

Abstract
Blood clotting reactions, such as those catalyzed by the tissue factor/factor VIIa complex (TF:VIIa), assemble on membrane surfaces containing anionic phospholipids such as phosphatidylserine (PS). In fact, membrane binding is critical for the function of most of the steps in the blood clotting cascade. In spite of this, our understanding of how the membrane contributes to catalysis, or even how these proteins interact with phospholipids, is incomplete. Making matters more complicated, membranes containing mixtures of PS and neutral phospholipids are known to spontaneously form PS-rich membrane microdomains in the presence of plasma concentrations of calcium ions, and it is likely that blood clotting proteases such as TF:VIIa partition into these PS-rich microdomains. Unfortunately, little is known about how membrane microdomain composition influences the activity of blood clotting proteases, which is typically not under experimental control even in “simple” model membranes. Our laboratories have developed and applied new technologies for studying membrane proteins to gain insights into how blood clotting protease-cofactor pairs assemble and function on membrane surfaces. This includes using a novel, nanoscale bilayer system (Nanodiscs) that permits assembling blood clotting protease-cofactor pairs on stable bilayers containing from 65 to 250 phospholipid molecules per leaflet. We have used this system to investigate how local (nanometer-scale) changes in phospholipid bilayer composition modulate TF:VIIa activity. We have also used detailed molecular dynamics simulations of nanoscale bilayers to provide atomic-scale predictions of how the membrane-binding domain of factor VIIa interacts with PS in membranes.
Most steps in the blood clotting cascade require the assembly of a serine protease together with its cognate regulatory protein on a membrane surface [1]. Both the coagulation serine protease (for example, factors VIIa, IXa or Xa) and its protein cofactor (tissue factor, factor VIIIa or factor Va, respectively) contain specific membrane-interactive domains that direct these proteins to assemble and function on membrane surfaces. Furthermore, the protein substrates (factors IX, X and prothrombin) of these membrane-bound enzymes also bind reversibly to membranes. Membrane surfaces can only support the binding and assembly of clotting factors if they contain exposed anionic phospholipids, with phosphatidylserine (PS) being the most active. But membrane binding does more than just localize these enzymes and substrates to regions of tissue trauma; the membrane also plays a critical role in catalysis, as blood clotting enzymes are thousands of times less active when released from the membrane surface. In spite of the critical importance of membrane binding, we still have a very incomplete understanding of how the membrane surface enhances blood clotting reactions. Furthermore, we lack a detailed understanding of how the membrane binding domains of blood clotting proteins actually interact with anionic phospholipids.
Binding of blood clotting proteins to anionic phospholipids on membrane surfaces is considerably more complex than it may seem at first. Biological membranes are inhomogeneous, with the plasma membrane of cells sequestering both specific proteins and specific lipid types into a variety of membrane microdomains, including but not limited to lipid rafts. In fact, even simple liposomes composed of binary mixtures of neutral phospholipids (such as phosphatidylcholine, PC) with anionic phospholipids such as phosphatidic acid or PS will spontaneously create anionic phospholipid-rich membrane microdomains when exposed to plasma concentrations of Ca2+ [2,3]. These membrane microdomains become even more pronounced in the presence of PS-binding proteins. Experiments with giant unilamellar vesicles have shown that anionic phospholipids can move over fairly long distances (several μm) to coalesce into membrane microdomains large enough to be visualized by light microscopy [2,3]. Such studies have taught us that local membrane microenvironments can differ dramatically from the average composition of the membrane even in relatively “simple” model membranes.
Since blood clotting proteins bind to anionic phospholipids like PS, and since anionic phospholipids tend to cluster in the presence of plasma concentrations of Ca2+, it follows that local differences in the composition of membrane microdomains will mean that clotting factors will favor binding to those microdomains that are enriched in PS. Even within membrane microdomains, there may be localized differences in PS abundance at the nanometer scale that could influence the assembly and catalytic activity of protease-cofactor pairs, as well as the local abundance of membrane-bound protein substrates. We therefore hypothesize that blood clotting reactions such as the activation of factor X by the tissue factor/factor VIIa complex (TF:VIIa), or the activation of prothrombin by the Va:Xa complex, will partition into, and have highest activity within, membrane microdomains that have locally very high PS content.
