

Abstract
During cortical development, when NR2B subunit is the major component of the NMDA glutamate receptors (NMDARs), moderate NMDAR activity supports neuronal survival at least in part by regulating gene transcription. We report that, in cultured cortical neurons from newborn rats, the NMDARs activated the calcium-responsive transcription regulator nuclear factor of activated T cells (NFAT). Moreover, in developing rat cortex, the NFAT isoforms c3 and c4 (NFATc3 and NFATc4) were expressed at relatively higher levels at postnatal day 7 (P7) than P21, overlapping with the period of NMDAR-dependent survival. In cultured cortical neurons, NFATc3 and NFATc4 were regulated at least in part by the NR2B NMDAR. Conversely, knockdown of NFATc4 but not NFATc3 induced cortical neuron apoptosis. Likewise, NFATc4 inhibition prevented antiapoptotic neuroprotection in response to exogenous NMDA. Expression of the brain-derived neurotrophic factor (BDNF) was reduced by NFATc4 inhibition. NFATc4 regulated transcription by the NMDAR-responsive bdnf promoter IV. In addition, NMDAR blockers including NR2B-selective once reduced BDNF expression in P7 cortex and cultured cortical neurons. Finally, exogenous BDNF rescued from the proapoptotic effects of NFATc4 inhibition. These results identify bdnf as one of the target genes for the antiapoptotic signaling by NMDAR–NFATc4. Thus, the previously unrecognized NMDAR–NFATc4–BDNF pathway contributes to the survival signaling network that supports cortical development.
Introduction
In the developing nervous system, neuronal survival requires extracellular signals (Oppenheim, 1991; Snider, 1994). In the cortex, those include the excitatory neurotransmitter glutamate and the neurotrophin brain-derived neurotrophic factor (BDNF). The glutamate NMDA receptors (NMDARs) are calcium-permeable ion channels that are critical for glutamate-mediated survival (for review, see Hardingham, 2006; Hetman and Kharebava, 2006). Thus, NMDAR antagonists induced cortical neuron apoptosis when administered to rodent pups at postnatal day 7 (P7) but not P1 or P21 (Ikonomidou et al., 1999). The NMDAR are formed by two molecules of the constant NR1 subunit and two molecules of the variable NR2 subunits (Cull-Candy and Leszkiewicz, 2004). In developing forebrain neurons, NR2B is the predominant NR2 subunit and thus a major mediator of glutamate-dependent survival of cortical or hippocampal neurons (Cull-Candy and Leszkiewicz, 2004; Habas et al., 2006; Papadia et al., 2008).
Influx of calcium is critical for the neuronal responses to NMDAR stimulation, including survival (Nakanishi, 1992). In addition, NMDAR-mediated protection has been proposed to involve transcriptional regulation of gene expression (Marini and Paul, 1992; Gonzalez-Zulueta et al., 2000; Hardingham et al., 2002; Lee et al., 2005; Papadia et al., 2005, 2008). The identified transcriptional regulatory events that contribute to NMDAR-dependent antiapoptotic response include cAMP response element-binding protein (CREB)/nuclear factor κB (NFκB)-mediated upregulation of the neurotrophin BDNF, CCAAT/enhancer-binding protein (c/EBP)-mediated, or AP1-mediated upregulation of antioxidant enzymes Sesn2 or Srxn1, and suppression of the pro-oxidant protein Txbp1 by inhibiting Foxo (Lipsky et al., 2001; Hardingham et al., 2002; Papadia et al., 2008). Although NR2B has been implicated in regulation of Sesn2, Srxn1, and Txbp1 (Papadia et al., 2008), its contribution to regulation of other NMDAR-dependent survival genes including bdnf has not been yet reported. Because BDNF and NR2B provide major survival signals during forebrain development, their mutual regulation offers a possibility for a prosurvival positive feedback loop. Such regulatory interaction is predicted by modeling studies of extracellular signal-dependent survival of developing neurons (Deppmann et al., 2008).
The nuclear factors of activated T-cells (NFATs) represent a family of at least five transcription factors, all of one of which are regulated by the Ca2+-activated protein phosphatase-2B/calcineurin (PP2B) (Crabtree and Olson, 2002; Hogan et al., 2003). The PP2B-regulated NFAT isoforms including the neuron-expressed NFATc4/NFAT3 establish partially redundant pathways coupling calcium signaling to the nuclear transcription (Graef et al., 1999; Benedito et al., 2005; Bradley et al., 2005; Seybold et al., 2006; Vihma et al., 2008; Nguyen et al., 2009). In cultured cortical or hippocampal neurons, NFAT-driven transcription is regulated by the L-type voltage-gated calcium channels, basal NMDAR activity, and BDNF (Graef et al., 1999; Groth and Mermelstein, 2003). In cultured rat cerebellar granule neurons (CGNs), NFATc4 has been implicated in antiapoptotic effects of depolarizing concentrations of KCl (Benedito et al., 2005). However, the NFAT role in NMDAR-mediated neuronal responses including survival of developing cortical neurons has not been reported before this study. Therefore, we set out to (1) determine the mediators of the NMDAR-stimulated NFAT-driven transcription, (2) evaluate its role in NMDAR-mediated neuronal survival, and (3) identify which of NMDAR-regulated survival genes are targeted by NFAT.
