

Abstract
It has been proposed that the carboxyl terminus of the smooth muscle myosin light chain kinase is expressed as an independent protein. This protein has been purified from tissues and named telokin (Ito, M., Dabrowska, R., Guerriero, V., Jr., and Hartshorne, D. J. (1989) J. Biol. Chem. 264, 13971–13974). In this study we have isolated and characterized cDNA and genomic clones encoding telokin. Analysis of a genomic DNA clone suggests that the mRNA encoding telokin arises from a promoter which appears to be located within an intron of the smooth muscle myosin light chain kinase (MLCK) gene. This intron interrupts exons encoding the calmodulin binding domain of the kinase. The amino acid sequence deduced from the cDNA predicts that telokin is identical to the carboxyl-terminal 155 residues of the smooth muscle MLCK. Unlike the smooth muscle MLCK which is expressed in both smooth and non-muscle tissues, telokin is expressed in some smooth muscle tissues but has not been detected in aortic smooth muscle or in any non-muscle tissues.
Phosphorylation of the 20,000-Da light chain subunit of myosin by the Ca2+/calmodulin-dependent MLCK1 is a key event in the initiation of contraction in smooth muscle cells. Phosphorylation of the myosin light chains increases the actin-activated myosin MgATPase and leads to increases in tension development (Kamm and Stull, 1985; Hai and Murphy, 1989). Accumulating evidence suggests that phosphorylation of myosin light chain by MLCK in non-muscle cells and tissues may also have an important physiological function. For example, myosin light chain phosphorylation has been implicated in secretory vesicle movement, cellular locomotion, and changes in cellular morphology (Adelstein et al., 1975; Bissonnette et al., 1989; Cande and Ezzell, 1986; Goldfine et al., 1981; Holzapfel et al., 1983; Masuda et al., 1984; Wysolmerski and Lagunoff, 1990).
Smooth muscle MLCKs are highly conserved, modular enzymes having several well characterized domains (Gallagher et al., 1991) (Olson et al., 1990). They are comprised of an amino-terminal “tail” of unknown function and a central catalytic core which is homologous to the catalytic core of other protein kinases (Hanks et al., 1988). Immediately carboxyl-terminal to the catalytic core are located the regulatory and calmodulin binding domains, which are involved in the activation of the kinase in response to increasing Ca2+. The extreme carboxyl terminus of smooth muscle MLCK is comprised of a highly acidic region, the function of which is presently unknown. A similar domain organization exists in the skeletal muscle MLCKs (Herring et al., 1990; Roush et al., 1988), except that the skeletal muscle MLCKs do not contain the acidic carboxyl-terminal region.
Removal of the unique carboxyl-terminal domain of the smooth muscle MLCK as a result of limited proteolysis (Ikebe et al., 1987) or by genetic manipulation (Bagchi et al., 1989; Ito et al., 1991) does not appear to alter significantly the catalytic properties of these molecules in vitro. Other studies have shown that the activity of smooth muscle MLCK is modulated by phosphorylation of two specific sites within the carboxyl-terminal region. In the absence of Ca2+/calmodulin, cAMP-dependent protein kinase phosphorylates two sites on the kinase (sites A and B, serine 992 and serine 1005 respectively, of the rabbit uterine MLCK) whereas in the presence of Ca2+/calmodulin, only one site (B, serine 1005) is phosphorylated. Phosphorylation of site A decreases the affinity of MLCK for Ca2+/calmodulin and, therefore, would decrease MLCK activity at low internal calcium (Conti and Adelstein, 1981; Payne et al., 1986; Adelstein et al., 1978). Phosphorylation at site B alone has no known effect on enzyme activity. Several other protein kinases such as cGMP-dependent protein kinase, protein kinase C, and the multifunctional Ca2+/calmodulin-dependent protein kinase II can also phosphorylate site A in vitro and produce similar changes in the activation properties of the kinase (Ikebe and Reardon, 1990; Ikebe et al., 1985; Nishikawa et al., 1984, 1985; Hashimoto and Soderling, 1990). In addition to the phosphorylation at sites A and B, Stull et al. (1990) have shown that there are several other sites of phosphorylation within the carboxyl terminus of MLCK; however, these sites have not yet been characterized.
Recently, a 24-kDa acidic protein named “telokin” has been purified from turkey gizzard (Ito et al., 1989). Sequencing of proteolytic fragments of telokin has established that this protein is highly related to the carboxyl terminus of smooth muscle MLCK. It has been predicted that the amino terminus of telokin begins between M816 and M818 of the chicken gizzard MLCK (equivalent to M993 and M995 of the rabbit uterine MLCK (Gallagher et al., 1991). Purified turkey telokin was found to be phosphorylated at one site (presumably equivalent to site B of the MLCK) by the catalytic subunit of cAMP-dependent protein kinase. The presence of a blocked amino terminus suggested that this protein is expressed independently of MLCK and was not simply a proteolytic fragment of the kinase (Ito et al., 1989). In support of this proposal a 2.6–2.7-kb mRNA is detected by Northern analysis of mRNA from smooth muscle tissues using cDNA probes corresponding to the 3′ end of the gizzard (Guerriero et al., 1986) and rabbit (Gallagher et al., 1991) smooth muscle MLCKs.
In this study, using molecular techniques we have confirmed that the carboxyl terminus of MLCK is expressed as an independent protein, telokin. The telokin mRNA is transcribed from a promoter which appears to be located within an intron of the MLCK gene. We have also determined that telokin expression is limited to some but not all smooth muscle tissues.
EXPERIMENTAL PROCEDURES
Preparation and Screening of a Specifically Primed Rabbit Uterine cDNA Library
RNA was isolated from uteri from retired New Zealand White female breeding rabbits. Poly(A+) RNA was prepared by affinity chromatography (Sambrook et al., 1989). A specific, oligonucleotide-primed library was prepared using an antisense oligonucleotide primer (5′-GTAACACGACTTACAAACTG) corresponding to bp 3768–3787 of the rabbit uterine smooth muscle MLCK (Gallagher et al., 1991). The resulting λgt10 library was screened with a 633-bp probe corresponding to bp 3665–3998 of the rabbit uterine smooth muscle MLCK.2 λDNA was prepared from positive plaques, digested with EcoRI, sized, subcloned, and sequenced (Fig. 1).