But how can one study the assembly and function of enzymes within such microdomains? Membrane-binding enzymes will partition into the microdomain for which they have the greatest affinity, which is unfortunately not under the control of the experimenter. In order to control the local membrane nanoenvironment surrounding coagulation protease-cofactor complexes, we have begun studying blood clotting reactions assembled on nanometer-scale phospholipid bilayers. This is accomplished using a novel supported bilayer system termed Nanodiscs [4,5]. Nanodiscs are water-soluble particles which self-assemble into nanoscale phospholipid bilayers encircled and stabilized by two copies of an engineered amphipathic helical protein termed membrane scaffold protein (MSP). The self-assembly process is highly reproducible and efficient, yielding monodisperse preparations of stable, supported lipid bilayers that exhibit bilayer fluidities comparable to liposomes [6]. The smallest Nanodiscs encircle a lipid bilayer approximately 8 nm in diameter, containing about 67 phospholipid molecules per leaflet. Larger versions of MSP have recently been engineered, allowing the formation of larger Nanodiscs (containing up to about 250 phospholipid molecules per leaflet). Importantly, membrane-spanning proteins such as tissue factor can efficiently be incorporated into Nanodiscs during self-assembly [7,8]. By using Nanodiscs to assemble protease-cofactor pairs in the blood clotting system, we can preclude long-distance (μm scale) recruitment of PS molecules into membrane subdomains. Rather, the experimenter retains complete control over the membrane composition immediately (i.e., within a few nm) around the protease-cofactor complex.
We recently employed the Nanodisc system to study the effect of changing the local PS content on the assembly and function of the TF:VIIa complex [9]. We found that the binding affinity of factor VIIa for TF in these nanoscale bilayers increased as a function of local PS content. Perhaps more importantly, the catalytic efficiency of TF:VIIa was dramatically altered when TF was incorporated into Nanodiscs of increasing PS content, with maximal catalytic efficiencies requiring 70% PS or higher. In fact, comparable rates of factor X activation were observed with TF-Nanodiscs containing 70% or more PS, compared to TF-liposomes containing 30% PS. This was paralleled by Biacore studies of the affinity of factor X binding to Nanodiscs of varying PS content (from 0 to 100% PS). We found that the Kd for factor X binding to these nanoscale bilayers decreased monotonically as the % PS was increased, reaching maximal binding affinity at >80% PS. These studies argue that extremely high local PS content is required for optimal assembly and function of the TF:VIIa complex.
Membrane-bound protease-cofactor pairs such as TF:VIIa will encounter membrane-bound protein substrates such as factor X, because factor X binds reversibly to membrane surfaces containing PS via its γ-carboxyglutamate-rich domain (GLA domain). Once a factor X molecule has bound to the membrane surface it may translocate on the membrane via lateral diffusion, effectively skating or hopping along the surface. One can therefore imagine at least two different mechanisms of substrate presentation to the TF:VIIa complex: Solution-phase factor X might bind directly to the membrane-bound TF:VIIa complex and be activated via limited proteolysis. Alternatively, membrane-bound factor X molecules might randomly move on the membrane surface via lateral diffusion or hopping to encounter the TF:VIIa complex. Several studies have addressed this question using TF incorporated into liposomes, concluding that either the membrane-bound pool of factor X [10], or solution-phase factor X is the preferred substrate [11,12].