Materials and Methods
Materials.
The following plasmids have been described previously: pON260 (Cherrington and Mocarski, 1989); hemagglutinin (HA) or green fluorescent protein (GFP) -tagged expression vectors for wild type (wt) NFATc4 cloned in pBJ5 or enhanced GFP (EGFP) mammalian expression vectors, respectively (Graef et al., 1999); expression vector for wtNFATc1 cloned in pBJ5 plasmid (Beals et al., 1997); NFAT–luciferase reporter plasmid (Graef et al., 1999); the flag-tagged wt and R474A/N475A/T541G NFATc4 expression plasmids (Yang and Chow, 2003); EF1αLacZ β-galactosidase (β-gal) expression vector and CRE–luciferase reporter plasmid (Impey et al., 1998); rBDNF IV 4.5–chloramphenicol acetyltransferase (CAT) containing a fragment of the rat BDNF promoter IV (from −4100 through 285 relative to the transcription start) cloned 5′ to a chloramphenicol acetyltransferase reporter gene in pBLCAT2 (Shieh et al., 1998); dominant-negative p53 expression vector cytomegalovirus–p53–DD (Shaulian et al., 1992); pSUPER vector (Brummelkamp et al., 2002); and pSuper-based small interfering hairpin RNA (shRNA) constructs targeting GFP and MKL1 (Kalita et al., 2006). The 5xSRF–luciferase reporter was purchased from Stratagene. The pcDNA3-based expression vector for GFP-tagged wt NFATc3 was kindly provided by Dr. Yuriy Usachev (University of Iowa, Iowa City, IA). The following antibodies and reagents were obtained from commercial sources: rabbit polyclonal anti-GFP (MBL); rabbit polyclonal anti-β-gal (MP Biomedicals); the Texas-Red- or HRP-conjugated goat antibodies to rabbit IgG (Calbiochem); BDNF (Alomone Labs); tacrolimus (FK506; A.G. Scientific); tetrodotoxin (TTX) (Ascent Scientific); and NMDA, dizocilpine maleate (MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]), ifenprodil, Ro-25-6981 [R-(R,S)- α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol], dl-amino-5-phosphonovalerate (APV), LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one], U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene], and Hoechst 33258 (Sigma or Calbiochem).
Cell culture and transfection.
Cortical neurons were prepared from newborn Sprague Dawley rats at postnatal day 0 as described previously (Habas et al., 2006). The same methodology was used to culture mouse cortical neurons isolated from the previously reported NFAT–luciferase transgenic mice that were bred on the FVBN background (Wilkins et al., 2004). Briefly, culture medium was basal medium Eagle supplemented with 10% heat-inactivated bovine calf serum (Hyclone), 35 mm glucose, 1 mm l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Cytosine arabinoside (2.5 μm) was added to cultures on the second day after seeding [day in vitro 2 (DIV2)] to inhibit the proliferation of non-neuronal cells. Cells were used for experiments on DIV6–DIV7 unless indicated otherwise. Transient transfections were performed on DIV3–DIV4 using the Lipofectamine 2000 reagent (Invitrogen) as described previously (Hetman et al., 2002). Electroporation of freshly dissociated newborn rat cortical neurons was conducted using a rat neuron nucleofection reagent kit (Amaxa).
Animal treatment.
Sprague Dawley rats were housed with their siblings under a 12 h light/dark cycle and with ad libitum access to water and food. All animals were treated in accordance with the guidelines of the National Institutes of Health and the University of Louisville Guidelines for the Care and Use of Laboratory Animals. MK801 and ifenprodil were dissolved in sterile saline and administered as a single subcutaneous injection at P7. To collect the tissues for RNA extraction, rats were killed with CO2.
Cultured neuron treatments.
LY294002, MK801, ifenprodil, Ro-25-6981, FK506, and U0126 were dissolved in dimethylsulfoxide (DMSO). Bicuculline/4AP, APV, NMDA, and TTX were dissolved in culture media and saline, respectively. BDNF was diluted in PBS containing 0.1% bovine serum albumin. BDNF or NMDA were added 30 min before LY294002. NMDAR blockers or FK506, U0126, or TTX were added 30 min before NMDAR or synaptic activity stimulations. Drug treatments were performed in regular culture media containing 10% bovine calf serum. The final concentration of DMSO in the medium was 0.2–0.4%.
