

Abstract
Insulin is used to control pro-inflammatory hyperglycemia in critically ill patients. However, recent studies suggest that insulin-induced hypoglycemia may negate its beneficial effects in these patients. It is noteworthy that recent evidence indicates that insulin has anti-inflammatory effects that are independent of controlling hyperglycemia. To date, the mechanism by which insulin directly reduces inflammation has not been elucidated. It is well established that insulin activates phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling in many cell types. We and others have shown that this pathway negatively regulates LPS-induced signaling and pro-inflammatory cytokine production in monocytic cells. We hypothesized that insulin inhibits inflammation during endotoxemia by activation of the PI3K/Akt pathway. We used a nonhyperglycemic mouse model of endotoxemia to determine the effect of continuous administration of a low dose of human insulin on inflammation and survival. It is noteworthy that insulin treatment induced phosphorylation of Akt in muscle and adipose tissues but did not exacerbate lipopolysaccharide (LPS)-induced hypoglycemia. Insulin decreased plasma levels of interleukin-6, tumor necrosis factor-α, monocyte chemotactic protein 1 (MCP1)/JE, and keratinocyte chemoattractant, and decreased mortality. The PI3K inhibitor wortmannin abolished the insulin-mediated activation of Akt and the reduction of chemokine and interleukin-6 levels. We conclude that insulin reduces LPS-induced inflammation in mice in a PI3K/Akt-dependent manner without affecting blood glucose levels.
During Gram-negative sepsis, lipopolysaccharide (LPS) induces the expression of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6), and chemokines, such as monocyte chemotactic protein 1 (MCP1)/JE and keratinocyte chemoattractant (KC) (Casey et al., 1993). However, over-reaction of the pro-inflammatory response to infection and injury can contribute to the development of sepsis, septic shock, multiple organ failure, and death. Hyperglycemia and insulin resistance followed by hypoglycemia and hypoinsulinemia can also occur during sepsis as a consequence of the metabolic effects of stress hormone and cytokine production (Maitra et al., 2000; Marik and Raghavan, 2004; Van den Berghe, 2004; van Waardenburg et al., 2006). Despite considerable progress in our understanding of the pathologic pathways that contribute to sepsis and septic shock, pharmacologic interventions are currently limited to insulin and activated protein C (Shapiro et al., 2006). Insulin is administered to septic patients with hyperglycemia to normalize glucose levels (Russell, 2006; Shapiro et al., 2006). Reducing glucose levels with insulin therapy is associated with decreased inflammation and endothelial cell damage (van den Berghe et al., 2001, 2006; Van den Berghe, 2004; Marik and Raghavan, 2004; Langouche et al., 2005). Recently, however, it has been shown that insulin-induced hypoglycemia may counteract the beneficial effects of aggressive insulin therapy in patients with severe sepsis (Brunkhorst et al., 2008).
Currently, insulin is not initiated in normoglycemic septic patients or to correct hypoinsulinemia during sepsis (Mitchell et al., 2006). However, results from animal studies indicate that insulin may have direct anti-inflammatory effects. In nonhyperglycemic animal models of endotoxemia, continuous infusion of insulin while maintaining blood glucose in the normal range with dextrose or administering a bolus injection of insulin results in decreased inflammation and morbidity (Brix-Christensen et al., 2004; Jeschke et al., 2004, 2005). However, the mechanism by which insulin reduces inflammation in the absence of hyperglycemia is unknown. One possibility is that insulin reduces inflammation by activating anti-inflammatory signaling pathways, such as the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway. It is well established that insulin activates PI3K/Akt signaling in many cell types (Taniguchi et al., 2006). It is noteworthy that we and others (Guha and Mackman, 2002; Schabbauer et al., 2004; Liew et al., 2005; Martin et al., 2005) have shown that this pathway negatively regulates LPS-induced signaling and pro-inflammatory cytokine production in monocytic cells. Furthermore, activation of PI3K enhanced survival, whereas inhibition of PI3K reduced survival of endotoxemic mice (Schabbauer et al., 2004; Williams et al., 2004). Taken together, these studies suggest that the protective effects of insulin in endotoxemia models may be mediated by activation of the PI3K/Akt pathway.
In this study, we used a pharmacological inhibitor of PI3K to test the hypothesis that exogenous insulin decreases inflammation in endotoxemic mice by activating the PI3K/Akt pathway and that this anti-inflammatory effect is independent of its affect on blood glucose levels.
Materials and Methods
Mice
All studies were approved by The Scripps Research Institute Animal Care and Use Committee and comply with Institute of Laboratory Animal Resources (1996). C57BL/6J male mice of 6 to 16 weeks of age were used for all experiments.