Fig. 1. Nucleotide and deduced amino acid sequence of the cDNA encoding the rabbit uterine telokin.

The nucleotide sequence of the 676-bp cDNA clone identified in an MLCK-specific, oligonucleotide-primed library is shown. The 5′-noncoding region has been translated and is shown in lowercase letters. Nucleotides that are overlined (bp 9–116) are unique to the telokin cDNA; the remainder of the sequence is identical to that present in the rabbit smooth muscle MLCK (bp 3237–3787) (Gallagher et al., 1991), excluding the linkers (bp 1–8, 668–676). Residues that are underlined are within the coding region of the rabbit uterine smooth muscle MLCK. The nucleotides for translational initiation and termination are shown in boldface. An asterisk indicates the positions of introns which have been identified from sequence of the telokin gene. Numbers on the right side correspond to nucleotide sequence of the telokin cDNA (above) and amino acids of rabbit telokin/smooth muscle MLCK cDNAs (below).
Isolation of the Rabbit Smooth Muscle Telokin/MLCK Gene
A rabbit genomic DNA library (Clontec, Palo Alto, CA) was screened with a portion of the rabbit uterine smooth muscle MLCK cDNA (bp 3665–3998). λDNA was prepared from positive plaques, and the inserts were excised by digestion with SalI and subcloned into pGEM. The genomic DNA inserts were mapped by digestion with restriction endonucleases and subcloned for DNA sequencing using synthetic oligonucleotides.
Primer Extension
Primer extension was performed essentially as described (Sambrook et al., 1989). The oligonucleotide used in this analysis corresponds to bp 585–619 of the rabbit smooth muscle telokin/MLCK genomic sequence (Fig. 2). The oligonucleotides were labeled with T4 polynucleotide kinase and [32P]ATP, purified, and hybridized to 50 μg of total RNA from rabbit uterus. The primer was then extended in a first strand cDNA reaction by reverse transcriptase. The length of the resulting end-labeled cDNA was estimated by electrophoresis through a denaturing polyacrylamide gel and comparison with radioactive molecular weight standards.
Fig. 2. Nucleotide sequence of a portion of the rabbit telokin gene.

The nucleotide and deduced amino acid sequence of the rabbit telokin gene are shown. Residues shown in boldface capital letters are included in the coding region of telokin and the smooth muscle MLCK. Residues in lowercase letters and underlined are within the coding region of the rabbit smooth muscle MLCK and are within the predicted 5′-noncoding region in the telokin cDNA. An asterisk indicates the position of an intron in the rabbit telokin gene. Nucleotides that are overlined correspond to a primer used in the primer extension analysis. Nucleotides that are in boldface are those that are proposed as comprising the TATA box and transcriptional start site for the 2.6-kb mRNA encoding telokin.
DNA Sequencing
Fragments of the cDNA or genomic clone were subcloned into pGEM and M13 vectors for double-stranded (Mierendorf and Pfeffer, 1987) and single-stranded sequencing by the dideoxy method (Sanger et al., 1977) using Sequenase version 2.0 and pyrophosphatase (U. S. Biochemical Corp.). Convenient restriction sites as well as synthetic oligonucleotide sequencing primers were used to resolve the DNA sequence. All DNA sequences were determined more than once, either on separate strands or from different end points on the same strand. Approximately 50% and 10% of the cDNA and genomic sequences, respectively, were obtained from both strands. Much of the DNA sequence was read from 8% acrylamide, 40% formamide, 7 M urea gels (U. S. Biochemical Corp.). pGEM vectors are commercially available from Promega (Madison, WI).
Oligonucleotide-directed Mutagenesis
Oligonucleotide-dimutagenesis was carried out as described by Zoller and Smith (1987). The oligonucleotide 5′-GACTGTCCTCTATGGCAATG-3′ was used to introduce an NdeI restriction endonuclease site at bp 3285–3290 of the rabbit uterine smooth muscle MLCK (Gallaghar et al., 1991). The mutation was confirmed by DNA sequencing.
Bacterial Expression and Purification of the Carboxyl Terminus of MLCK
A 724-bp NdeI-EcoRI fragment from the mutagenized rabbit uterine smooth muscle MLCK (representing bp 3285–4009) was subcloned into the NdeI-EcoRI sites of the bacterial expression vector pET3a (Studier and Moffatt, 1986). This construction, pET3a-CT, was used to express the carboxyl-terminal portion of the MLCK from residues 993 to 1147 in the Escherichia coli strain HMS174(DE3). The deduced sequence of the cDNA encoding telokin suggests that these carboxyl-terminal residues are identical to the predicted telokin protein, and this bacterially expressed protein will be called telokin for the rest of this paper. High levels of expression of telokin were found after induction by isopropyl 1-thio-β-D-galactoside (Fig. 3). The expressed protein was purified as follows. One liter of NZCYM (per liter, 10 g of NZ amine, 5 g of NaCl, 5 g of Bacto-yeast extract, 1 g of casamino acids, 2 g of MgSO4, 7H2O) was inoculated with 5 ml of an overnight culture of HMS174(DE3) containing the pET3a-CT plasmid; this was grown for 2 h at 37 °C; isopropyl 1-thio-β-D-galactoside was added to a final concentration of 1 mM. After 3 h bacterial cells were pelleted by centrifugation at 500 × g for 20 min. The remaining steps were carried out at 4 °C unless otherwise indicated. The cells were washed and then resuspended in a lysis buffer (50 mM Tris, pH 8.0, 1 mM EDTA, 20 μg/ml PMSF, 20 ng/ml aprotinin, and 500 μg/ml DNase). The cells were lysed by freezing and thawing six times. The bacterial lysate was clarified by centrifugation at 5,000 × g for 20 min, and the supernatant was dialyzed against 50 mM MOPS, pH 6.6, 200 mM NaCl, 20 ng/ml aprotinin, 20 μg/ml PMSF, 1 mM dithiothreitol. After dialysis the supernatant was adsorbed on DEAE-cellulose (preequilibrated in the same buffer) by stirring for 2 h and then washed with 0.2 M NaCl in 50 mM MOPS, pH 6.6. The adsorbed telokin protein was eluted from the DEAE-cellulose with 0.3 M NaCl in 50 mM MOPS, pH 6.6, then concentrated by Amicon filtration. E. coli HMS174(DE3) used for bacterial expression and pET3a plasmid were the gift of W. Studier (Brookhaven National Laboratories, Upton, NY).