Design and cloning of shRNA and reporter plasmids.
To generate NFATc4 shRNA constructs, we selected two sequences corresponding to nucleotides 564–582 (shNFATc4-N) and 2306–2324 (shNFATc4-C) of rat NFATc4, both of which were validated previously as suitable small interfering RNA targets (Benedito et al., 2005). Comparison of mouse, rat, and human mRNA sequences demonstrated that the NFATc4-N target sequence was completely conserved between these species, whereas the NFATc4-C target sequence was unique for rodents. Oligonucleotides were designed (shNFATc4-N, gatccccgggacggctctcctagagattttcaagagaaatc- tctaggagagccgtccctttttggaaa; NFATc4-C, gatccccggcggaggaagcgcagtccttcaagagaggactgcg-cttcctccgcctttttggaaa) together with their complementary counterparts, followed by annealing and subcloning into pSUPER vector digested with BglII and HindIII (Oligoengine). Also, the NFATc3 shRNAs (shNFATc3) constructs were prepared in a similar way after selecting the target regions using the shRNA design freeware (http://sonnhammer.cgb.ki.SE/). The shNFATc3-1 targets nucleotide sequence 113–131 of rat NFATc3, AGATGATTGTGCATCCATT, which is rodent/human conserved; shNFATc3-2 targets nucleotide sequence 1243–1261 of rat NFATc3, CATCTTCATTACCTCCATT, which is rodent specific. The shNFATc3 consisted of an equimolar mix of shNFATc3-1 and shNFATc3-2. The hBDNF IV–luciferase plasmid was constructed by placing a fragment of the human BDNF promoter IV 5′ from a luciferase reporter gene in pGL4.15 vector (Promega). The fragment included position −205 through 337 relative to the major transcription start site (Pruunsild et al., 2007). Site-directed mutagenesis was used to introduce base substitutions in the composite NFAT/MEF2 consensus site at positions 140–156: wild type, 5′ATTTCCACTATCAAATA3′; mutant, 5′ATTggCACgcTCAAATA3′ (base substitutions in the NFAT/MEF2 binding element are in lowercase).
Reporter gene assays.
Luciferase and β-gal activities were assayed using commercial assay kits (Promega); CAT protein levels were determined using ELISA (Roche Diagnostics). For experiments with transiently transfected reporter plasmids, neurons were cultured on 24-well plates (5 × 105 per well) and transfected with Lipofectamine 2000; transcriptional activity was determined as a luciferase activity or CAT expression normalized to β-gal activity and compared with unstimulated controls. In experiments with neurons from the NFAT–luciferase reporter mouse line, luciferase activity was normalized against the total protein concentration.
RNA analysis.
RNA was isolated from 2–5 × 106 cells or from newborn rat tissues using TRI Reagent (Sigma). The remaining DNA was removed by digestion with DNase I (Promega), and RNA was reverse transcribed with AMV First-Strand cDNA Synthesis kit (Invitrogen) in the presence of random hexamers. For quantitative real time (qRT)-PCR, RT2 Real-Time SYBR Green mix (SuperArray Bioscience Corporation) and the ΔΔCT method of quantification were applied. The reference RNA was 18S rRNA. The qRT-PCR primers were as follows: 18S (sense, gttggttttcggaactgaggc; antisense, gtcggcatcgtttatggtcg), NFATc1 (sense, agatggtgctgtctggccataact; antisense, tgcggaaaggtggtatctcaacca), NFATc2 (sense, tcacagctgagtccaaggttgtgt; antisense, agcatgttaggctggctcttgtct), NFATc3 (sense, tggcatcaacagtatggacctgga; antisense, tttaccacaaggagaagtgggcct), NFATc4 (sense, atcactggcaagatggtggctaca; antisense, agcttcaggattccagcacagtca), BDNF (sense, gagaaagtcccggtatcaaa; antisense, ccagccaattctcttttt), alivin (sense, aaacctgtctaaggtgcctgggaa; antisense, gttgttgtggcgaacaatcagggt), and L1 (sense, tctgcttcatcaaacgcagcaagg; antisense, attgtcactgtactcgccgaaggt). For reverse-transcriptase-PCR, the qRT-PCR primers for NFATc1-4 were applied together with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (sense, catcaccatcttccaggagc; antisense, ctaagcagttggtggtgc).
Immunofluorescence and Western blot analysis.
Transfected cells were detected by immunostaining with the rabbit antibody against β-gal using standard immunofluorescence methodology. For Western blot analysis, cortical neurons were washed twice with PBS and lysed in SDS-PAGE sample buffer. SDS-PAGE and blotting with the anti-GFP antibody were performed according to standard procedures.
Quantification of apoptosis.
Cell nuclei were visualized with Hoechst 33258. The transfected, β-gal-positive cells with uniformly stained nuclei were scored as viable. The transfected cells displaying condensed or fragmented nuclei were scored as apoptotic. At least 200 cells were evaluated for each condition in each independent experiment.