Mouse Endotoxemia Model and Insulin Administration
We used a mouse model of endotoxemia consisting of an intraperitoneal injection of LPS (Escherichia coli serotype O111:B4; Sigma-Aldrich, St Louis, MO). Doses of 5 or 10 mg/kg were used in experiments. These doses corresponded to the LD50 for two different lots of LPS. Accordingly, LPS produced similar levels of cytokine expression and lethality in each experiment. Human insulin (Humulin 70/30; Eli Lilly, Indianapolis, IN) was administered to mice either as a bolus intraperitoneal injection or via Alzet micro-osmotic pumps (model 1003D; Durect Corp, Cupertino, CA) that were implanted subcutaneously 16 h before administration of LPS. These pumps deliver 1 μl/h for 72 h. Human insulin was used to allow us to measure low levels of exogenous insulin and to distinguish it from endogenous mouse insulin in the endotoxemic mice. Furthermore, human insulin has been used previously in studies examining the effect of acute insulin treatment in endotoxemic rodents (Jeschke et al., 2004). To inhibit PI3K activity, mice were given wortmannin (Sigma-Aldrich) at a dose of 0.06 mg/kg or its vehicle [Ringer’s solution containing 10% (v/v) dimethyl sulfoxide] three times by retro-orbital injection at −90, +90, and + 360 min relative to LPS administration. Blood samples were collected from the retro-orbital sinus (final concentration 0.32%) at various times after LPS administration (0–24 h). Plasma was collected and stored at −80°C until analysis.
Measurement of Glucose, Insulin, and Cytokine Levels
Glucose levels in the plasma were determined using a Glucometer Elite XL (Bayer Corporation, Elkhart, IN). Human insulin levels in the plasma were determined using a commercial ELISA that detects only human insulin (10-1132-01; Mercodia, Uppsala, Sweden). Total insulin (mouse + human) levels in the plasma were determined using a different commercial ELISA kit (10–1150–01; Mercodia). The levels of TNF-α, IL-6, JE, and KC in plasma were determined using commercial ELISA kits (R&D Systems, Minneapolis, MN).
Western Blotting
Muscle and epididymal adipose tissue were collected and quick-frozen in liquid nitrogen and stored at −80°C until processed. Tissues were homogenized in 1.5 ml of a buffer [10 mM HEPES, 10 mM KCl, 300 mM sucrose, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM PMSF, half of a tablet, and complete EDTA-free pro-tease inhibitor cocktail (Roche, Indianapolis, IN)], spun at 550g for 2 min at 4°C, and supernatants were frozen. Protein concentration was measured using a colorimetric assay according to manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA). Proteins were separated by SDS-polyacrylamide gel electrophoresis and then transferred to Millipore polyvinylidene fluoride membranes (Millipore, Bedford, MA). Levels of phosphorylated (Ser473) and nonphosphorylated Akt were detected using primary rabbit antibodies (Cell Signaling, Beverly, MA) and a secondary antibody conjugated to horseradish peroxidase. Antibodies were developed using a chemiluminescence reagent assay (Super Signal West Femto; Pierce, Rockford, IL), according to manufacturer’s instructions, and exposed to radiographic film (Kodak, Rochester, NY).
Statistical Analysis
Data were analyzed by one-way analysis of variance with Tukey’s test for multiple comparisons or by Student’s t test. Non-normal data were analyzed by the Mann-Whitney rank sum or the Kruskal-Wallis one way analysis of variance on ranks and Dunn’s method of multiple comparisons. Survival data were analyzed using the log-rank test. The criterion for significance for all studies was p < 0.05.
Results
Effect of Exogenous Insulin on Plasma Insulin and Glucose Levels in Untreated and LPS-Treated Mice
Two different bolus doses of human insulin were used to determine a dose that did not affect plasma glucose levels in fasted male mice. A 1 U/kg dose of human insulin (Humulin 70/30) administered intraperitoneally to mice significantly reduced plasma glucose levels, whereas a 0.1 U/kg dose of insulin had no effect (Table 1). Therefore, the 0.1 U/kg dose of human insulin was selected as the dose for all further studies. We used micro-osmotic infusion pumps to continuously infuse insulin throughout the study period. These pumps delivered insulin at a rate of 2.5 mU/h/mouse (approximately 0.1 U/kg/h) subcutaneously for 72 h. Low levels of human insulin were detected in insulin-treated mice but not in saline-treated controls, using an ELISA specific for human insulin (Fig. 1A). Human insulin levels were maintained in insulin-treated mice at a constant level for the first 8 h of endotoxemia. Total insulin (mouse + human) was measured using a second ELISA. There was no significant difference in total insulin levels between insulin-treated and saline-treated mice during endotoxemia (Fig. 1B). It is noteworthy that insulin treatment did not exacerbate LPS-induced hypoglycemia (Fig. 1C).
