+/calmodulin regulation of smooth muscle myosin light chain kinase activity remains unresolved.
Several investigators have proposed that the smooth muscle myosin light chain kinase contains a regulatory region located carboxyl-terminal to the catalytic core, which includes both an inhibitory domain and a calmodulin binding domain (Kemp et al., 1987; Ikebe et al., 1989). Based on similarities in the spatial distribution of basic residues within the regulatory region of smooth muscle myosin light chain kinase and the smooth muscle myosin regulatory light chain, Kemp et al. (1987) proposed that a portion of the calmodulin binding domain acted as a pseudosubstrate inhibitor. Specific basic residues within the regulatory region of the smooth muscle myosin light chain kinase were predicted to mimic the basic substrate determinants in the myosin light chain and bind to the active site in the absence of Ca2+/calmodulin, thereby inhibiting enzyme activity. Similarly, protein kinase A, protein kinase C, and the Ca2+/calmodulin-dependent protein kinase II also are believed to be regulated by an analogous autoinhibitory domain, thereby maintaining low basal activity in the absence of an allosteric activator (Soderling, 1990).
In support of this hypothesis for smooth muscle myosin light chain kinase, synthetic peptides, modeled after the proposed pseudosubstrate region in smooth muscle myosin light chain kinase, were found to be competitive inhibitors with respect to the regulatory light chain substrate (Kemp et al., 1987). Additional support for an autoinhibitory region in smooth muscle myosin light chain kinase was obtained from limited proteolysis studies. Proteolysis of the gizzard smooth muscle myosin light chain kinase results in an inactive form of the enzyme (64 kDa), which upon further digestion becomes active and calmodulin-independent (61 kDa) (Foster et al., 1986; Ikebe et al., 1987; Pearson et al., 1988). It is the removal of carboxyl-terminal sequences of smooth muscle myosin light chain kinase, including the calmodulin binding domain that is believed to be critical for generating the calmodulin-independent species of smooth muscle myosin light chain kinase, as only a single cleavage site within the amino-terminal region has been identified in these limited proteolysis studies (Ikebe et al., 1989). Construction of a smooth muscle myosin light chain kinase with multiple mutations in the calmodulin binding domain resulted in an intramolecular phosphorylation of an inserted serine (Bagchi et al., 1992b). These results indicate that this portion of the kinase has access to the catalytic site.
At the present time, neither the critical residues participating in the pseudosubstrate inhibition nor the extent of the autoinhibitory domain has been unambiguously determined. It has been reported that the autoinhibitory region of gizzard myosin light chain kinase includes a portion of the calmodulin binding domain (Ito et al., 1991; Olson et al., 1990); however, residues amino-terminal to the calmodulin binding domain also have been implicated in autoinhibition (Ikebe et al., 1989). Shoemaker et al. (1990) have concluded that the putative inhibitory region of a truncated non-muscle myosin light chain kinase may not be acting simply as a pseudosubstrate inhibitor.
The main objective of the present study was to assess the role of specific basic residues, residing between Lys961 and Lys979, of the rabbit smooth muscle myosin light chain kinase (Gallagher et al., 1991) in activation of the enzyme. Site-directed mutagenesis was used to determine whether any of these residues participate in enzyme regulation.
Materials and Methods
Oligonucleotide-directed Mutagenesis
A 1,660-base pair 5′-SalI/EcoRI-3′ cDNA fragment, representing the carboxyl-terminal half of a smooth muscle myosin light chain kinase (Gallagher et al., 1991), was subcloned into M13. Oligonucleotide-directed mutagenesis (Sambrook et al., 1989) was performed using oligonucleotides designed to produce 14 mutant cDNAs having the desired substitutions indicated in Fig. 1. For each mutant cDNA the desired nucleotide substitutions were verified by DNA sequencing (Sanger et al., 1977).
Fig. 1. Schematic representation of site-directed mutants within the regulatory region of smooth muscle myosin light chain kinases.

A, charge reversal mutants; B, mutants at residues 965, 966, and 967. The amino acid sequence within a portion of the regulatory region of rabbit smooth muscle myosin light chain kinase (Leu940 to Ser992) is shown below the sequence for smooth muscle light chain. This region contains a pseudosubstrate domain (Ser964 to Val984) and calmodulin binding domain (Ala973 to Leu990). The specific amino acid substitutions are indicated below the sequence of the regulatory region. Basic residues in the pseudosubstrate region are aligned with important substrate determinants of the light chain.
Expression of Wild-type and Mutant Smooth Muscle Myosin Light Chain Kinases
Wild-type and mutant myosin light chain kinase cDNAs were subcloned into a pCMV5 expression vector as described previously (Gallagher, et al., 1991; Andersson et al., 1989). Subsequent expression was accomplished by transfection into COS cells using DEAE-dextran and chloroquine as described previously (Herring et al., 1990).