Statistical analysis.
Statistical analysis of the data was performed using one-way ANOVA, followed by post hoc comparisons.
Results
To determine whether the NMDARs regulate NFAT-driven transcription, we evaluated effects of NR2B-selective or nonselective NMDAR antagonists on the transiently transfected luciferase reporter gene whose promoter contained five repeats of the consensus NFAT binding sites from the 5′ regulatory region of the human Il-2 gene. At DIV7, a 6 h treatment with either NR2B-selective ifenprodil or the nonselective dizocilpine (MK801) reduced NFAT activity by 35 or 45%, respectively (vehicle controls vs ifenprodil or MK801, p < 0.01 or <0.001, respectively) (Fig. 1A). The effects of dizocilpine were similar to those of ifenprodil (ifenprodil vs MK801, p > 0.05) (Fig. 1A). In contrast, neither ifenprodil nor dizocilpine affected CRE- or serum response factor (SRF)-mediated transcription (Fig. 1A). In cortical neurons, which were isolated from newborn NFAT–luciferase transgenic mice, ifenprodil or dizocilpine also lowered NFAT-mediated transcription (vehicle controls vs ifenprodil or MK801, p < 0.001, respectively) (Fig. 1A). Either drug induced similar decrease of NFAT activity (ifenprodil vs MK801, p > 0.05) (Fig. 1A). Thus, NR2B-mediated regulation of NFAT-driven transcription occurred at the levels of episomal plasmid- or chromatin-integrated NFAT reporter genes.
Figure 1.

Regulation of NFAT-driven transcription by NMDAR. A, At DIV4, rat cortical neurons were transfected with the indicated reporter plasmids and EF1αLacZ (0.2 + 0.125 μg of plasmid DNA/5 × 105 cells, respectively). Forty-eight hours after transfection, cells were treated for 6 h with 10 μm ifenprodil (IFN), 10 μm MK801, or vehicle (Veh, 0.2% DMSO). Cortical neurons from newborn transgenic mice carrying an NFAT-driven luciferase reporter gene underwent the same pharmacological treatment at DIV7. A specific decrease of NFAT-driven transcription after ifenprodil or MK801 treatments suggests NFAT dependence on the basal NR2B NMDAR activity. B, Rat cortical neurons were transfected as in A. To enhance synaptic activity, bicuculline/4AP (50 μm + 2.5 mm, Bicc/4AP) was applied for 16 h at DIV6. After blocking neuronal electrical activity with 1 mm TTX, bicuculline/4AP failed to activate NFAT-driven transcription. In addition, the synaptic activity-induced NFAT-driven transcription was reduced after blocking NR2B NMDAR. In A, data represent means ± SEM of triplicate determinations from at least three independent experiments; in B, triplicate determinations from sister cultures ± SD are depicted. **p < 0.01; ***p < 0.001. Results similar to those in B were obtained in at least two independent experiments.
Although in some experiments, low prosurvival concentrations (1–15 μm) of NMDA moderately increased endogenous NFAT activity, increasing synaptic activity with the 16 h bicuculline + 4AP treatment consistently stimulated NFAT-driven transcription (data not shown and Fig. 1B). Moreover, that effect was sensitive to NR2B-selective or nonselective NMDAR blockade (Fig. 1B). Therefore, our results indicate that, at least in cultured rat cortical neurons, NFAT-driven transcription is regulated by the NR2B NMDAR.
To determine which NFATc isoforms may be regulated by NMDAR and contribute to NMDAR-mediated neuronal survival, we studied their expression in cultured rat cortical neurons and in developing rat cortex. In DIV7 cultures, mRNAs for NFATc1, NFATc2, NFATc3, and NFATc4 were detected (Fig. 2A). However, significant temporal differences were observed in relative abundance of those mRNAs during cortical development in vivo. Expression of NFATc1 mRNA increased 3.5-fold at P7 compared with P1 (p < 0.001) (Fig. 2B). It remained elevated at P21 (P21 vs P1, p < 0.05; P21 vs P7, p > 0.05) (Fig. 2B). NFATc2 mRNA levels were 6- or 11-fold higher at P7 or P21 than at P1, respectively (P7 or P21 vs P1, p < 0.001; P21 vs P7, p < 0.001) (Fig. 2B) NFATc3 mRNA levels were similar at P1 and P7 but declined by half at P21 (P1 vs P7, p > 0.05; P21 vs P7, p < 0.05) (Fig. 2C). At P7, cortical NFATc4 mRNA levels were at least twofold higher than at P1 or P21 (p < 0.05) (Fig. 2C). Thus, the relatively higher expression of NFATc3 and NFATc4 at P7 than P21 overlaps with the period of the predominant cortical presence of NR2B NMDAR and the NMDAR-dependent cortical neuron survival (Ikonomidou et al., 1999; Cull-Candy and Leszkiewicz, 2004). Consequently, we focused our additional studies on these two members of the NFAT family.