TABLE 1.
Establishment of a dose of insulin that does not affect blood glucose
| Insulin Dose | 0.1 U/kg | 1.0 U/kg | ||||||
|---|---|---|---|---|---|---|---|---|
| Time after insulin (min) | 0 | 20 | 60 | 120 | 0 | 20 | 60 | 120 |
| Plasma glucose (mg/dl) | 99.8 | 83.0 | 94.3 | 95.3 | 99.8 | 69.7 | 54.3* | 79.0 |
| Mean ± S.E.M. | ± | ± | ± | ± | ± | ± | ± | ± |
| n = 3–6 mice/group | 10.8 | 13.3 | 6.9 | 7.1 | 10.8 | 8.0 | 9.0 | 14.5 |
Significantly different from time zero, P < 0.05.
Fig. 1.
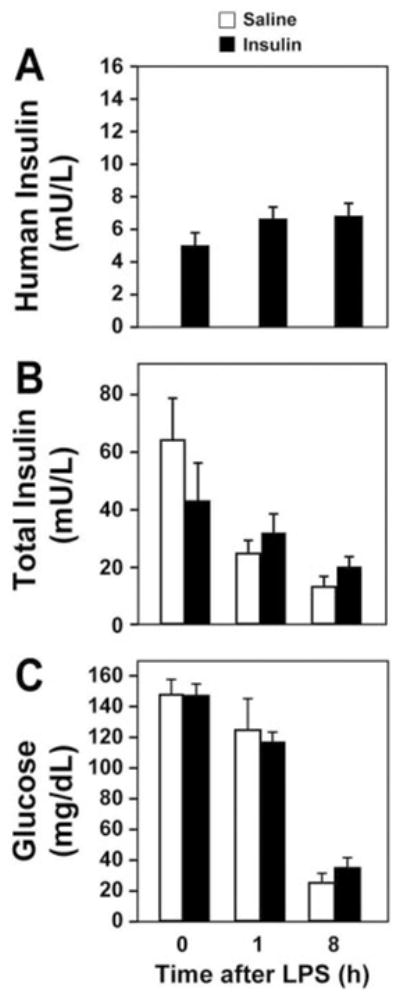
Measurement of human insulin, total insulin, and glucose in normal and endotoxemic mice. Plasma levels of human insulin (n = 12 per group) (A), total insulin (mouse + human) (n = 14 mice per group) (B), and plasma glucose (n = 20 mice per group) (C) were measured by ELISA (insulin) or glucometer (glucose) at various times (0–8 h) after LPS. Results are displayed as mean ± S.E.M.
Exogenous Insulin Decreases Inflammation and Mortality in Endotoxemic Mice
Insulin treatment significantly reduced plasma levels of TNF-α, IL-6, JE, and KC and improved the survival of endotoxemic mice compared with mice treated with saline (Fig. 2, A–F). It is interesting that we observed a negative correlation between plasma levels of human insulin and IL-6 (data not shown).
Fig. 2.
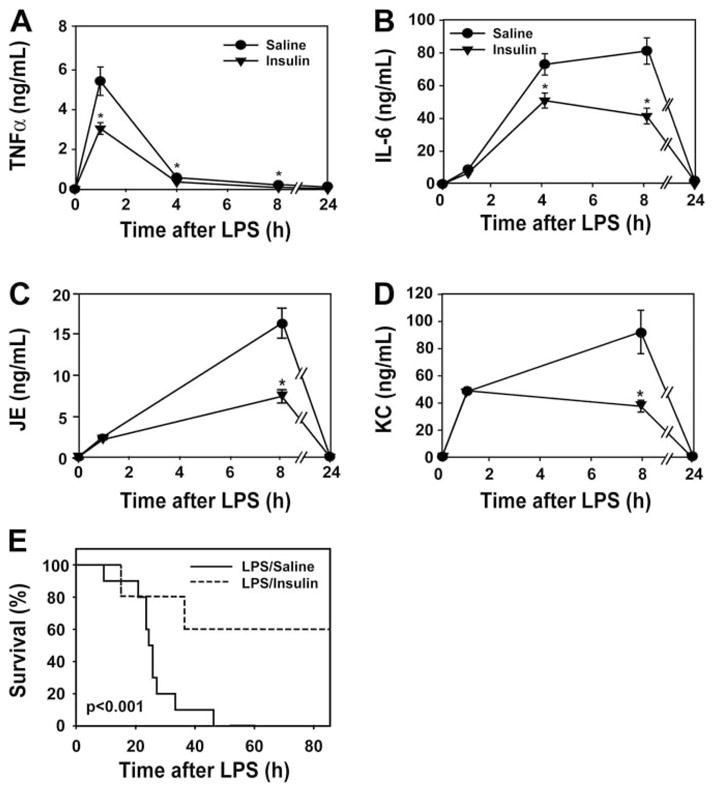
Administration of human insulin decreases inflammation and mortality in endotoxemic mice. Human insulin (2.5 mU/μl/h) or saline (1 μl/h) was administered continuously to mice using a subcutaneously implanted pump. Plasma levels of TNF-α (n = 6–10 mice per group) (A), IL-6 (n = 10–12 mice per group) (B), JE (n = 5–6) (C), and KC (n = 5–7) (D) were measured by ELISA before and 1, 8, and 24 h after LPS. Results are shown as mean ± S.E.M. E, survival was evaluated for 4 days. Results are presented as a Kaplan-Meier plot (n = 10 mice/group). *, p < 0.05.