Protein Purification and Myosin Light Chain Kinase Assays
Chicken gizzard myosin regulatory light chain was purified by the method of Hathaway and Haeberle (1983). Calmodulin was purified from bovine testes (Blumenthal and Stull, 1982). Smooth muscle myosin light chain kinase was purified from bovine trachea (Stull et al., 1990b). COS cell lysates, prepared as described by Gallagher et al. (1991), were used to determine recombinant wild-type and mutant myosin light chain kinase activity. For all assays, COS cell lysates were diluted 50–500-fold into the reaction mixture. The Ca2+/calmodulin-dependent activity of wild-type and mutant smooth muscle myosin light chain kinases was measured by 32P incorporation into chicken gizzard myosin regulatory light chain (Blumenthal and Stull, 1980). Maximal activity was determined in reaction mixtures having 50 mm MOPS,1 10 mm magnesium acetate, 1 mm dithiothreitol, 1 μm calmodulin, 300 μm CaCl2, 1 mm [γ-32P]ATP (200–300 cpm/pmol), and 20 μm gizzard regulatory light chain at 30 °C. The Ca2+/calmodulin-independent activity of wild-type and mutant smooth muscle myosin light chain kinases was measured as radioactivity incorporated in the presence of 3 mm EGTA in the absence of exogenous Ca2+ and calmodulin. When assayed at a 1:10 dilution, mock transfected COS cell extracts exhibited < 5% of the total kinase activity, with no detectable activity observed in the presence of 3 mm EGTA.
To examine the calmodulin activation properties of the mutant smooth muscle myosin light chain kinase in COS cell lysates, Ca2+ activation assays were performed as described by Herring (1991), in the following reaction mixture: 50 mm MOPS, 10 mm magnesium acetate, 1 mm dithiothreitol, 1 mm [γ-32P]ATP (200–300 cpm/pmol), 15 μm purified gizzard regulatory light chain, and 1.05 μm calmodulin at pH 7.0. The free Ca2+ concentration was varied over a range from 100 nm to 100 μm with a Ca2+/EGTA buffer system (Potter and Gergely, 1975). The free Ca2+ concentration determines the concentration of Ca2+/calmodulin. To assess the quantitative changes in the calmodulin activation properties (KCaM) of mutant myosin light chain kinases, the ratio of activities at Ca2+ concentrations that result in less than maximal activity to the maximal activity measured at 100 μm Ca2+ were determined as described previously (Miller et al., 1983; Stull et al., 1990b). The ratio of activity at a specific Ca2+ concentration that is less than that required for maximal activity decreases quantitatively as the KCaM value for a mutant myosin light chain increases relative to the wild-type enzyme. Likewise, if the ratio of activity increases, the KCaM value decreases. It is assumed in this analysis that the Ca2+ binding properties of calmodulin are not changed and that the kinase is activated by a single Ca2+/calmodulin complex. Although the ratio of activities does not allow determination of the absolute value of KCaM, it may be used to calculate the fold change in KCaM based upon the quantitative relationship derived previously
| (Eq. 1) |
where KCaM is the concentration of Ca2+/calmodulin required for half-maximal activation, υ is the activity at a specific Ca2+ concentration that results in less than maximal activity, and Vmax is the activity measured at 100 μm Ca2+. The primed (′) portion of the equation refers to mutated kinase compared with wild-type kinase (non-primed). An average KCaM value for the native smooth muscle myosin light chain kinase is 1 nm (Stull et al., 1990a).
Limited Tryptic Proteolysis of Wild-type and Mutant Smooth Muscle Myosin Light Chain Kinases
COS cell lysates containing smooth muscle myosin light chain kinases expressed at approximately 10 ng/μl were diluted 1:5 into the following 25-μl reaction mixture: 6 mm MOPS, 1 mm CaCl2, 0.6 mm dithiothreitol, and 1 μm calmodulin at pH 7.5. The mixture was incubated at room temperature for 5 min. Digestion was initiated by the addition of varying amounts of trypsin (type I bovine pancreas, Sigma), and stopped after 10 min by the addition of soybean trypsin inhibitor (Boehringer Mannheim) in a 2-fold excess relative to the amount of trypsin. An aliquot of each digest was then diluted into protein gel sample buffer and the resultant proteolytic digestion pattern examined following sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting. Fragments of smooth muscle myosin light chain kinase were detected using a polyclonal anti-bovine tracheal smooth muscle myosin light chain kinase antibody as described previously (Gallagher et al., 1991). Myosin light chain kinase activity was measured at room temperature following 4-fold dilution of the proteolyzed extracts into reaction mixtures containing either Ca2+ or EGTA as described above.
Immunoblot and Calmodulin Overlay Analyses
The quantity of wild-type and mutant smooth muscle myosin light chain kinases present in COS cell lysates was determined by immunoblotting with purified bovine tracheal myosin light chain kinase as a standard (Gallagher et al., 1991). Wild-type and mutant smooth muscle myosin light chain kinases were probed with a monoclonal antibody raised against purified bovine tracheal smooth muscle myosin light chain kinase (Kamm et al., 1987). Biotinylated calmodulin overlay blots were performed as described previously (Herring, 1991) using a modification of Hubbard and Klee (Hubbard and Klee, 1987). Biotinylated calmodulin was prepared according to the method of Billingsley et al. (1985).
Results
Immunoblot and Calmodulin Overlay Analyses of Recombinant Smooth Muscle Myosin Light Chain Kinases
Several groups of basic amino acids which are potentially involved in the autoinhibition are located between residues Lys961 and Lys979 in the rabbit smooth muscle myosin light chain kinase (Gallagher et al., 1991). This region is analogous to the previously characterized pseudosubstrate or autoinhibitory region of the smooth muscle myosin light chain kinase (Kemp et al., 1987). Oligonucleotide-directed mutagenesis was utilized to alter these basic residues to either the acidic amino acids, Asp and Glu (basic charge reversal mutants, Fig. 1A) or Ala (charge-to-Ala mutants, Fig. 1B). In addition, Asp966 was altered to either Lys or Ala (Fig. 1B). To analyze the phenotype of these mutant kinases, the full-length cDNA was constructed in the vector pCMV5 (Gallagher et al., 1991; Andersson, et al., 1989) for transient expression in COS cells.