Figure 2.
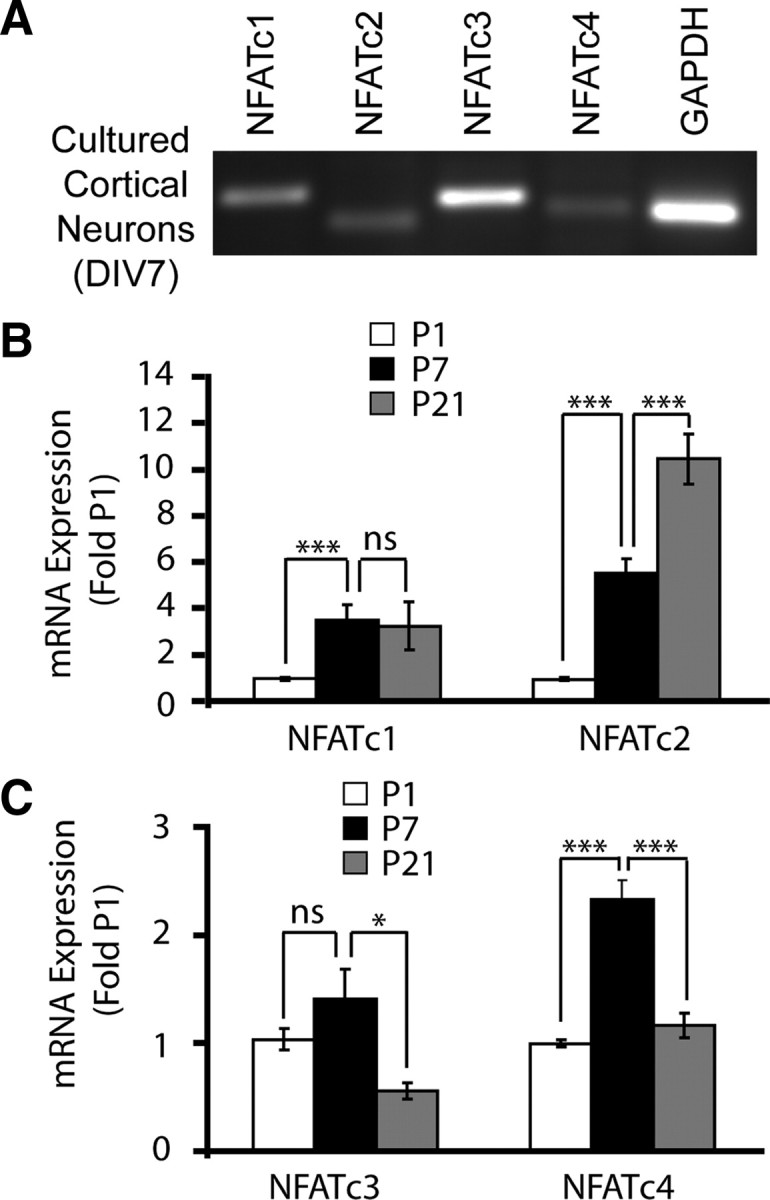
NFAT isoform expression in cultured rat cortical neurons and in developing rat cortex. A, Reverse transcription-PCR analysis of NFAT isoform mRNA expression in cultured rat cortical at DIV7. GAPDH mRNA levels were also determined. B, C, The qRT-PCR was used to determine the NFAT isoform mRNA levels in developing rat neocortex at P1, P7, and P21. The 18S rRNA levels were used for normalizations. Note that expression of NFATc3 and NFATc4 peaks at P7 and declines at P21, overlapping with the maximum expression of the NR2B NMDAR and the survival dependence of cortical neurons on NMDAR (Sheng et al., 1994; Ikonomidou et al., 1999). In B and C, each data point represents six to seven animals ± SEM. *p < 0.05; ***p < 0.001.