Unlike the situation on liposomes, TF:VIIa on a Nanodisc cannot access a large pool of membrane-bound factor X molecules moving over long distances by lateral diffusion or hopping. Instead, the nanoscale membrane surface can bind at most five or six factor X molecules at a time, which should be completely converted to factor Xa within two or three seconds. But we have observed sustained, linear rates of factor X activation over 20 minute time courses, during which time at least 2400 factor X molecules were activated per TF:VIIa complex [9]. This means that the TF:VIIa complex assembled on Nanodiscs relies on continuous binding of substrate (factor X) to, and dissociation of, product (factor Xa) from the nanobilayer in order to allow such sustained rates of fX activation. The fact that kcat values obtained with TF-Nanodiscs rival those of TF-liposomes demonstrates that the TF:VIIa complex is not absolutely dependent on a large, preexisting pool of membrane-bound fX to serve as substrate.
We are now extending our studies using nanoscale bilayers to investigate the mechanism of synergy between phospholipid types, such as how the synergistic interaction between PS and phosphatidylethanolamine (PE) enhances the activity of TF:VIIa [13]. One hypothesis for the synergy between PE and PS is that PE promotes the formation of PS-rich microdomains, into which the TF:VIIa complex partitions. By rigorously controlling the PS and PE content in the nanoscale bilayers with TF-Nanodiscs, we are investigating whether or not PE and PS can still synergize when confined to nanodomains of defined composition. We are also extending our nanoscale bilayer studies to investigating the phospholipid dependence of the activity of other protease-cofactor pairs in blood clotting. Preliminary studies in our lab have shown that highly active prothrombinase complexes (Va:Xa complexes) can be assembled on the surface of Nanodiscs that have been engineered to encompass 12 nm-diameter phospholipid bilayers (Pureza and Morrissey, unpublished observations).
In order to provide a detailed view of the modes and the extent of interaction between PS and coagulation factors, the above experimental efforts are complemented by computational modeling and simulation. Through combining various molecular modeling techniques and an extensive set of molecular dynamics simulations we have developed for the first time at full atomic detail a model for the membrane bound form of the GLA domain of factor VIIa in the presence of an explicit representation of a PS bilayer [14]. The model captures somewhat unexpected features of the membrane-bound state of the GLA domain. In contrast to all previously suggested models for membrane association of the GLA domain in which Ca2+ ions are depicted as sandwiched between the protein and the surface of the negative membrane, our model shows that the structurally bound Ca2+ ions are completely and deeply immersed within the PS head-group layer, sometimes reaching even the phosphate groups of the lipids (see Fig. 1). Consequently, the ω-loop is also fully inserted into the bilayer and establishes significant hydrophobic interaction with the tail region of the bilayer [14]. Another unexpected result of the present study is the observation that some of the structurally bound Ca2+ ions do not directly participate in membrane binding of the GLA domain. Supported by the simulations of the GLA domain in solution at different Ca2+-bound states, we propose that Ca2+ ions play a dual role in binding of the GLA domain to negative lipid bilayers, a bracing and an anchoring role; a fraction of Ca2+ ions play their role primarily through maintaining the conformation of the GLA domain that is needed for membrane insertion, while others directly participate in anchoring the GLA domain to the membrane. Once inserted in the membrane, the GLA domain is also stabilized through direct interactions of certain amino acid side chains that are positioned optimally to reach the membrane head groups [14]. This model is currently being used to investigate structural determinants of membrane binding affinity of various GLA domains.
Figure 1. Membrane-bound model of the complex of tissue factor and factor VIIa.