Fig. 3. Characterization of anti-telokin polyclonal antibodies.

Panel A, Coomassie stained, SDS-polyacrylamide gel electrophoresis (15% acrylamide gel) of the bacterial expressed and purified telokin. Lane 1, bacterial lysate of E. coli HMS 174(DE3)-pET3A-TEL; lane 2, purified telokin protein. Panel B, immunoblot analysis with anti-telokin antibodies to detect the 152-kDa MLCK in a transfected COS cell extract expressing rabbit smooth muscle MLCK (lane 1) and rabbit uterine tissue (lane 2). Panel C, immunoblot analysis with anti-telokin polyclonal antibodies to detect rabbit telokin protein in extracts prepared from COS cells transfected with CMV5-TEL. Lane 1, purified, bacterial expressed telokin, 1 ng; lane 2, rabbit uterine tissue (19 μg of total protein); lanes 3 and 4, 5 and 10 μl of a COS cell extract expressing CMV5-Tel; lane 5, 10 μl of a COS cell extract expressing rabbit smooth muscle MLCK
Preparation of a Polyclonal Antibody to Telokin
Antiserum to telokin was produced in New Zealand White rabbits. The initial injection was 100 μg of telokin purified from bacteria, in complete Freund's adjuvant. Subsequent boosts were made at 3 and 6 weeks, with 150 μg of purified protein in incomplete Freund's adjuvant. The specificity of the antiserum for telokin and the carboxyl terminus of MLCK was shown by Western blot experiments using both the expressed recombinant MLCK and MLCK present in tissues as well as recombinant telokin and telokin present in tissues (Figs. 3 and 4)
Fig. 4. Immunoblot analysis of smooth and non-muscle rabbit, chicken, and bovine tissues.
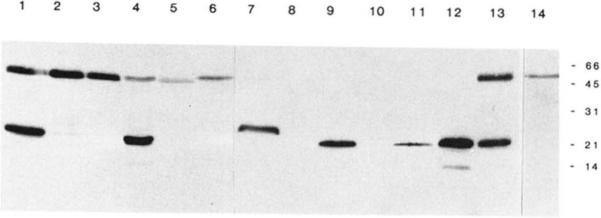
Immunoblot analysis of tissue extracts using polyclonal anti-telokin antibodies. The amount of total protein loaded per lane varies as indicated and is not representative of the relative amounts of immunoreactive protein present in these tissues. The following tissues were used to prepare extracts: lanes 1 and 13, rabbit uterus (20 μg); lane 2, rabbit trachea (89 μg); lane 3, rabbit aorta (86 μg); lane 4, rabbit ileum (42 μg); lane 5, rabbit kidney (84 μg); lane 6, rabbit adrenal gland (110 μg); lane 7, chicken gizzard (60 μg); lane 8, chicken liver (76 μg); lane 9, bovine trachea (44 μg); lane 10, bovine adrenal gland (264 μg); lanes 11 and 12, purified telokin (1 and 5 ng, respectively); lane 14 represents an immunoblot performed using preimmune antiserum and 20 μg of rabbit uterine tissue.
Expression of the Telokin cDNA in COS Cells
A 676-bp telokin cDNA which was isolated from the primed library was subcloned into the EcoRI site of pCMV5 to generate the vector (pCMV5-TEL) used for the expression of telokin in COS cells. Purified DNA (2 μg) was then used to transfect a 60-mm dish which had been seeded with 6 × 105 COS cells using a DEAE-dextran transfection procedure (Sambrook et al., 1989). After transfection the cells were incubated for 48 h before being lysed in 0.4 ml of lysis buffer (10% glycerol, 1% Nonidet P-40, 10 mM MOPS, 1.25 mM EGTA, 10 mM dithiothreitol, 50 mM MgCl2, 120 μg/ml TPCK and TLCK, 100 μg/mL PMSF, 10 μg/ml each of pepstatin, APMSF, leupeptin, and 4 μg/ml aprotinin). The detergent lysate was clarified by centrifugation (2 min, 15,000 × g at 4 °C), the supernatant fraction was aliquoted, rapidly frozen in liquid nitrogen, and maintained at −70 °C until electrophoresis. pCMV5 DNA is a derivative of pCMV2 (Andersson et al., 1989).
Preparation of Tissue and Cultured Cell Extracts
All tissues used in this study except rabbit tracheal tissue were dissected to remove adventitia tissue and immediately frozen in liquid nitrogen. Rabbit trachea, including cartilage, were frozen and homogenized without dissection. Frozen tissues (10 mg) were homogenized in 400 μl of homogenization buffer (20% sucrose, 125 mM Tris-HCl, pH 6.8, 10% SDS, 120 μg/ml TPCK and TLCK, 100 μg/ml PMSF, 10 μg/ml each of pepstatin, APMSF, and leupeptin, and 4 μg/ml aprotinin), centrifuged (2 min, 15,000 × g, 4 °C), and the supernatant fraction was aliquoted and immediately frozen in liquid nitrogen. The fractions were maintained at −70 °C until electrophoresis. Samples were prepared for electrophoresis by the addition of dithiothreitol to a final concentration of 50 mM, and bromophenol blue was added to a final concentration of 0.01% and then boiled for 2 min. Protein determinations were performed on samples which did not contain dithiothreitol with the bicinchoninic acid protein assay method (BCA Protein Assay; Pierce Chemical Co.) according to the manufacturer's directions.