Immunoblot analysis of COS cell lysates with a monoclonal antibody raised to bovine tracheal smooth muscle myosin light chain kinase demonstrated that all mutant smooth muscle myosin light chain kinases were full-length and expressed at high levels (5–15 ng/μl). All of the mutant smooth muscle myosin light chain kinases comigrated with the 152-kDa wild-type recombinant smooth muscle myosin light chain kinase (Fig. 2A). Biotinylated calmodulin overlay analyses were performed to visualize the calmodulin binding properties of the mutant kinases (Fig. 2B). As expected, the wild-type smooth muscle myosin light chain kinase bound biotinylated calmodulin, as did all mutants described in Fig. 1, except mutant RRK974–976EED (Fig. 2B).
Fig. 2. Immunoblot and calmodulin overlay analyses of wild-type and mutant smooth muscle myosin light chain kinases.
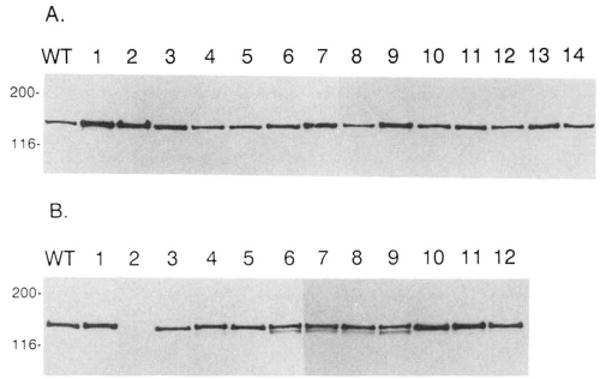
Panel A, Western immunoblot analysis of full-length smooth muscle myosin light chain kinase measured in COS cell extracts by a monoclonal antibody directed against the bovine tracheal myosin light chain kinase. Approximately 20–40 ng of smooth muscle myosin light chain kinase was present in each lane. Molecular mass markers (kDa) are listed on the far left of the blot. Panel B, calmodulin overlay of smooth muscle myosin light chain kinases. Biotinylated calmodulin and horseradish peroxidase conjugated to avidin was used to detect smooth muscle myosin light chain kinases in COS cell extracts as described under “Materials and Methods.” Approximately 40 ng of smooth muscle myosin light chain kinase was loaded per lane. Molecular mass markers (kDa) are listed on the far left of the blots. For both panels A and B, WT is the wild-type, recombinant rabbit smooth muscle myosin light chain kinase; lanes 1–12 are K979E, RRK974–76EED, KK969–970EE, K965E-R967D, KK961–962EE, KK961–962EE/KK969–970EE, K965E-R967D/KK969–970EE, KK961–962EE/K965E-R967D, KK961–962EE/K965E-R967D/KK969–970EE, R967A, K965A, K965A-R967A, respectively. In panel A, lanes 13 and 14 are D966K and D966A, respectively. D966K and D966A also bind calmodulin using the identical overlay technique (data not shown).
Kinetic Analyses of the Charge Reversal Mutants
The catalytic activity of the group of basic charge reversal mutants (Fig. 1A) was determined in kinase assays by measuring the rate of 32P incorporation into gizzard myosin regulatory light chain. All active mutant smooth muscle myosin light chain kinases were completely dependent on Ca2+/calmodulin for catalytic activity, with no significant Ca2+/calmodulin-independent activity detected (Table I). Of the five active mutant smooth muscle myosin light chain kinases, KK969–970EE and KK961–962EE had specific activities (24.5 and 20.3 pmol min−1 ng−1, respectively) similar to that of the wild-type kinase (28.8 pmol min−1 ng−1). Interestingly, the combined mutant KK961–962EE/KK969–970EE had a reduced specific activity. K979E and RRK974–976EED also exhibited reduced specific activities (10.9 and 3.0 pmol min−1 ng−1, respectively) (Table I). Four of the mutant kinases (K965E-R967D; K965E-R967D/KK969–970EE; KK961–962EE/K965E-R967D, and KK961–962EE/K965E-R967D/KK969–970EE) were catalytically inactive (Table I), even though they bound biotinylated calmodulin (Table I and Fig. 2B). All four of these inactive kinases share a common mutation in which residues Lys965 and Arg967 have been changed to Glu and Asp, respectively. The measured Km values for myosin light chain of the active mutant kinases were similar to the wild-type enzyme (Table I). The differences in Vmax values were reflected in the differences noted in the specific activities (Table I).
Table I. Kinetic properties of recombinant smooth muscle myosin light chain kinases.