Exogenous Insulin Activates Akt in Insulin-Sensitive Tissues in Endotoxemic Mice
We hypothesized that the protective effects of insulin during endotoxemia are mediated by activation of PI3K/Akt. We chose to analyze the activation of Akt in muscle and adipose tissue because glucose transport in theses tissues is regulated by insulin, and they express inflammatory mediators during endotoxemia (Chang et al., 2004; Brix-Christensen et al., 2005; Bultinck et al., 2006). It is noteworthy that Akt phosphorylation was increased in the muscle and maintained in adipose tissue of insulin-treated endotoxemic mice (Fig. 3, A and B).
Fig. 3.
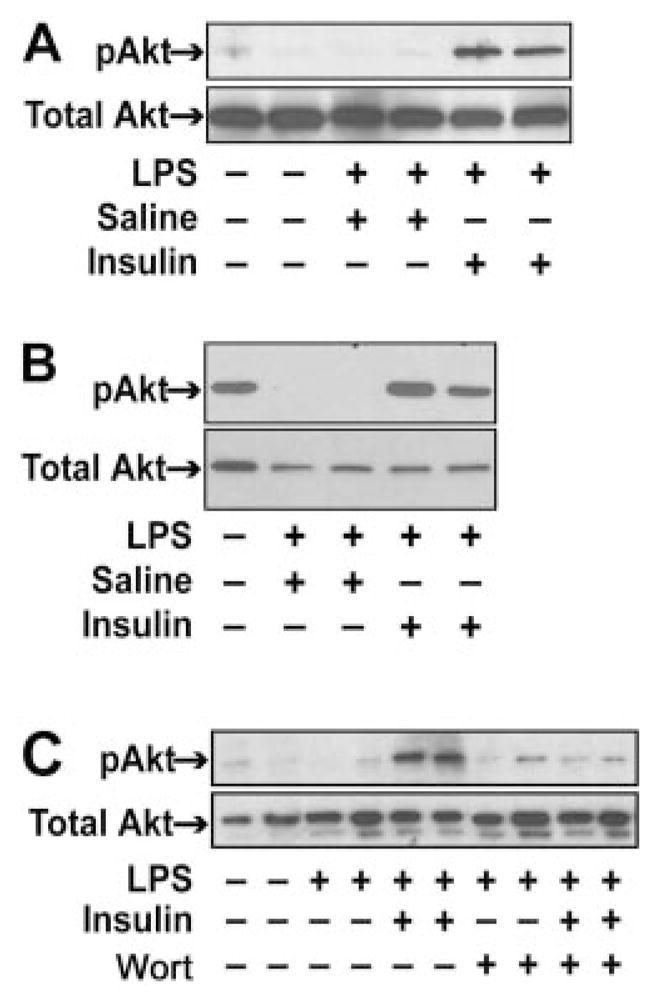
Effect of human insulin and wortmannin on Akt phosphorylation in muscle and adipose tissue in endotoxemic mice. Human insulin (2.5 mU/μl/h) or saline (1 μl/h) was administered continuously to mice using a subcutaneously implanted pump before treating the mice with LPS with or without wortmannin. The left quadriceps muscle and adipose tissue were harvested 8 h after administration of LPS. Nonfasted, untreated mice served as controls. Levels of phosphorylated Akt and total Akt were detected by Western blotting in muscle (A and C) and adipose tissue (B). Representative data from one to three independent mice in each group is shown.