Western Immunoblots
Low molecular weight proteins in tissue and cell extracts were separated by electrophoresis through discontinuous SDS-polyacrylamide gels (15% acrylamide). After electrophoresis, the proteins were transferred to a polyvinyldene difluoride membrane at 80 V for 40 min in transfer buffer (10 mM CAPS, pH 11, 10% methanol). After transfer, the membrane was treated with 0.4% glutaraldehyde diluted in phosphate-buffered saline (0.058 M Na2HPO4, 0.017 M NaH2PO4, 0.068 M NaCl) for 30 min then washed in phosphate-buffered saline. The membrane was treated with a blocking solution composed of 6% casein, 1% polyvinylpyrridoline, 10 mM EDTA in phosphate-buffered saline for 2 h, then reacted with a polyclonal antibody raised against the bacterially expressed, purified telokin, diluted 1:5,000 in TBST (10 mM Tris, 7.5, 150 mM NaCl, 0.05% Tween 20) containing 1% gelatin. After several washes in TBST (15 min total) the blots were reacted for 30 min with goat anti-rabbit IgG conjugated with horseradish peroxidase second step antibody diluted 1:7,500 in TBST containing 1% gelatin. After extensive washing in TBST (four changes for a total of 60 min) the immunoreactive proteins were visualized on film with the enhanced chemiluminescent detection system (ECL Western Blotting detection system, Amersham Corp.). ECL detection was performed according to the manufacturer's directions. Immunoblotting of high molecular weight proteins was performed as described previously (Gallagher et al., 1991).
Protein Purification
MLCK from bovine trachea were purified as described previously (Stull et al., 1990).
Northern Blot Analysis
Poly(A+) RNA was prepared as described previously (Herring et al., 1990). The blots were probed with a 32P-labeled, antisense RNA probe which included the entire telokin cDNA (Fig. 5). Hybridization, washing, and autoradiography were as described (Sambrook et al., 1989). Hybridization was performed at 68 °C, and final high stringency wash conditions were in 1.25 mM sodium phosphate, pH 7.4, 30 mM NaCl, 0.2 mM EDTA (0.1 × SSPE), and 0.1% SDS at 68 °C.)
Fig. 5. Northern analysis of RNA isolated from smooth muscle tissues.
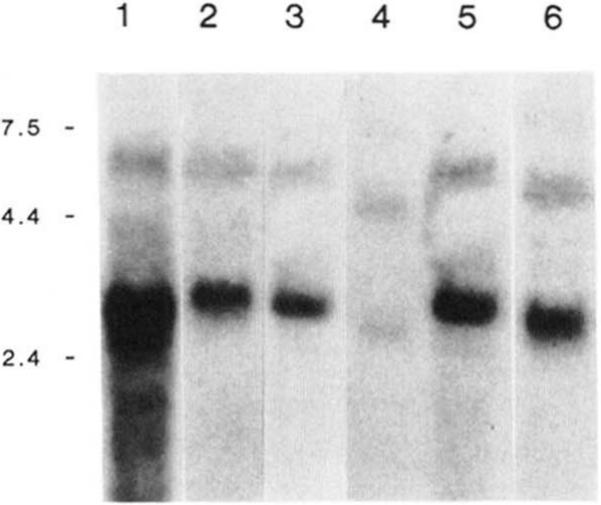
Panel A, Northern analysis of RNA isolated from rabbit uteri (lane 1, 15 μg), rabbit ileum (lane 2, 20 μg), chicken gizzard (lane 3, 20 μg), human myometrial (lane 4, 20 μg), bovine trachea (lane 5, 30 μg), rat uteri (lane 6, 20 μg). The blot was probed with a 32P-labeled antisense RNA probe corresponding to the telokin cDNA. The positions of the RNA molecular weight standards are shown in kb on the left side of the figure. The lane representing rabbit uterine tissue was exposed for 24 h, and the other lanes were exposed for 72–96 h.
RNase Protection
Antisense RNA probes were transcribed from the telokin cDNA fragment which was subcloned into the EcoRI site of pGEM 7Z vector. The vector was linearized with the BamHI site present in the polylinker. T7 RNA polymerase was used to transcribe a 32P-labeled, 766-bp, antisense RNA probe (Fig. 6). RNase protection assays were performed as described previously (Little and Jackson, 1987). Briefly, total RNA from various tissues was hybridized in solution to the labeled antisense RNA probe in the presence of 80% formamide at 45 °C for 18 h. Optimal conditions for RNase treatment (6 μg/ml RNase A, 0.3 μg/ml RNase T1 for 1 h at 37 °C) were determined from a series of control experiments.
Fig. 6. Ribonuclease protection analysis of rabbit smooth and non-muscle RNA.
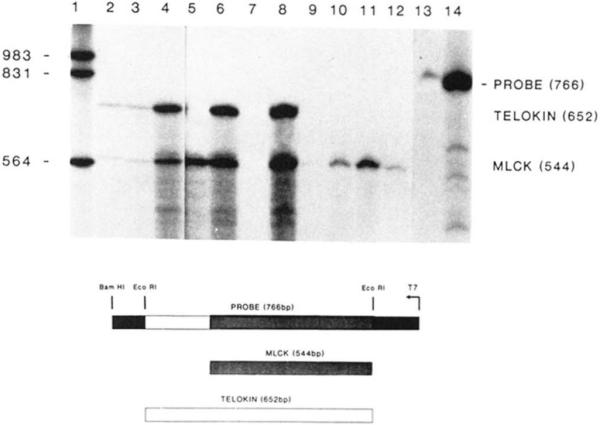
Hybridization of total RNA to the 32P-labeled antisense RNA was performed in solution (37 °C), and unprotected probe was digested with RNase as described under “Experimental Procedures.” Fragments that were protected by mRNAs present in the tissue were separated by electrophoresis on denaturing polyacrylamide gels and visualized by autoradiography. Lanes 2, 3, and 4, 2, 10 and 50 μg of rabbit uterine RNA, respectively; lane 5, 50 μg of rabbit aorta RNA; lanes 6 and 8, 10 and 50 μg of rabbit ileum RNA, respectively, lane 7 is blank; lanes 9, 10, and 11, 10, 50 and 100 μg of rabbit liver RNA, respectively; lane 12, 100 μg of rabbit kidney RNA; lanes 13 and 14, undigested probe. The sizes of end-labeled DNA molecular weight fragments (lane 1) are shown in bp on the left side of the figure. Indicated to the right side of the figure are the positions of the undigested probe, telokin, and MLCK fragments. A schematic below the figure illustrates the two protected fragments derived from the 766-bp riboprobe. The lightly shaded box represents the fragment protected by MLCK mRNA, and the open box represents the fragment protected by telokin mRNA.