Recombinant and mutant smooth muscle myosin light chain kinases were expressed in COS cells, and activities were measured in lysates for the various kinetic parameters as described under “Materials and Methods.” The values are means ± S.E. for three to six experiments.
| Kinase | Ca2+/CaMa | EGTAa | [Ca2+].0.5b | KCaMc | Kmd | Vmaxd |
|---|---|---|---|---|---|---|
| pmol/min/mg | μm | nm | ||||
| Wild-type | 28.8 ± 3.1 | 0.0 | 0.66 ± 0.04 | 1.0 | 5.3 ± 1.2 | 36.9 ± 1.7 |
| K979E | 10.9 ± 2.6 | 0.0 | 3.50 ± 0.05 | 15 ± 1.0 | ||
| RRK974-976EED | 3.0 ± 0.3 | 0.0 | 8.53 ± 1.3 | 41 ± 5 | 4.3 ± 0.7 | 4.3 ± 0.4 |
| KK969-970EE | 24.5 ± 1.5 | 0.0 | 0.80 ± 0.05 | 4 ± 3 | 3.4 ± 0.4 | 33.0 ± 1.7 |
| K965E-R967De | 0.0 | 0.0 | ||||
| KK961-962EE | 20.3 ± 2.1 | 0.0 | 0.29 ± 0.01 | 0.20 ± 0.12 | 3.1 ± 0.6 | 24.8 ± 2.2 |
| KK961-962EE/KK969-970EE | 5.1 ± 0.1 | 0.0 | 0.51 ± 0.06 | 0.55 ± 0.15 | ||
The specific activities measured in the presence of Ca2+ and calmodulin or EGTA are expressed as pmol of 32P incorporated/min/ng of kinase.
The concentration of Ca2+ (μm) required for half-maximal activation of smooth muscle myosin light chain kinase.
KCaM values (nm) were calculated from the data presented in Fig. 3 as described under “Materials and Methods.”
Kinetic values (Km and Vmax) were determined from double-reciprocal plots (Lineweaver-Burk) of data obtained from experiments with varying concentrations of myosin light chain.
Mutants K965E-R967D/KK969-970EE, KK961-962EE/K965E-R967D, and KK961-962EE/K965E-R967D/KK969-970EE were also catalytically inactive.
The Ca2+/calmodulin activation properties of the wild-type and mutant smooth muscle myosin light chain kinases were determined by performing assays at a high calmodulin concentration with a Ca2+/EGTA buffer used to vary the free Ca2+ concentration (Miller et al., 1983). The added calmodulin is in excess of the endogenous calmodulin contributed by the COS cell lysate and therefore provides controlled conditions to establish the relative concentration of Ca2+/calmodulin required for kinase activation. K979E and RRK974–976EED required greater concentrations of Ca2+ (3.5 and 8.53 μm, respectively) for half-maximal activation than the wild-type kinase (0.66 μm) (Table I and Fig. 3). These values represent 15- and 41-fold increases, respectively, in KCaM for the two mutant kinases compared with the wild-type kinase. The much greater KCaM value obtained for the RRK974–976EED mutant is consistent with the observation that this enzyme did not demonstrate high affinity binding on a calmodulin blot assay (Fig. 2B). In contrast to these results, mutant KK961–962EE required significantly less Ca2+ (0.29 μm) for half-maximal activation relative to the wild-type kinase with a decrease in KCaM from 1 to 0.2 nm. Both the KK969–970EE and KK961–962EE/KK969–970EE kinases, required similar concentrations of Ca2+ (0.80 and 0.51 μm, respectively) for half-maximal activation as the wild-type kinase.
Fig. 3. Ca2+/calmodulin activation of wild-type and charge reversal mutant smooth muscle myosin light chain kinases.
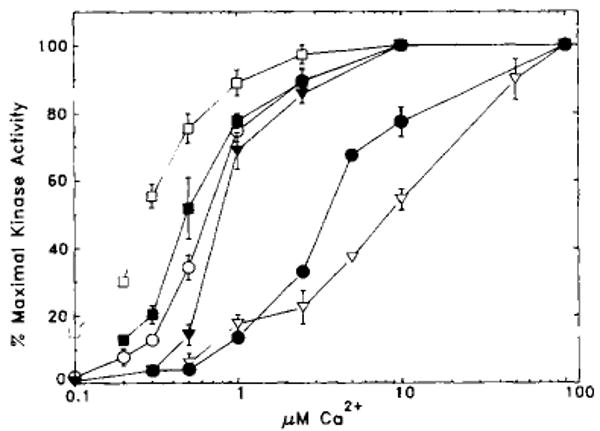
Kinase assays were performed as described under “Materials and Methods.” The percentage of maximal kinase activity is plotted versus free Ca2+ concentrations. The data are presented as mean ± S.E. from three or four independent assays, each performed in duplicate. If no error bar is visible, the S.E. is less than the size of the symbol. A summary of the data is presented in Table I. ○, wild type; ●, K979E; ▽, RRK974–976EED; ▼, KK969–970EE; □, KK961–962EE; ■, KK961–962EE/KK969–970EE.
Kinetic Analyses of Charge to Ala Mutants
Substitution of basic residues Lys965 and Arg967 with oppositely charged acidic residues by themselves or in combination with other mutants was consistently associated with loss of catalytic activity (Table I). Therefore, these basic residues were replaced either singularly or in combination with alanine residues. The resulting charge-to-alanine mutants (R967A, K965A, and K965A-R967A, Fig. 1B) were catalytically active and Ca2+/calmodulin-dependent. The specific activities were slightly lower than the wild-type kinase for the single mutants and decreased from 28.8 to 8.6 pmol of 32P incorporated per min per ng of kinase for the double mutant K965A-R967A (Table II). Similar to the results obtained with the KK961–962EE kinase, all three charge-to-Ala mutants required less Ca2+ for half-maximal activation, compared with the wild-type kinase (Table II and Fig. 4). For the single mutants the KCaM values decreased to 0.25 and 0.11 nm, whereas for the double mutant the KCaM value decreased to 0.04 nm compared with the 1 nm value for the wild-type kinase. These results indicate that there is a greater sensitivity to activation by Ca2+/calmodulin with these mutations with a maximal 25-fold decrease in KCaM. The Km value for myosin light chain was not significantly different from the wild-type kinase (2.6 ± 0.03 versus 5.3 ± 1.2 μm, respectively) although the Vmax value was decreased (10.7 ± 1.3 versus 36.9 ± 1.7 pmol of 32P incorporated per min per ng of kinase, respectively). There is an acidic residue (Asp966) between the two basic residues Lys965 and Arg967, and the possibility was considered that it may also play a role in the activation of myosin light chain kinase by Ca2+/calmodulin. This acidic residue was mutated to Ala or Lys, and the kinetic properties of the expressed kinase were examined (Table II). Both mutant kinases were dependent upon Ca2+/calmodulin for activity. Furthermore, there was no significant change in the concentration of Ca2+ required for half-maximal activation and no significant changes in KCaM. There was a slight increase in the specific activities of D966K and D966A compared with wild-type kinase. These results show that Asp966 probably plays no significant role in the activation of myosin light chain kinase in contrast to Lys965 and Arg967.