Effect of Wortmannin on the Anti-Inflammatory Activity of Insulin
To investigate the role of the PI3K/Akt pathway in the anti-inflammatory effects of insulin during endotoxemia, we treated mice with the PI3K inhibitor wortmannin. The dose of wortmannin used in these experiments did not affect plasma glucose levels (data not shown) but abolished insulin-dependent Akt phosphorylation in the muscle of endotoxemic mice (Fig. 3C). Wortmannin also abolished the insulin-dependent decrease in IL-6, JE, and KC levels at 8 h (Fig. 4, A–C). Interestingly, wortmannin increased KC expression during endotoxemia (Fig. 4B). However, wortmannin did not inhibit the insulin-dependent reduction in TNF-α levels at 1.5 h (Fig. 4D).
Fig. 4.
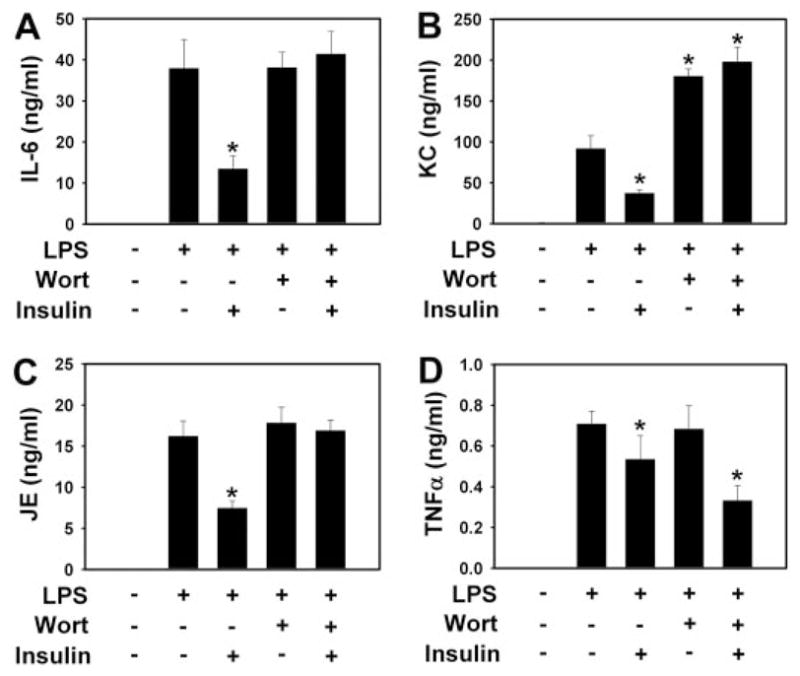
Effect of wortmannin on the anti-inflammatory activity of insulin in endotoxemic mice. Human insulin (2.5 mU/μl/h) or saline (1 μl/h) was continuously administered to mice using subcutaneously implanted pumps. Mice were treated with LPS and/or wortmannin. Plasma levels of IL-6 (A), JE (B), KC (8 h after LPS) (C), and TNF-α (1.5 h) (D) were measured in the four groups. Results are displayed as the mean ± S.E.M. (n = 5–12 mice per group). *, p < 0.05. Wort, wortmannin.
Discussion
In this study, we analyzed the mechanism by which insulin reduces LPS-induced inflammation in mice. We used a non-hyperglycemic model of endotoxemia and continuously infused insulin at a dose and rate that did not worsen LPS-induced hypoglycemia. We showed that a low dose of human insulin decreased levels of TNF-α, IL-6, JE, and KC and decreased mortality in endotoxemic mice without exacerbating hypoglycemia. We then used a pharmacologic approach to analyze the role of PI3K/Akt signaling in the anti-inflammatory effects of insulin. The low dose of human insulin used in this study enhanced phosphorylation of Akt in muscle and adipose tissue during the hypoglycemic, hypoinsulinemic phase of endotoxemia. Wortmannin abolished the insulin-induced phosphorylation of Akt in these mice indicating inhibition of PI3K. It is noteworthy that inhibition of insulin-mediated activation of PI3K/Akt with wortmannin reversed the insulin-dependent reduction in LPS-induced IL-6, JE, and KC expression. These data indicate that insulin activation of the PI3K/Akt pathway inhibits the production of these cytokines in endotoxemic mice. However, because wortmannin inhibits other cell signaling pathways, we cannot exclude the possibility that additional mechanisms also contribute to the anti-inflammatory effects of insulin in these mice (Ding et al., 1995).
It is interesting to note that insulin-dependent reduction in TNF-α production was not reversed by wortmannin. This observation may, in part, relate to the complexity of TNF-α release in endotoxemia. For example, previous studies have shown that the cleavage of the pro-form of TNF-α from the membrane by TNF-α-converting enzyme contributes significantly to plasma TNF-α levels in endotoxemic mice (Zhang et al., 2004). Further studies are required to determine whether insulin-mediated activation of PI3K plays a role in reducing TNF-α levels in endotoxemic mice.