RESULTS
Identification of a cDNA Encoding Telokin
A 676-bp cDNA was isolated from a rabbit uterine cDNA library enriched for MLCK clones by the use of a specific primer. This library was screened with a 333-bp probe corresponding to bp 3665–3998 within the carboxyl terminus of the rabbit smooth muscle MLCK cDNA (Gallagher et al., 1991). The sequence of the cDNA isolated from this library was identical to that of the rabbit uterine MLCK except for 108 bp of unique sequence located at the 5′ end of the cDNA (Fig. 1). Inspection of this cDNA sequence showed that the first ATG occurs at bp 162–164 and that an additional 54 bp immediately 5′ to this ATG are also identical to sequence present in the rabbit smooth muscle MLCK cDNA (Gallagher et al., 1991). The longest open reading frame, beginning with the methionine located at bp 162–164, predicts a protein of 155 amino acids with a calculated mass of 17,181 Da, in agreement with previous data (Ito et al., 1989).
This cDNA, when used as a probe for Northern blots, specifically hybridizes to two mRNAs of 5.8 and 2.6 kb, respectively, which are present in rabbit uterine tissue (Fig. 5). The 5.8-kb mRNA encodes the MLCK and the smaller 2.6-kb mRNA encodes telokin. Three probes corresponding to bp 3365–3998, 3998–5349, and 5427–5608 of the rabbit smooth muscle MLCK, all of which are located within the 3′-coding and 3′-untranslated regions of this clone, hybridize to a 2.6-kb mRNA present in rabbit uterus in Northern blotting experiments (results not shown). These data suggest that the 1862 bp of 3′-untranslated sequence present at the 3′ end of the 5.8-kb rabbit smooth muscle MLCK mRNA are also present at the 3′ end of the 2.6-kb telokin mRNA. In addition to these data, screening of oligo(dT)-primed rabbit uterine smooth muscle MLCK cDNA libraries has resulted in the isolation of six cDNA clones, which have been partially sequenced and characterized (results not shown). Five of these clones all have the same 3′ end, which is identical to the 3′ end of the rabbit uterine MLCK cDNA sequence. The sixth clone encompasses bp 3998–5349, and its entire sequence is identical to the rabbit uterine smooth muscle MLCK cDNA (Gallagher et al., 1991). The 3′ end of this clone occurs within a potential polyadenylation signal (AATAA), and it is not known if this clone represents an alternatively spliced and polyadenylated mRNA. Together these data suggest the mRNA for telokin is comprised of the sequence derived from the cDNA clone together with an additional 1800 bp of 3′-untranslated region which is identical to the MLCK mRNA.
Characterization of Promoter/Regulatory Region
An approximately 17-kb DNA fragment representing the 3′ portion of the rabbit smooth muscle MLCK gene was isolated and mapped, and portions of this DNA were sequenced (Fig. 7). A 10-kb EcoRI-SalI fragment representing the 5′ portion of this genomic DNA clone was found to contain the entire coding region of the telokin cDNA. Sequence data from the ends of this fragment revealed that the 3′ EcoRI site corresponded to the EcoRI site at bp 4004–4009 of the rabbit smooth muscle MLCK cDNA (Gallagher et al., 1991). This sequence, together with that obtained from specific primers derived from the cDNA sequence, revealed that the telokin coding sequence is divided into at least three exons (comprised of bp 1–249, 250–381, and 382–661 within the telokin cDNA sequence) as shown schematically in Fig. 7. The complete nucleotide sequence of the first exon of telokin, together with its promoter region, was obtained (Fig. 2). Base pairs 1–635 of this sequence correspond to part of an intron in the MLCK gene which is predicted to interrupt sequence encoding the calmodulin binding domain of the smooth muscle MLCK. However, this genomic DNA clone terminates within this intron and does not extend into the 5′ exon encoding the amino-terminal half of the calmodulin binding domain. Therefore, we are unable to confirm that this clone comprises the 3′ end of the MLCK gene. Base pairs 636–768 of the gene sequence represent an exon encoding part of the calmodulin binding domain MLCK. Overlap of this gene sequence with the 108 bp of unique sequence identified at the 5′ end of the telokin cDNA begins at bp 462 and extends to bp 768 (Fig. 2). Within this 108 bp of of overlap, and immediately 5′ to the predicted translational start for telokin at bp 680, are 54 bp of sequence corresponding to 15 amino acids located with the calmodulin binding domain of MLCK. The conservation of this sequence in 5′-noncoding region of telokin supports the proposal that this genomic DNA clone is the 3′ portion of the MLCK gene and not a separate gene.
Fig. 7. Schematic representation of the rabbit smooth muscle MLCK gene.

This schematic illustrates the proposed gene organization of the rabbit smooth muscle MLCK gene. The proposed structure has been based upon sequence obtained from portions of the rabbit MLCK gene and from cDNAs encoding the rabbit MLCK, and telokin and primer extension data. The upper portion of the figure shows a partial map of the two genomic DNA clones which have been characterized for this proposal. Portions of the gene which have been sequence are indicated by dotted lines. The genomic DNA comprising the extreme 5′ region of the gene has been described previously (Gallagher et al., 1991). The relative positions of the transcriptional start sites for the 5.8- and 2.6-kb mRNAs are indicated by TATAA. MLCK exons are represented by shaded boxes (not all exons are indicated), and telokin exons are represented by open boxes. Part of the amino acid sequence within the calmodulin (CaM) binding domain is shown above the linear schematic; residues indicated by lowercase letters are those present in MLCK, and residues in capital letters represent sequence at the translational start site for telokin.
Primer extension experiments (results not shown) using an antisense primer located between bp 585 and 619 and corresponding to unique sequence present in the cDNA have shown that this cDNA lacks approximately 170 bp of sequence at the 5′ end. These data suggest that a transcriptional start site would be located at bp 417–420. Between bp 356 and 391 are located a potential CAAT and TATA sequence element (Fig. 2) consistent with a mRNA cap site between bp 417 and 420.
Within the genomic sequence reported here two additional ATG codons located at bp 404–406 and 443–445 have been identified. Both of these potential translational start sites maintain the open reading fame present in the cDNA and the carboxyl terminus of the smooth muscle MLCK. One of these located at bp 404–406 occurs between the predicted TATA box and mRNA start site at bp 417–426. The second ATG codon occurs after the predicted transcriptional start site and would be located 43–45 bp from the 5′ end of the mRNA. The close proximity to the 5′ end of the mRNA together with it having a weak similarity to a consensus translational initiation site make it unlikely that this ATG is used as a start site (Kozak, 1989).