To determine NFATc3/c4 responsiveness to NMDAR stimulation, DIV4 neurons were cotransfected with an expression plasmid for either NFAT together with the NFAT–luciferase reporter plasmid. After 48 h, cells were stimulated with 10 μm NMDA for 6 h. The transcriptional activity of the overexpressed NFATs increased in response to NMDA treatment (9.2- or 5.8-fold of unstimulated controls for NFATc3 or NFATc4, respectively) (Fig. 3A). In addition, the NMDA-mediated NFATc3/c4 activation was disrupted by the NMDAR antagonists, including the NR2B-selective ifenprodil and Ro-25-6981, as well as the nonselective MK801 and APV (Fig. 3A). NMDA stimulation induced nuclear translocation of the EGFP-tagged NFATc3/c4 (Fig. 3B and data not shown). This translocation was abolished by the PP2B inhibitor FK506 but not by the MKK1/2 inhibitor U0126 (Fig. 3B and data not shown). However, either inhibitor reduced the NMDAR-mediated transcriptional activation of the overexpressed NFATc3/c4 (Fig. 3C). Also, NMDA failed to activate the R474A/N475A/T541G mutant form of NFATc4 that is deficient in interactions with the transcription factors AP1 and c/EBP (Yang and Chow, 2003). For instance, in cortical neurons stimulated with 10 μm NMDA for 6 h, activities of the overexpressed wt or the R474A/N475A/T541G mutant NFATc4 were 3.35 ± 0.32-fold or 1.2 ± 0.11-fold of the unstimulated controls, respectively. Thus, as in the case of other NFATc3/c4-activating stimuli, NMDAR-mediated activation of NFATc3/4 required PP2B-dependent nuclear translocation, ERK1/2 activation, and interactions with other partner transcription factor(s) (Crabtree and Olson, 2002; Hogan et al., 2003).
Figure 3.

Regulation of NFATc3/c4 by the NR2B NMDAR. A, The DIV4 cortical neurons were cotransfected with NFAT–luciferase reporter plasmid, EF1αLacZ, and wild-type NFATc3 or NFATc4 (0.2 + 0.125 + 0.2 μg of plasmid DNA/5 × 105 cells, respectively). Forty-eight hours after transfection, cells were treated with NMDA for 6 h in the presence of the NMDAR inhibitors: 10 μm ifenprodil (IFN), 0.5 μm Ro-25-6981 (Ro), 100 μm APV, or 10 μm MK801. Vehicle control was 0.2% DMSO (Veh). NFATc3/c4 activation by exogenous NMDA required NR2B. B, The DIV4 cortical neurons were cotransfected with the expression plasmid for wild-type NFATc4 that was tagged with GFP (0.4 μg of plasmid DNA/5 × 105 cells). Forty-eight hours after transfection, cells were treated with NMDA in the presence of the PP2B inhibitor FK506 or the ERK1/2 pathway blocker U0126 for 6 h. The z-stack confocal images of GFP–NFATc4 revealed NMDAR-mediated nuclear translocation of NFATc4. This translocation was blocked by PP2B but not extracellular signal-regulated kinase inhibition. Similar results were obtained in three independent experiments using either GFP–NFATc3 or GFP–NFATc4. C, Forty-eight hours after transfection that was performed as in A, cells were treated with NMDA in the presence of FK506 or U0126 for 20 h as indicated. NMDAR-mediated activation of NFATc3/c4 was antagonized by blocking either PP2B or the ERK1/2 pathway. In A and C, averages ± SD of triplicate determinations from sister cultures are shown. Similar results were obtained in at least two independent experiments.
To further determine the NFATc3/c4 contributions to NFAT-driven transcription in cortical neurons, we used shRNA plasmids targeting these NFAT isoforms. The shNFATc4-N plasmid targeted a sequence in the 5′ coding region of the NFATc4 mRNA that was used before for a successful knockdown of rat NFATc4 (Benedito et al., 2005). The shNFATc3 consisted of a pool of two shRNAs that were designed to target rat NFATc3. The shNFATc3/c4 disrupted activity of their respective targets in NMDAR-stimulated cortical neurons (Fig. 4A,B). Additional validation experiments using NMDAR-stimulated neurons and evaluating effects of (1) shNFATc3 on activation of the overexpressed NFATc4, (2) shNFATc4-N on activation of the overexpressed NFATc1, and (3) shNFATc4-N on activation of the endogenous CRE-driven transcription, demonstrated that neither shRNA reduced NMDAR activity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Also, shNFATc4-N reduced levels of the overexpressed NFATc4 (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Therefore, these results indicate efficient knockdown of NFATc3 or NFATc4 using shNFATc3 or shNFATc4-N, respectively.
Figure 4.