Other studies indirectly support our observation that activation of PI3K/Akt signaling mediates many of the anti-inflammatory effects of insulin during endotoxemia. For instance, insulin has been shown to increase the expression of mitogen-activated protein kinase phosphatase (MKP-1) in a PI3K-dependent manner (Desbois-Mouthon et al., 2000; Takehara et al., 2000), and this phosphatase negatively regulates IL-6 and TNF-α production in endotoxemic mice (Chi et al., 2006). Furthermore, a recent study showed that insulin and a glycogen synthase kinase 3β inhibitor had similar anti-inflammatory effects in a rat model of sepsis (Dugo et al., 2006). It is noteworthy that Akt-dependent phosphorylation decreases the activity of glycogen synthase kinase 3β (Guha and Mackman, 2002; Martin et al., 2005). These studies, together with the findings reported here, support the notion that activation of the PI3K/Akt pathway mediates the protective effects of insulin during endotoxemia.
Both increased levels of IL-6 and hypoinsulinemia have been associated with increased morbidity and decreased survival during endotoxemia and sepsis (Ciancio et al., 1991; Casey et al., 1993; van Waardenburg et al., 2006). In this study, endogenous insulin levels decreased dramatically at a time when IL-6 levels were maximal. In insulin-treated mice, the maximal amount of human insulin in plasma 8 h after LPS was 8.9 mU/liter. However, as a consequence of LPS-induced hypoinsulinemia, this small amount of insulin constituted a large portion of total insulin levels 8 h after the administration of LPS. It is noteworthy that we observed a strong negative correlation between human insulin levels and plasma IL-6 levels in insulin-treated mice. These data suggest that small amounts of insulin can significantly affect the inflammatory response without affecting plasma glucose levels.
Glucose transport into muscle and adipose is partially dependent on insulin-mediated activation of the PI3K/Akt pathway. Insulin-mediated effects on glucose metabolism in the liver are also dependent on activation of this pathway. The dose of insulin used in this study increased levels of phosphorylated Akt in muscle and adipose tissue during endotoxemia. It is interesting that we did not observe increased levels of phosphorylated Akt in the liver (data not shown). Therefore, tissue-specific differences in the activation of the PI3K/Akt pathway may explain how a low dose of insulin can affect the inflammatory response without affecting glucose metabolism.
The cellular target(s) mediating the protective effects of insulin in this model remain to be determined. Insulin-dependent tissues, such as skeletal muscle and adipose tissue, contribute to IL-6 expression during endotoxemia (Brix-Christensen et al., 2005; Bultinck et al., 2006), suggesting that the anti-inflammatory effects of insulin on IL-6 production during endotoxemia may be the result of direct activation of PI3K in these tissues. Hematopoietic cells, such as monocytes and macrophages, have been shown to be the major source of cytokine production during endotoxemia (Michalek et al., 1980). Insulin may directly activate PI3K in monocytes and thereby reduce inflammatory cytokine production in response to LPS in vivo. Insulin signaling has not been studied extensively in macrophages and mononuclear cells. Although they do not require insulin for glucose transport, these cells express insulin receptors and insulin increases glucose transport (Daneman et al., 1992; Estrada et al., 1994; Welham et al., 1997). A recent study showed that high concentrations of insulin (104 μM) decreased TNF-α and IL-1β production and inhibited apoptosis in LPS-treated THP-1 cells in vitro (Leffler et al., 2007). The anti-apoptotic effects of insulin were mediated by activation of PI3K. However, this study did not determine whether the anti-inflammatory effects of insulin were PI3K-dependent.
Apoptosis of cells, particularly endothelial cells, may contribute to inflammation and organ damage during endotoxemia (Power et al., 2002; Bannerman and Goldblum, 2003). It is interesting that insulin inhibits apoptosis in a variety of cell types in a PI3K/Akt-dependent manner and may reduce endothelial apoptosis in endotoxemic mice (Hermann et al., 2000; Conejo and Lorenzo, 2001; Leffler et al., 2007). Further studies are required to determine the cellular targets of insulin during endotoxemia.
In summary, we demonstrated that continuous administration of small amounts of insulin decreased inflammation in endotoxemic mice in a PI3K-dependent manner without affecting blood glucose levels. These findings may ultimately have important implications regarding the use of insulin for treating normoglycemic, hypoinsulinemic patients with sepsis.
Acknowledgments
We thank Cheryl Johnson for preparing the manuscript and Dr. T. Combs for critical reading of the manuscript.