Table II. Kinetic properties of recombinant smooth muscle myosin light chain kinases.
Recombinant and mutant smooth muscle myosin light chain kinases were expressed in COS cells, and activities were measured in lysates for the various kinetic parameters as described under “Materials and Methods.” The values are means ± S.E. for three to six experiments.
| Kinase | Ca2+/CaMa | EGTAa | [Ca2+]0.5b | KCaMc |
|---|---|---|---|---|
| pmol/min/ng | μm | nm | ||
| Wild-type | 28.8 ± 3.1 | 0.0 | 0.66 ± 0.04 | 1.0 |
| R967A | 18.9 ± 1.9 | 0.0 | 0.36 ± 0.02 | 0.25 ± 0.02 |
| K965A | 15.8 ± 1.3 | 0.0 | 0.24 ± 0.02 | 0.11 ± 0.03 |
| K965A-R967A | 8.6 ± 1.2 | 0.0 | 0.12 ± 0.01 | 0.04 ± 0.01 |
| D966K | 51.6 ± 7.6 | 0.0 | 0.56 ± 0.03 | 0.71 ± 0.16 |
| D966A | 42.7 ± 2.9 | 0.0 | 0.78 ± 0.05 | 1.5 ± 0.27 |
The specific activities measured in the presence of Ca2+/calmodulin or EGTA are expressed as pmol of 32P incorporated/min/ng of kinase.
The concentration of Ca2+ (μm) required for half-maximal activation of smooth muscle myosin light chain kinase.
KCaM values (nm) were calculated from the data presented in Fig. 4 as described under “Materials and Methods.”
Fig. 4. Ca2+/calmodulin activation of wild-type and mutant smooth muscle myosin light chain kinases.
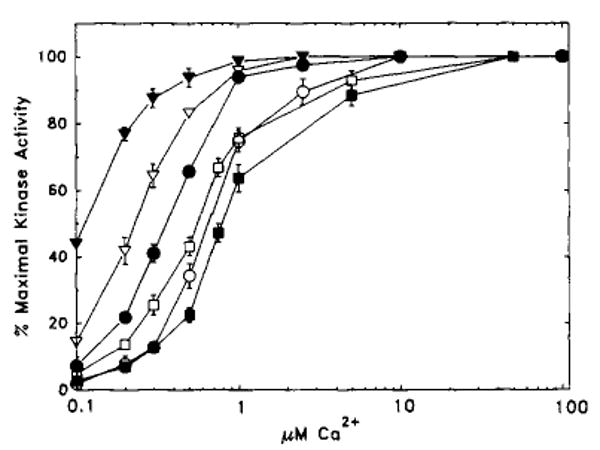
Kinase assays were performed as described under “Materials and Methods.” The relative percentage of maximal kinase activity is plotted versus free Ca2+ concentrations. The data are presented as mean ± S.E. from three or four independent assays, each performed in duplicate. If no error bar is visible, the S.E. is less than the size of the symbol. A summary of the data is presented in Table II. ○, wild type; ●, R967A; ▽, K965A; ▼, K965A-R967A; □, D966K; ■, D966A.
Detection of Structural Alterations in Mutant Smooth Muscle Myosin Light Chain Kinases by Limited Tryptic Proteolysis
Alterations to the native structure of proteins may lead to significant increases in the susceptibility to digestion by proteases (Gallagher et al., 1988). The possibility that the charge reversal of basic residues Lys965 and Arg967 may have altered either the native structure or the activation pathway of the mutant kinase was examined using limited proteolysis. Previous studies have shown that limited trypsin proteolysis of the avian enzyme generates a Ca2+/calmodulin-independent form of the enzyme.
Limited trypsin proteolysis was used to determine if the inactive charge reversal mutant K965A-R967A could be activated and to examine by Western blotting procedures the proteolytic patterns that were produced. For both wild-type and mutant K965A-R967A, Ca2+/calmodulin-independent activity was produced upon incubation at various trypsin concentrations (Fig. 5A). Furthermore, the changes in kinase activities were more prominent at the lower trypsin concentrations for K965A-R967A. Mutant K965E-R967D was not activated by this procedure.
Fig. 5. Limited proteolysis of wild-type and mutant smooth muscle myosin light chain kinases.