Characterization of Anti-telokin Antibodies
Fig. 3 shows the expression and subsequent purification of telokin from bacteria. The purified protein migrates aberrantly on SDS-polyacrylamide gel electrophoresis having an apparent molecular mass of 22 kDa although its calculated mass is 17 kDa. A similar phenomenon was observed with telokin purified from turkey gizzard (Ito et al., 1989). Polyclonal antibodies raised against the bacterial expressed telokin have been shown to recognize a single, high molecular weight, immunoreactive protein of 152 kDa (smooth muscle MLCK) which is present both in rabbit uterine tissue as well as COS cells which have been transfected with pCMV5-SMMLCK, an expression vector for the full-length rabbit uterine smooth muscle MLCK (Gallagher et al., 1991)(Fig. 3). Immunoblot analysis of low molecular weight proteins (Fig. 3) has demonstrated that the antisera detects a single protein of 22 kDa in rabbit uterine tissue. This protein co-migrates with the purified bacterial protein which was used to prepare the polyclonal antibodies. A second, 48-kDa protein detected only in rabbit tissues (Figs. 3 and 4) was also detected by preimmune serum (Fig. 4). Two small proteins of 22 and 20 kDa are detected in extracts of COS cells transfected with pCMV5-TEL. It is likely that these two proteins represent initiation from M1 and M3 within the sequence M1-A-M3-I-… (Fig. 1), and from the relative immunoreactivity of the two proteins it is apparent that initiation from M3 is favored. However, we cannot rule out the possibility that these two proteins are the result of premature termination. The 22-kDa recombinant protein detected in COS cells co-migrates with the protein detected in rabbit uterine tissues and the recombinant purified protein. No smaller molecular mass proteins are detected in extracts prepared from COS cells transfected by pCMV5-SMMLCK, suggesting that the two proteins that have been detected in COS cell extracts are not proteolytic products of the expressed, recombinant protein or endogenous proteins (Fig. 3).
Tissue-specific Expression of Telokin
Extracts of smooth and non-muscle tissues were analyzed by an immunoblot procedure with the telokin polyclonal antibodies. In all mammalian smooth muscle tissues which were tested, except the rabbit aorta, a single immunoreactive protein of 22 kDa was detected. Similarly, in chicken gizzard smooth muscle tissue a 26-kDa protein was detected. No telokin protein was detected in any of the non-muscle tissues that were tested (Fig. 4). The amounts of total protein which have been analyzed on the immunoblot in Fig. 4 represent amounts that are between one-half to the equivalent amount required to detect MLCK (Gallagher et al., 1991) in rabbit tissues and 2–20 times the amount required to detect MLCK in chicken and bovine tissues. No telokin has been detected in rabbit aorta, kidney, or adrenal tissues, chicken liver, or bovine adrenal tissue, even when 3 times the amount of total protein represented in Fig. 4 has been immunoblotted. In all of the smooth and non-muscle tissue preparations the presence of unproteolyzed MLCK has been verified by immunoblotting, thus making it unlikely that these small molecular mass proteins are a proteolytic product of the MLCK detected in these tissues.
It is interesting that the telokin detected in chicken tissues has a greater molecular mass (24 kDa) on SDS-polyacrylamide gel electrophoresis than telokin detected in rabbit and bovine tissues (22 kDa). A comparison of the sequences of the rabbit telokin (Gallagher et al., 1991) and chicken gizzard telokin (Ito et al., 1989; Olson et al., 1990) predicts that the telokin protein present in avian tissues will have two additional amino acids. The avian and mammalian telokin proteins are 84% identical, and it is possible that differences in amino acid composition and number might account for the greater apparent molecular mass of the avian telokin protein, even though the calculated masses of each of these proteins is nearly identical (17 kDa).
RNA Analysis
Northern blots of RNA prepared from various smooth muscle tissues were probed with an antisense RNA corresponding to the telokin cDNA. Under stringent hybridization conditions two mRNAs are detected by this probe, a 5.8-kb mRNA encoding MLCK and a 2.6-kb mRNA encoding telokin (Fig. 5). In similar experiments using both total RNA (30 μg) or poly(A+) RNA (10 μg) prepared from non-muscle tissues (kidney, adrenal, liver) we have been unable to detect any mRNAs that hybridize to this probe. Probes corresponding to a region within the amino terminus of MLCK do not detect the smaller 2.6-kb mRNA (Gallagher et al., 1991).
RNase protection analysis was used as a more sensitive method for detecting mRNA for telokin and MLCK in various rabbit smooth and non-muscle tissues. An antisense RNA probe (766 bp) containing the entire telokin cDNA was prepared. RNA isolated from rabbit uterus, aorta, ileum, kidney, and liver was hybridized in solution to the antisense probe, and the unprotected regions of the probe were digested with RNase (Little and Jackson, 1987). As the telokin cDNA has an addition 108 bp of unique 5′ sequence, this analysis has allowed the detection of telokin mRNA as well as RNA corresponding to MLCK. The size of the probe protected by telokin and MLCK mRNAs is predicted to be 652 and 544 bp, respectively. In rabbit uterus and ileum both a 652- and a 544-bp fragment are detected. However, in rabbit aorta, kidney, and liver, the 544-bp MLCK fragment but no 652-bp telokin fragment is detected, even after extended exposures of the autoradiogram. These results suggest that the mRNA for telokin is not present in aorta, kidney, or liver and are, therefore, consistent with the immunoblotting experiments.
DISCUSSION
This report describes the isolation and characterization of a cDNA encoding the mammalian homolog of telokin. The cDNA corresponds to a 2.6-kb mRNA present in rabbit uterus and other smooth muscle tissues. The nucleotide and deduced amino acid sequence of this cDNA suggest that the protein that it encodes is identical to the 155 carboxyl-terminal amino acids of the rabbit uterine smooth muscle MLCK from residue M993 to residue E1147 (Gallagher et al., 1991). The presence of unique sequence within the 5′-noncoding region of the telokin cDNA clone is strong evidence that this cDNA clone corresponds to an independent mRNA. Together these data substantiate the finding that the carboxyl terminus of MLCK is expressed as an independent protein called telokin and that telokin is not a proteolytic fragment of MLCK.