This work is supported by National Institutes of Health Grant HL48872 (to N.M.), National Institutes of Health Grant NSRA F32 HL085983 (to J.P.L.), Austrian Science Fund FWFP19850 (to G.S.), and National Institutes of Health Grant NRSA T32 5 P32 HL007195-30 (to L.K.).
ABBREVIATIONS
- LPS
lipopolysaccharide
- PI3K
phosphatidylinositol 3-kinase
- ELISA
enzyme-linked immunosorbent assay
- Akt
protein kinase B
- KC
keratinocyte chemoattractant
- TNF-α
tumor necrosis factor-α
- IL-6
interleukin-6
References
- Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L899–L914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- Brix-Christensen V, Andersen SK, Andersen R, Mengel A, Dyhr T, Andersen NT, Larsson A, Schmitz O, Orskov H, Tonnesen E. Acute hyperinsulinemia restrains endotoxin-induced systemic inflammatory response: an experimental study in a porcine model. Anesthesiology. 2004;100:861–870. doi: 10.1097/00000542-200404000-00016. [DOI] [PubMed] [Google Scholar]
- Brix-Christensen V, Gjedsted J, Andersen SK, Vestergaard C, Nielsen J, Rix T, Nyboe R, Andersen NT, Larsson A, Schmitz O, et al. Inflammatory response during hyperglycemia and hyperinsulinemia in a porcine endotoxemic model: the contribution of essential organs. Acta Anaesthesiol Scand. 2005;49:991–998. doi: 10.1111/j.1399-6576.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- Brunkhorst F, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, Moerer O, Gruendling M, Oppert M, Grond S, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125–139. doi: 10.1056/NEJMoa070716. [DOI] [PubMed] [Google Scholar]
- Bultinck J, Brouckaert P, Cauwels A. The in vivo contribution of hematopoietic cells to systemic TNF and IL-6 production during endotoxemia. Cytokine. 2006;36:160–166. doi: 10.1016/j.cyto.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Casey LC, Balk RA, Bone RC. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann Intern Med. 1993;119:771–778. doi: 10.7326/0003-4819-119-8-199310150-00001. [DOI] [PubMed] [Google Scholar]
- Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancio MJ, Hunt J, Jones SB, Filkins JP. Comparative and interactive in vivo effects of tumor necrosis factor alpha and endotoxin. Circ Shock. 1991;33:108–120. [PubMed] [Google Scholar]
- Conejo R, Lorenzo M. Insulin signaling leading to proliferation, survival, and membrane ruffling in C2C12 myoblasts. J Cell Physiol. 2001;187:96–108. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1058>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Daneman D, Zinman B, Elliott ME, Bilan PJ, Klip A. Insulin-stimulated glucose transport in circulating mononuclear cells from nondiabetic and IDDM subjects. Diabetes. 1992;41:227–234. doi: 10.2337/diab.41.2.227. [DOI] [PubMed] [Google Scholar]
- Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Caron M, Atfi A, Cherqui G, Capeau J. Insulin-mediated cell proliferation and survival involve inhibition of c-Jun N-terminal kinases through a phosphatidylinositol 3-kinase- and mitogen-activated protein kinase phosphatase-1-dependent pathway. Endocrinology. 2000;141:922–931. doi: 10.1210/endo.141.3.7390. [DOI] [PubMed] [Google Scholar]
- Ding J, Vlahos CJ, Liu R, Brown RF, Badwey JA. Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein kinases in neutrophils. J Biol Chem. 1995;270:11684–11691. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- Dugo L, Collin M, Allen DA, Murch O, Foster SJ, Yaqoob MM, Thiemermann C. Insulin reduces the multiple organ injury and dysfunction caused by coad-ministration of lipopolysaccharide and peptidoglycan independently of blood glucose: role of glycogen synthase kinase-3beta inhibition. Crit Care Med. 2006;34:1489–1496. doi: 10.1097/01.CCM.0000215457.83953.E3. [DOI] [PubMed] [Google Scholar]
- Estrada DE, Elliott E, Zinman B, Poon I, Liu Z, Klip A, Daneman D. Regulation of glucose transport and expression of GLUT3 transporters in human circulating mononuclear cells: studies in cells from insulin-dependent diabetic and nondiabetic individuals. Metabolism. 1994;43:591–598. doi: 10.1016/0026-0495(94)90201-1. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:402–409. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals. 7. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council; Washington DC: 1996. [Google Scholar]