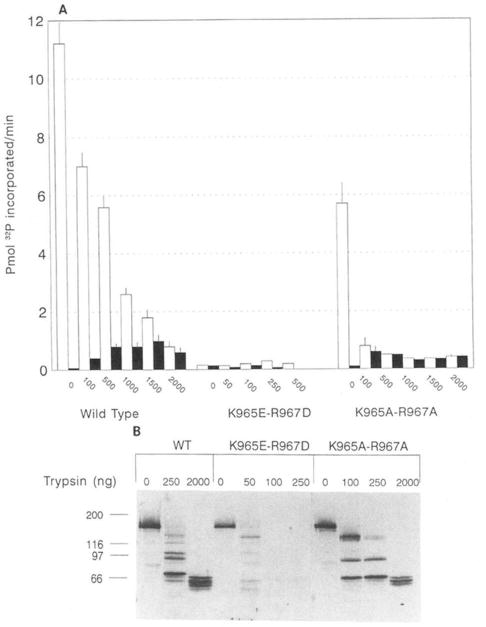
COS cells extracts having approximately the same concentration of expressed wild-type or mutant (K965E-R967D and K965A-R967A) enzymes were treated as described under “Materials and Methods” with the indicated concentrations of trypsin. Samples were aliquoted for kinase activity measurements (A) or Western blotting analysis (B). Activity was measured in the presence of EGTA (filled bars) or in the presence of Ca2+/calmodulin (open bars).
Western blotting showed differences in the proteolytic digestion pattern produced for both mutants compared with the wild-type enzyme (Fig. 5B). Such alteration would be predicted because of the removal of sites for proteolysis by trypsin. However, at each concentration of trypsin used in this experiment there is significant reduction in the total amount of protein remaining for mutant K965E-R967D compared with both mutant K965A-R967A and the wild-type enzyme. For example, measurements of the densities of the blotted proteins at 50 ng of trypsin relative to no digestion (0 ng of trypsin) show a recovery of 31% for K965E-R967D. In contrast, trypsin digestion of both wild-type and K965A-R967A generated stable, low molecular peptides which represented a substantial fraction of the original protein. For example, at 250 ng of trypsin 89 and 67% of the wild-type kinase and K965A-R967A, respectively, remained. These values were 84 and 47% at 2,000 ng of trypsin. Mutant K965E-R967D was completely digested in trypsin concentrations higher than 50 ng. Thus, the inability to detect a Ca2+/calmodulin-independent species when Lys965 and Arg967 are mutated to acidic residues appears to be caused by an alteration in the mutant protein structure. Mutation of Lys965 and Arg967 to Ala appears not as disruptive as the acidic substitutions as evidenced by the expression of a Ca2+/calmodulin-dependent active mutant that can be converted to a Ca2+/calmodulin-independent form by limited proteolysis.
Discussion
Basic residues within the myosin regulatory light chain are important determinants for binding to smooth muscle myosin light chain kinase (Kemp et al., 1983; Kemp and Pearson, 1985). To assess the importance of positively charged basic residues within the pseudosubstrate region of smooth muscle myosin light chain kinase, we have altered groups of residues within the pseudosubstrate inhibitory region and calmodulin binding domain to negatively charged acidic amino acids. We hypothesized that these alterations would either weaken the binding of the inhibitory region to the catalytic core, thereby giving rise to kinases that were more easily activated by Ca2+/calmodulin (decrease in KCaM), or lead to kinases that were partially or completely Ca2+/calmodulin-independent. None of the catalytically active mutant kinases described in this study displayed any detectable Ca2+/calmodulin-independent kinase activity. Thus, these charge reversal mutants have not completely abolished autoinhibition, suggesting that the groups of basic residues between Lys961 and Lys979 by themselves are not absolutely required to maintain the smooth muscle myosin light chain kinases in an inactive state in the absence of Ca2+/calmodulin. These results indicate that a pseudosubstrate, autoinhibitory mechanism is more complex than originally proposed (Kemp et al., 1987).
Substitution of Lys965, Arg967 with Ala965, Ala967 (R967A; K965A; K965A-R967A) and Lys961 and Lys962 with Asp965 and Glu967 (KK961–962EE) resulted in smooth muscle myosin light chain kinase mutants that had significant decreases in KCaM compared with the wild-type enzyme. If the amino acid replacements at residues 965 and 967 were charge reversed, the mutant kinase was catalytically inactive (K965E-R967D). It is unlikely that they are involved directly in binding to calmodulin since the defined calmodulin binding domain is 6–23 residues carboxyl-terminal (Bagchi et al., 1992a, 1992b). Our observations are consistent with these residues being structural determinants involved in kinase activation. In the native enzyme these basic residues are probably binding to other acidic residues in the kinase. With the mutations, these ionic interactions would be disrupted, resulting in an increase in the rate of isomerization of the kinase from the inactive to the active form after calmodulin binding (Bowman et al., 1992).
Charge reversal substitution of Arg974, Arg975, Lys976 (RRK974–976EED), and Lys979 (K979E) generated mutant smooth muscle myosin light chain kinases which increased KCaM. These results indicate that the affinity of both K979E and RRK974–976EED kinases for Ca2+/calmodulin has been reduced by these specific substitutions. Furthermore, the affinity of RRK974–976EED is sufficiently reduced (41-fold) so that it did not demonstrate high affinity binding with the calmodulin overlay procedure. Similarly, Bagchi et al. (1992a) have concluded that residues analogous to Arg974, Arg975, Lys976 in the chicken smooth muscle myosin light chain kinase are important for calmodulin binding. Mutating these residues to Ala increase the Ka for calmodulin 10-fold. In contrast to RRK974–976EED, the mutant chicken enzyme (R797A-R798A-K799A) was demonstrated to bind calmodulin by a similar blot overlay technique using 125I-calmodulin. Thus, alteration of these residues to acidic charges results in a mutant enzyme with a greater decrease in calmodulin affinity. These data are consistent with other studies describing the relative importance of analogous residues in both chicken non-muscle myosin light chain kinase (Shoemaker et al., 1990) and rabbit skeletal muscle myosin light chain kinase (Herring, 1991) for conferring high affinity calmodulin binding. In fact, Arg974, Arg975, Lys976, Lys979 have been postulated to interact with acidic residues in calmodulin (Shoemaker et al., 1990; Van Berkum and Means, 1991). In addition, peptide studies by Lukas et al. (1986) suggest that these amino-terminal basic residues in the calmodulin binding domain are important for calmodulin binding.