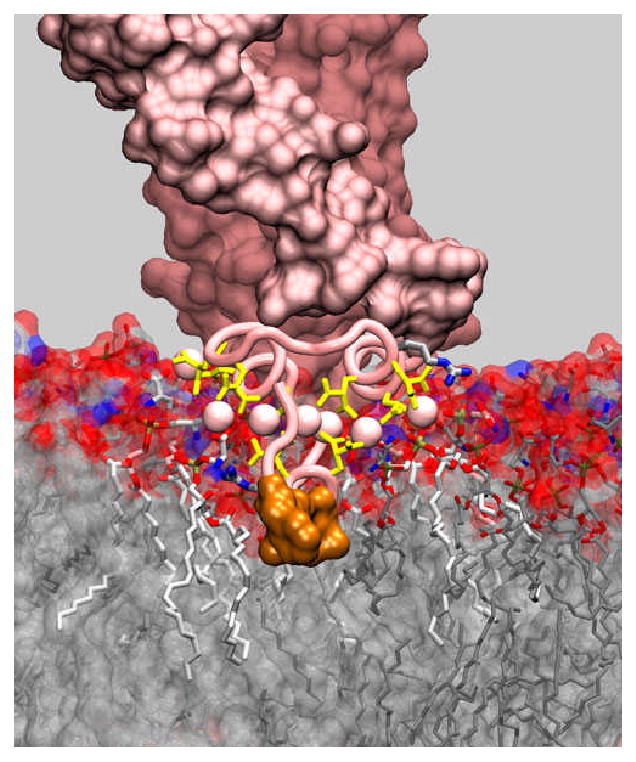
The model is constructed by superimposing the X-ray structure of the TF:VIIa complex (PDB code 1DAN) on a recently developed model of the GLA domain of factor VIIa bound to a DOPS lipid bilayer [14]. The GLA domain is shown using a tube representation, and structurally bound Ca2+ ions are drawn in spheres. Gla residues, lipid molecules and a number of resides that directly participate in interaction with the membrane are drawn using a stick representation. The hydrophobic tip of the GLA domain that inserts completely into the core of the bilayer is shown in a surface representation. Lipid molecules that are directly involved in specific interactions with Ca2+ ions are draws using a thicker representation. Structurally bound calcium ions are immersed in the head group region of the lipid bilayer and play two distinct roles: central ions seem to be primarily involved in stabilization of the fold of the GLA domain, whereas peripheral ions are directly interacting with negative charges of lipids.
Footnotes
Conflict of interest: The authors report no conflicts of interest.
References
- 1.Zwaal RFA, Comfurius P, Bevers EM. Lipid-protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376:433–53. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 2.Haverstick DM, Glaser M. Visualization of Ca2+-induced phospholipid domains. Proc Natl Acad Sci U S A. 1987;84:4475–9. doi: 10.1073/pnas.84.13.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Glaser M. Formation of membrane domains during the activation of protein kinase C. Biochemistry. 1996;35:13966–74. doi: 10.1021/bi9610008. [DOI] [PubMed] [Google Scholar]
- 4.Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer Nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–69. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 5.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–87. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 6.Shaw AW, McLean MA, Sligar SG. Phospholipid phase transitions in homogeneous nanometer scale bilayer discs. FEBS Lett. 2004;556:260–4. doi: 10.1016/s0014-5793(03)01400-5. [DOI] [PubMed] [Google Scholar]
- 7.Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of Beta2-adrenergic receptors utilizing self-assembling Nanodisc technology. BioTechniques. 2006;40:601–2. 604, 606. doi: 10.2144/000112169. [DOI] [PubMed] [Google Scholar]
- 8.Bayburt TH, Sligar SG. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 2003;12:2476–81. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007;282:6556–63. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 10.Krishnaswamy S, Field KA, Edgington TS, Morrissey JH, Mann KG. Role of the membrane surface in the activation of human coagulation factor X. J Biol Chem. 1992;267:26110–20. [PubMed] [Google Scholar]
- 11.Forman SD, Nemerson Y. Membrane-dependent coagulation reaction is independent of the concentration of phospholipid-bound substrate: Fluid-phase factor X regulates the extrinsic system. Proc Natl Acad Sci USA. 1986;83:4675–9. doi: 10.1073/pnas.83.13.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bom VJ, Bertina RM. The contributions of Ca2+, phospholipids and tissue-factor apoprotein to the activation of human blood-coagulation Factor X by activated Factor VII. Biochem J. 1990;265:327–36. doi: 10.1042/bj2650327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: Enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34:13988–93. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 14.Ohkubo YZ, Tajkhorshid E. Distinct structural and adhesive roles of Ca2+ in membrane binding of blood coagulation factors. Structure. 2008;16:72–81. doi: 10.1016/j.str.2007.10.021. [DOI] [PubMed] [Google Scholar]