- Jeschke MG, Klein D, Bolder U, Einspanier R. Insulin attenuates the systemic inflammatory response in endotoxemic rats. Endocrinology. 2004;145:4084–4093. doi: 10.1210/en.2004-0592. [DOI] [PubMed] [Google Scholar]
- Jeschke MG, Rensing H, Klein D, Schubert T, Mautes AE, Bolder U, Croner RS. Insulin prevents liver damage and preserves liver function in lipopolysaccharide-induced endotoxemic rats. J Hepatol. 2005;42:870–879. doi: 10.1016/j.jhep.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Langouche L, Vanhorebeek I, Vlasselaers D, Vander PS, Wouters PJ, Skogstrand K, Hansen TK, Van den BG. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest. 2005;115:2277–2286. doi: 10.1172/JCI25385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler M, Hrach T, Stuerzl M, Horch RE, Herndon DN, Jeschke MG. Insulin attenuates apoptosis and exerts anti-inflammatory effects in endotoxemic human macrophages. J Surg Res. 2007;143:398–406. doi: 10.1016/j.jss.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Maitra SR, Wojnar MM, Lang CH. Alterations in tissue glucose uptake during the hyperglycemic and hypoglycemic phases of sepsis. Shock. 2000;13:379–385. doi: 10.1097/00024382-200005000-00006. [DOI] [PubMed] [Google Scholar]
- Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004;30:748–756. doi: 10.1007/s00134-004-2167-y. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek SM, Moore RN, McGhee JR, Rosenstreich DL, Mergenhagen SE. The primary role of lymphoreticular cells in the mediation of host responses to bacterial endotoxim. J Infect Dis. 1980;141:55–63. doi: 10.1093/infdis/141.1.55. [DOI] [PubMed] [Google Scholar]
- Mitchell I, Knight E, Gissane J, Tamhane R, Kolli R, Leditschke IA, Bellomo R, Finfer S. A phase II randomised controlled trial of intensive insulin therapy in general intensive care patients. Crit Care Resusc. 2006;8:289–293. [PubMed] [Google Scholar]
- Power C, Fanning N, Redmond HP. Cellular apoptosis and organ injury in sepsis: a review. Shock. 2002;18:197–211. doi: 10.1097/00024382-200209000-00001. [DOI] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. N Engl J Med. 2006;19;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- Shapiro NI, Howell MD, Talmor D, Lahey D, Ngo L, Buras J, Wolfe RE, Weiss JW, Lisbon A. Implementation and outcomes of the Multiple Urgent Sepsis Therapies (MUST) protocol. Crit Care Med. 2006;34:1025–1032. doi: 10.1097/01.CCM.0000206104.18647.A8. [DOI] [PubMed] [Google Scholar]
- Takehara N, Kawabe J, Aizawa Y, Hasebe N, Kikuchi K. High glucose attenuates insulin-induced mitogen-activated protein kinase phosphatase-1 (MKP-1) expression in vascular smooth muscle cells. Biochim Biophys Acta. 2000;1497:244–252. doi: 10.1016/s0167-4889(00)00050-1. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Van den Berghe G. How does blood glucose control with insulin save lives in intensive care? J Clin Invest. 2004;114:1187–1195. doi: 10.1172/JCI23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- van Waardenburg DA, Jansen TC, Vos GD, Buurman WA. Hyperglycemia in children with meningococcal sepsis and septic shock: the relation between plasma levels of insulin and inflammatory mediators. J Clin Endocrinol Metab. 2006;91:3916–3921. doi: 10.1210/jc.2006-0525. [DOI] [PubMed] [Google Scholar]
- Welham MJ, Bone H, Levings M, Learmonth L, Wang LM, Leslie KB, Pierce JH, Schrader JW. Insulin receptor substrate-2 is the major 170-kDa protein phosphorylated on tyrosine in response to cytokines in murine lymphohematopoietic cells. J Biol Chem. 1997;272:1377–1381. doi: 10.1074/jbc.272.2.1377. [DOI] [PubMed] [Google Scholar]
- Williams DL, Li C, Ha T, Ozment-Skelton T, Kalbfleisch JH, Preiszner J, Brooks L, Breuel K, Schweitzer JB. Modulation of the phosphoinositide 3-kinase pathway alters innate resistance to polymicrobial sepsis. J Immunol. 2004;172:449–456. doi: 10.4049/jimmunol.172.1.449. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu J, Levin J, Hegen M, Li G, Robertshaw H, Brennan F, Cummons T, Clarke D, Vansell N, et al. Identification and characterization of 4-[[4[(2-butynyloxy)phenyl]sulfonyl]-N-hydroxy-2,2-dimethyl-(3S) thiomorpholinecarbox-amide] (TMI-1), a novel dual tumor necrosis factor-alpha-converting enzyme/matrix metalloprotease inhibitor for the treatment of rheumatoid arthritis. J Pharmacol Exp Ther. 2004;309:348–355. doi: 10.1124/jpet.103.059675. [DOI] [PubMed] [Google Scholar]