There are three possible mechanisms by which the mRNA encoding telokin could be produced: it may be transcribed from an independent gene, from an independent promoter within the MLCK gene, or it may be generated by alternative splicing of the MLCK mRNA. The large distance of the first exon of telokin from the MLCK promoter, together with the tentative identification of a promoter immediately 5′ of the first exon of telokin, make it unlikely that the telokin mRNA is an alternatively spliced product of the MLCK mRNA. The genomic clone characterized in this study does not overlap a clone containing the promoter region of the MLCK gene (Gallagher et al, 1991) and does not extend into the exon encoding the amino-terminal half of the calmodulin binding domain (Fig. 7); thus, we cannot definitively determine whether these two genomic clones represent independent genes or fragments of the same gene. It is possible that the two clones represent two distinct genes that have arisen via a gene duplication and/or fusion event. However, 54 bp within the 5′-untranslated region of the telokin mRNA correspond identically to sequence within the coding region of the smooth muscle MLCK. It is unlikely that such strong sequence conservation would occur within the 5′-untranslated region of telokin if it were encoded by an independent gene. Thus, it is most likely that the 2.6-kb telokin mRNA arises from an independent promoter tentatively identified within an intron that interrupts the calmodulin binding domain of the smooth muscle MLCK (Fig. 7). In support of this model preliminary reports describing the chicken smooth muscle MLCK gene (Collinge et al., 1991; Shattuck et al., 1988) have also suggested that a second promoter is located within an intron that interrupts the calmodulin binding domain of the MLCK gene. Although the location of an additional promoter within the 3′ end of a gene is unusual, it may not be unique to the smooth muscle MLCK gene. Recently Means et al. (1991) have reported that calspermin, a small, acidic protein, represents the independent expression of the carboxyl terminus of the calcium/calmodulin-dependent protein kinase IV. It has not yet been determined if the expression of calspermin occurs via alternative splicing or a second promoter located within the kinase gene, but it may be significant that this protein kinase is also regulated by calmodulin. Further studies will be required to confirm this model and to demonstrate the functionality of the telokin promoter.
The experimental data presented in this report suggest that the ATG located at bp 162–164 (M1 in the sequence M1-A-M3-I..) of the cDNA is the translational start site for telokin. This is not in agreement with the previous studies of Ito et al. (1989), which suggest that M3 is the first methionine of the purified protein. This proposal was based upon the finding that the amino terminus of purified telokin was blocked and that there were 4 mol of methionine in the purified protein. If this is correct, then the penultimate residue in the mature telokin protein would be a modified isoleucine. However, it has been shown that the presence of an isoleucine in this position would inhibit co-translational modification (Arfin and Bradshaw, 1988). The most convincing evidence that the ATG at bp 162–164 is the start site is deduced from immunoblot analysis of recombinant and tissue forms of telokin. Polyclonal antibodies directed against telokin detect two small molecular mass proteins of 22 and 20 kDa in extracts prepared from COS cells transfected with the telokin cDNA. These two proteins presumably result from initiation at either M1 or M3 in the sequence (M1-A-M3-I-..). The larger 22 kDa form co-migrates with the purified telokin and the protein present in rabbit uterine tissue. Thus, assuming the initiating M has been removed during co-translational processing of all forms of the telokin, then the 22-kDa protein detected in COS cells likely represents expression from M1 of the telokin cDNA. Inspection of the gene and cDNA sequence have shown that there are two other in-frame ATG codons which could potentially encode a 22–24-kDa protein. They are located either within or very near to the proposed 2.6-kb mRNA start site, and neither ATG is a good consensus translational start site (Kozak, 1989). It is therefore unlikely that either sequence would efficiently initiate translation. However, additional studies will be required to resolve this issue.
Telokin expression was not detected in any non-muscle tissue examined. In contrast, we have shown previously that MLCK is expressed in both smooth muscle and non-muscle tissues (Gallagher et al, 1991). Telokin protein or mRNA was also not detected in aortic smooth muscle. This finding suggests that the expression of telokin is not only differentially regulated between smooth muscle and non-muscle tissues but also within different smooth muscle tissues. Thus, we conclude that the expression of telokin and MLCK is independently regulated in a tissue-specific manner. This supports and extends previous findings which demonstrated steroid induction of telokin mRNA but not MLCK mRNA in chicken oviduct (Russo et al., 1987).
The function of the carboxyl-terminal region of the smooth muscle MLCK or telokin is at present unknown. The presence of potential phosphorylation sites within telokin raises the possibility that its function may be modulated by phosphorylation. The differential distribution of telokin in smooth and non-muscle tissues, in contrast to the ubiquitous distribution of MLCK, together with previous reports describing steroid induction of the telokin, but not MLCK mRNA (Russo et al., 1987), suggests that the regulation of the relative abundance of the two proteins may play an important physiological role in their function. As removal of the carboxyl terminus of the kinase by partial proteolysis has no marked effect on enzyme catalysis in vitro (Ikebe et al., 1987), it is possible that the function of this region is only manifest in vivo. For example, this region may be important for intracellular localization or protein binding. If this region functions to localize the kinase to a particular region or protein within the cell, it would be reasonable to propose that telokin may regulate this binding by competition. This interaction may also be regulated by the phosphorylation state of telokin; thus, the relative concentrations of MLCK and telokin and the phosphorylation state of telokin may regulate the localization of the kinase.
Acknowledgments
We wish to thank Suzy Griffin for her expert technical assistance and Phyllis Foley for assistance in preparing this manuscript. We also wish to thank Jim Stull for his generous support of this project and for his helpful suggestions.
This work was supported in part by Biomedical Research Support Grant (BSRG 2 S07 RR 07175) (to P. J. G) and by National Institutes of Health Grant HL26043.
Footnotes
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EMBL Data Bank with accession number(s) M76181 and M76234.
The abbreviations used are: MLCK, myosin light chain kinase; SDS, sodium dodecyl sulfate; bp, base pair(s); kb, kilobase(s); PMSF, phenylmethylsulfonyl fluoride; APMSF, amidinophenylmethylsulfonyl fluoride; TLCK, Nα-p-tosyl-L-lysine chloromethyl ketone; TPCK, tosylphenylalanyl chloromethyl ketone; MOPS, 4-morpholine propanesulfonic acid; CAPS, 3-[cyclohexylamino]-1-propanesulfonic acid.