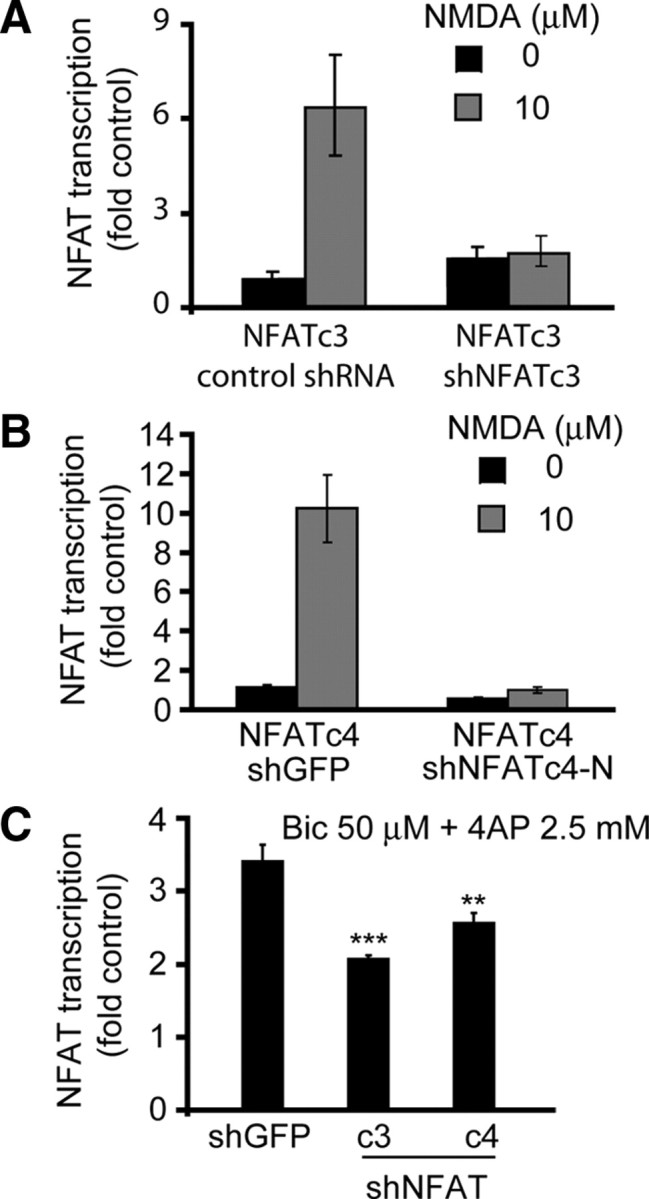
The shRNA-mediated inhibition of NFATc3/c4 reduces activation of NFAT-driven transcription in synaptically stimulated cortical neurons. A, B, DIV4 cortical neurons were cotransfected with expression plasmids for the GFP-tagged NFATc3 or the HA-tagged NFATc4 together with the NFAT–luciferase reporter, the β-gal expression plasmid EF1αLacZ, and the shNFATc3 or shNFATc4-N (0.2 + 0.2 + 0.125 + 1.1 μg of plasmid DNAs/5 × 105 cells, respectively). Control shRNAs included shRNA expression plasmids targeting SRF coactivator MKL1 (control shRNA for shNFATc3) or GFP (shGFP, used as a control for shNFATc4-N). Two days after transfection, cells were stimulated with 10 μm NMDA for 20 h. The shNFATc3 or shNFATc4 inhibited NMDAR-mediated activation of their respective targets. These results together with data from additional validation studies (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) suggest that shNFATc3 or shNFATc4 specifically inhibited NFATc3 or NFATc4, respectively. C, Neurons were transfected with shRNAs as described in A and B except omission of the NFATc3/c4 expression plasmids. At 72 h after transfection, cells were stimulated with bicuculline/4AP (50 μm + 2.5 mm, Bic/4AP) for 8 h. The synaptic activity-mediated increase of NFAT-driven transcription was reduced by shNFATc3 or shNFATc4-N. Because synaptic activity-mediated stimulation of NFAT is NMDAR dependent, these results indicate a role for NMDAR in regulating endogenous NFATc3/c4 in rat cortical neurons. In A and B, averages ± SD of triplicate determinations from sister cultures are depicted. Similar results were obtained in at least two independent experiments. In C, data represent averages ± SEM from six sister cultures in two independent experiments. **p < 0.01; ***p < 0.001.
To assess contributions by NFATc3/c4 to the NMDAR-mediated increase of NFAT-driven transcription, we selected bicuculine/4AP synaptic stimulation protocol that resulted in NR2B NMDAR-dependent activation of NFAT (Fig. 1B). Cortical neurons that received shNFATc3 or shNFATc4 displayed reduced activation of NFAT-driven transcription in response to an 8 h bicuculine/4AP treatment. In neurons receiving control shRNA (shGFP), there was a 3.5-fold activation of NFAT-driven transcription that declined to 2- or 2.4-fold of unstimulated controls after NFATc3 or NFATc4 knockdown, respectively (shGFP vs shNFATc3 or shNFATc4, p < 0.01 or p < 0.001, respectively) (Fig. 4C). Hence, NMDAR regulates activity of endogenous NFATc3/c4.