Using multidimensional NMR, Ikura et al. (1992) recently described the solution structure of the interaction of calmodulin with a peptide comprising the calmodulin binding domain of skeletal muscle myosin light chain kinases. The results indicated that calmodulin binding to this peptide primarily involves hydrophobic interactions. Residues in the target peptide analogous to Arg974 and Arg975 did not appear to interact with calmodulin in the solution structure since they were disordered. However, basic residues analogous to Lys976 and Lys979 were predicted to form electrostatic interactions with calmodulin, but the relative importance of these residues for a high affinity binding was not resolved in the solution structure. Other studies (Blumenthal and Stull, 1982; Bagchi et al., 1992a) have implicated both hydrophobic and electrostatic interactions in the interaction of calmodulin with myosin light chain kinases. It is possible that the structural interactions of calmodulin with the calmodulin binding domain in a peptide versus myosin light chain kinase are not identical. Blumenthal et al. (1985) noted that the peptide comprising the calmodulin binding domain of skeletal muscle myosin light chain kinase was not eluted from a calmodulin-Sepharose column in the presence of EGTA, whereas the kinase was. Urea plus EGTA was required for elution. Even if Lys976 does not bind directly to calmodulin, it is located next to the Trp residue which has been shown both in the solution structure and by site-directed mutagenesis studies to be a primary determinant in calmodulin binding (Ikura et al., 1992; Bagchi et al., 1992a). It is also possible that substitution of Lys979 to an acidic residue partially disrupts the central helix of the calmodulin binding domain thereby indirectly decreasing the affinity for calmodulin.
Ito et al. (1991) postulated that residues between Tyr794 and Trp800 in the gizzard smooth muscle myosin light chain kinase (equivalent to Tyr971 to Trp977 in the rabbit smooth muscle myosin light chain kinase) comprise at least part of the autoinhibitory domain. Results from our studies and those of Bagchi et al. (1992b) suggest that these residues are important for high affinity calmodulin binding. The possibility exists that these basic residues are involved both in autoinhibition of the kinase and calmodulin binding. However, kinases described by Ito et al. (1991) were truncated and could have been structurally altered relative to the wild-type, full-length molecule. Additional investigations will be necessary to examine these possibilities.
Shoemaker et al. (1990) reported that multiple charge reversal substitutions amino-terminal of the calmodulin binding domain resulted in Ca2+/calmodulin-independent activity. However, no specific activity was reported for the mutant (rMLCK17) which was a bacterially expressed, truncated form of the chicken myosin light chain kinase. It is possible that this particular mutant had low enzymatic activity. An analogous mutant in the rabbit smooth muscle myosin light chain kinase (KK961–962EE/K965E-R967D/KK969–970EE) resulted in no detectable kinase activity. The KK961–962EE/K965E-R967D/KK969–970EE mutant kinase, in contrast to rMLCK17, was assayed in COS cell lysates and not as a purified protein; hence, low levels of activity would not be detectable. The different experimental approaches may account for the observed differences in the effect of these alterations.
Collectively, the present data suggest that basic amino acids Arg974, Arg975, Lys976, Lys979 are important residues for high affinity calmodulin binding to smooth muscle myosin light chain kinase. Lys961, Lys962, Lys965, and Arg967 are involved in the activation of smooth muscle myosin light chain kinase and may participate in autoinhibition. The regulation of smooth muscle myosin light chain kinase activity is probably more complex than the simple pseudosubstrate inhibition mechanism originally proposed (Kemp et al., 1987) and may be more adequately described by an intrasteric autoinhibitory mechanism (Kemp and Pearson, 1991).
Acknowledgments
We gratefully appreciate Suzy Griffin for technical expertise and Phyllis Foley for assistance in preparing the manuscript. We also appreciate Li-Chu Hsu for assistance in the purification of many proteins used in this study.
Footnotes
This work was supported by a grant-in-aid from the American Heart Association, Texas Affiliate (to B. P. H.) and by National Institutes of Health Grant HL26043 (to J. T. S.).
The abbreviations used are: MOPS, 3-(N-morpholino)-propanesulfonic acid; KCaM, the concentration of calmodulin required for half-maximal activation of myosin light chain kinase.