REFERENCES
- Adelstein RS, Conti MA, Daniel JL, Anderson W., Jr. Biochemistry and Pharmacology of Platelets. Elsevier Science Publishing Co.; North Holland, The Netherlands: 1975. pp. 101–119. [Google Scholar]
- Adelstein RS, Conti MA, Hathaway DR, Klee CB. J. Biol. Chem. 1978;253:8347–8350. [PubMed] [Google Scholar]
- Andersson S, Davis DL, Dahlbäch H, Jörnvall H, Russell DW. J. Biol. Chem. 1989;264:8222–8229. [PubMed] [Google Scholar]
- Arfin SM, Bradshaw RA. Biochemistry. 1988;27:7979–7984. doi: 10.1021/bi00421a001. [DOI] [PubMed] [Google Scholar]
- Bagchi IC, Kemp BE, Means AR. J. Biol. Chem. 1989;264:15843–15849. [PubMed] [Google Scholar]
- Bissonnette M, Kuhn D, de Lanerolle P. Biochem. J. 1989;258:739–747. doi: 10.1042/bj2580739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cande WZ, Ezzell RM. Cell Motil. Cytoskeleton. 1986;6:640–648. doi: 10.1002/cm.970060612. [DOI] [PubMed] [Google Scholar]
- Collinge M, Matrisian P, Van Eldik LJ, Watterson DM. FASEB J. 1991;5:802. abstr. [Google Scholar]
- Conti MA, Adelstein RS. J. Biol. Chem. 1981;256:3178–3181. [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Griffin SA, Stull JT. J. Biol. Chem. 1991;266:23936–23944. [PMC free article] [PubMed] [Google Scholar]
- Goldfine SM, Schroter EH, Izzard CS. J. Cell Sci. 1981;50:391–405. doi: 10.1242/jcs.50.1.391. [DOI] [PubMed] [Google Scholar]
- Guerriero V, Jr., Russo MA, Olson NJ, Putkey JA, Means AR. Biochemistry. 1986;25:8372–8381. doi: 10.1021/bi00374a007. [DOI] [PubMed] [Google Scholar]
- Hai C-M, Murphy RA. Annu. Rev. Physiol. 1989;51:285–298. doi: 10.1146/annurev.ph.51.030189.001441. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Soderling TR. Arch. Biochem. Biophys. 1990;278:41–45. doi: 10.1016/0003-9861(90)90228-q. [DOI] [PubMed] [Google Scholar]
- Herring BP, Stull JT, Gallagher PJ. J. Biol. Chem. 1990;265:1724–1730. [PMC free article] [PubMed] [Google Scholar]
- Holzapfel G, Wehland J, Weber K. Exp. Cell Res. 1983;148:117–126. doi: 10.1016/0014-4827(83)90192-1. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Reardon S. J. Biol. Chem. 1990;265:8975–8978. [PubMed] [Google Scholar]
- Ikebe M, Inagaki M, Kanamaru K, Hidaka H. J. Biol. Chem. 1985;260:4547–4550. [PubMed] [Google Scholar]
- Ikebe M, Stepinska M, Kemp BE, Means AR, Hartshorne DJ. J. Biol. Chem. 1987;262:13828–13834. [PubMed] [Google Scholar]
- Ito M, Dabrowska R, Guerriero V, Jr., Hartshorne DJ. J. Biol. Chem. 1989;264:13971–13974. [PubMed] [Google Scholar]
- Ito M, Guerriero V, Jr., Chen X, Hartshorne DJ. Biochemistry. 1991;30:3498–3503. doi: 10.1021/bi00228a021. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Annu. Rev. Pharmacol. Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- Kozak M. J. Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little PFR, Jackson IJ. In: DNA Cloning: A Practical Approach. Glover DM, editor. IRL; New York: 1987. pp. 1–18. [Google Scholar]
- Masuda H, Owaribe K, Hayashi H, Hatano S. Cell Motil. 1984;4:315–331. doi: 10.1002/cm.970040503. [DOI] [PubMed] [Google Scholar]
- Means AR, Cruzalegui F, LeMagueresse B, Needleman DS, Slaughter GR, Ono T. Mol. Cell. Biol. 1991;11:3960–3971. doi: 10.1128/mcb.11.8.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierendorf RC, Pfeffer D. Methods Enzymol. 1987;152:556–562. doi: 10.1016/0076-6879(87)52061-4. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Sellers JR, Adelstein RS, Hidaka H. J. Biol. Chem. 1984;259:8808–8814. [PubMed] [Google Scholar]
- Nishikawa M, Shirakawa S, Adelstein RS. J. Biol. Chem. 1985;260:8978–8983. [PubMed] [Google Scholar]
- Olson NJ, Pearson RB, Needleman DS, Hurwitz MY, Kemp BE, Means AR. Proc. Natl. Acad. Sci. U. S. A. 1990;87:2284–2288. doi: 10.1073/pnas.87.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne ME, Elzinga M, Adelstein RS. J. Biol. Chem. 1986;261:16346–16350. [PubMed] [Google Scholar]
- Roush CL, Kennelly PJ, Glaccum MB, Helfman DM, Scott JD, Krebs EG. J. Biol. Chem. 1988;263:10510–10516. [PubMed] [Google Scholar]
- Russo MA, Guerriero V, Jr., Means AR. Mol. Endocrinol. 1987;1:60–67. doi: 10.1210/mend-1-1-60. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. rev. ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. p. 7. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. Proc. Natl. Acad. Sci. U. S. A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattuck RL, Zimmer WE, Lukas TJ. J. Cell Biol. 1988;107:747. abstr. [Google Scholar]
- Studier FW, Moffatt BA. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Stull JT, Hsu L-C, Tansey MG, Kamm KE. J. Biol. Chem. 1990;265:16683–16690. [PubMed] [Google Scholar]
- Wysolmerski RB, Lagunoff D. Proc. Natl. Acad. Sci. U. S. A. 1990;87:16–20. doi: 10.1073/pnas.87.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller MJ, Smith M. Methods Enzymol. 1987;154:329–351. doi: 10.1016/0076-6879(87)54083-6. [DOI] [PubMed] [Google Scholar]