References
- Andersson S, Davis DL, Dahlbäch H, Jörnvall H, Russell DW. J Biol Chem. 1989;264:8222–8229. [PubMed] [Google Scholar]
- Bagchi IC, Huang Q, Means AR. J Biol Chem. 1992a;267:3024–3029. [PubMed] [Google Scholar]
- Bagchi IC, Kemp BE, Means AR. Mol Endocrinol. 1992b;6:621–626. doi: 10.1210/mend.6.4.1584224. [DOI] [PubMed] [Google Scholar]
- Billingsley ML, Pennypacker KR, Hooever CG, Brigati DJ, Kincaid RL. Proc Natl Acad Sci U S A. 1985;82:7585–7589. doi: 10.1073/pnas.82.22.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal DK, Stull JT. Biochemistry. 1980;19:5608–5614. doi: 10.1021/bi00565a023. [DOI] [PubMed] [Google Scholar]
- Blumenthal DK, Stull JT. Biochemistry. 1982;21:2386–2391. doi: 10.1021/bi00539a017. [DOI] [PubMed] [Google Scholar]
- Blumenthal DK, Takio K, Edelman AM, Charbonneau H, Titani K, Walsh KA, Krebs EG. Proc Natl Acad Sci U S A. 1985;82:3187–3191. doi: 10.1073/pnas.82.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman BF, Peterson JA, Stull JT. J Biol Chem. 1992;267:5346–5354. [PubMed] [Google Scholar]
- Foster C, Van Fleet M, Marshak A. Arch Biochem Biophys. 1986;251:616–623. doi: 10.1016/0003-9861(86)90371-1. [DOI] [PubMed] [Google Scholar]
- Gallagher P, Henneberry J, Wilson I, Sambrook J, Gething MJ. J Cell Biol. 1988;107:2059–2073. doi: 10.1083/jcb.107.6.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Griffin SA, Stull JT. J Biol Chem. 1991;266:23936–23944. [PMC free article] [PubMed] [Google Scholar]
- Hathaway DR, Haeberle JR. Anal Biochem. 1983;135:37–43. doi: 10.1016/0003-2697(83)90726-1. [DOI] [PubMed] [Google Scholar]
- Herring BP. J Biol Chem. 1991;266:11838–11841. [PubMed] [Google Scholar]
- Herring BP, Fitzsimons DP, Stull JT, Gallagher PJ. J Biol Chem. 1990;265:16588–16591. [PMC free article] [PubMed] [Google Scholar]
- Hubbard MJ, Klee CB. J Biol Chem. 1987;262:15062–15070. [PubMed] [Google Scholar]
- Ikebe M, Stepinska M, Kemp BE, Means AR, Hartshorne DJ. J Biol Chem. 1987;262:13828–13834. [PubMed] [Google Scholar]
- Ikebe M, Maruta S, Reardon S. J Biol Chem. 1989;264:6967–6971. [PubMed] [Google Scholar]
- Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- Ito M, Guerriero V, Jr, Chen X, Hartshorne DJ. Biochemistry. 1991;30:3498–3503. doi: 10.1021/bi00228a021. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Leachman SA, Michnoff CH, Nunnally MH, Persechini A, Richardson AL, Stull JT. Prog Clin Biol Res. 1987;245:183–193. [PubMed] [Google Scholar]
- Kemp BE, Pearson RB. Biochim Biophys Acta. 1991;1094:67–76. doi: 10.1016/0167-4889(91)90027-u. [DOI] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB. J Biol Chem. 1985;260:3355–3359. [PubMed] [Google Scholar]
- Kemp BE, Pearson RB, House C. Proc Natl Acad Sci U S A. 1983;80:7471–7475. doi: 10.1073/pnas.80.24.7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB, Guerriero V, Jr, Bagchi IC, Means AR. J Biol Chem. 1987;262:2542–2548. [PubMed] [Google Scholar]
- Lukas TJ, Burgess WH, Prendergast FG, Lau W, Watterson DM. Biochemistry. 1986;25:1458–1464. doi: 10.1021/bi00354a041. [DOI] [PubMed] [Google Scholar]
- Miller JR, Silver PJ, Stull JT. Mol Pharmacol. 1983;24:235–242. [PubMed] [Google Scholar]
- Olson NJ, Pearson RB, Needleman DS, Hurwitz MY, Kemp BE, Means AR. Proc Natl Acad Sci U S A. 1990;87:2284–2288. doi: 10.1073/pnas.87.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RB, Wettenhall REH, Means AR, Hartshorne DJ, Kemp BE. Science. 1988;241:970–973. doi: 10.1126/science.3406746. [DOI] [PubMed] [Google Scholar]
- Potter JD, Gergely J. J Biol Chem. 1975;250:4628–4633. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker MO, Lau W, Shattuck RL, Kwiatkowski AP, Matrisian PE, Guerra-Santos L, Wilson E, Lukas TJ, Van Eldik LJ, Watterson DM. J Cell Biol. 1990;111:1107–1125. doi: 10.1083/jcb.111.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. J Biol Chem. 1990;265:1823–1826. [PubMed] [Google Scholar]
- Stull JT, Nunnally MH, Moore RL, Blumenthal DK. Adv Enzyme Regul. 1985;23:123–140. doi: 10.1016/0065-2571(85)90043-3. [DOI] [PubMed] [Google Scholar]
- Stull JT, Bowman BF, Gallagher PJ, Herring BP, Hsu LC, Kamm KE, Kubota Y, Leachman SA, Tansey M. In: Progress in Clinical and Biological Research, Vol. 327: Frontiers in Smooth Muscle Research. Sperelakis N, Wood JD, editors. Alan R. Liss; New York: 1990a. pp. 107–126. [Google Scholar]
- Stull JT, Hsu LC, Tansey MG, Kamm KE. J Biol Chem. 1990b;265:16683–16690. [PubMed] [Google Scholar]
- Van Berkum MFA, Means AR. J Biol Chem. 1991;266:21488–21495. [PubMed] [Google Scholar]