Because the NR2B NMDAR-regulated NFATc3/4 are expressed in the cortex during the period when NMDAR antagonists induce cortical neuron apoptosis (Fig. 2C) (Ikonomidou et al., 1999), NFATc3/c4 may promote neuronal survival by suppressing apoptosis. Indeed, in cultured cortical neurons, inhibition of NFATc4 induced apoptosis as indicated by nuclear chromatin condensation (Fig. 5A). Thus, in neurons transfected with a control shRNA that targeted GFP, 15, 24, and 30% apoptosis were present at 48, 72, and 96 h after transfection, respectively (Fig. 5B). At each of these time points, neurons that were transfected with shNFATc4-N displayed significantly more apoptosis (20, 45, and 52%, respectively) (Fig. 5B). To ensure that this proapoptotic effect is not caused by off-target effects of the shNFATc4-N, we also tested another shRNA construct that targeted a 3′ coding region of the rodent NFATc4 mRNA (shNFATc4-C). The shNFATc4-C induced cortical neuron apoptosis whose extent and kinetics were similar to those in shNFATc4-N-receiving cells (Fig. 5B,C). Conversely, knockdown of NFATc3 did not induce neuronal apoptosis (Fig. 5B,C). Therefore, at least under culture conditions, NFATc4 but not NFATc3 is required for cortical neuron survival.
Figure 5.
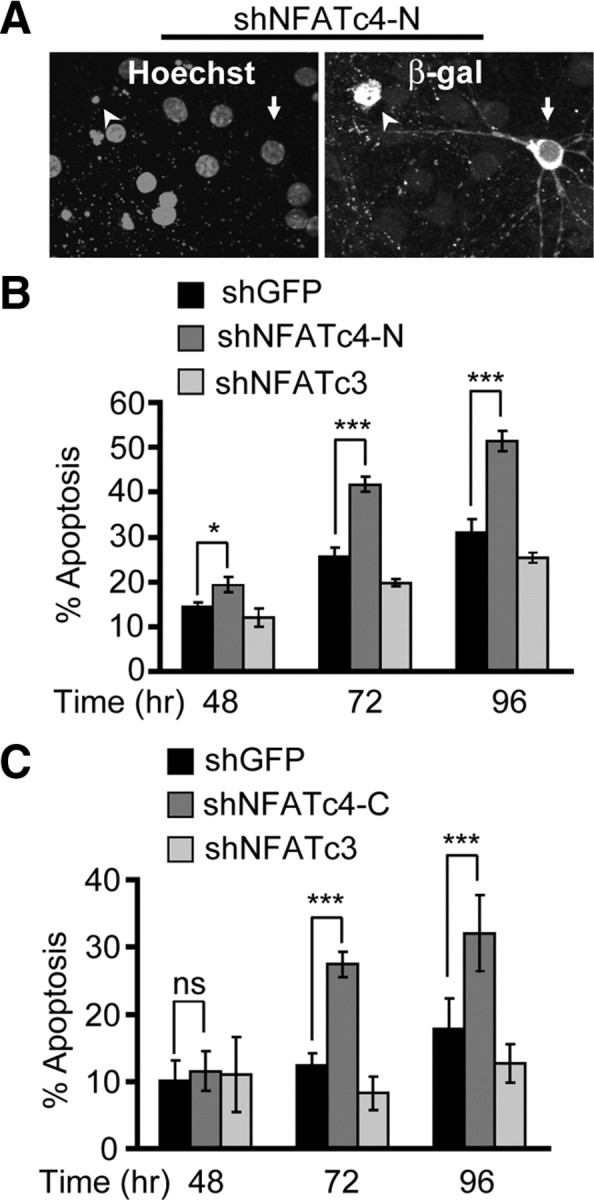
Cortical neuron apoptosis in response to NFATc4 knockdown. The DIV4 cortical neurons were cotransfected with control shRNA (shGFP), shNFATc4-N, or shNFATc4-C together with the β-gal expression plasmid pON260 (1.0 + 0.2 μg of plasmid DNA/5 × 105 cells, respectively). The shNFATc3 was used as an additional control. A, Representative micrographs of shNFATc4-N-transfetced neurons at 96 h after transfection. Immunofluorescence for β-gal was used to identify transfected neurons. Counterstaining with Hoechst 33258 revealed the noncondensed chromatin of live cells (arrow) or chromatin condensation accompanying apoptosis (arrowhead). B, C, The shNFATc4-N or the shNFATc4-C but not shNFATc3 induced cortical neuron apoptosis. Data represent duplicate determinations from at least three independent experiments ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; nsp > 0.05.
To determine whether NFATc4 contributes to NMDAR-dependent survival, DIV4 cortical neurons were transfected with shNFATc4-N or shGFP. At 48 h after transfection, cells were exposed for 24 h to 30 μm LY294002, which is a drug inhibitor of the phosphatidylinositol-3-kinase (PI3K). We have reported previously that such treatment induces cortical neuron apoptosis that can be suppressed by NR2B stimulation with moderate concentrations of exogenous NMDA (Habas et al., 2006). In concert with our published results, in shGFP- or shNFATc4-transfected neurons, LY294002 increased apoptosis from 21 to 45% or 30 to 65%, respectively (p < 0.01) (Fig. 6). In shGFP-receiving neurons, cotreatment with LY294002 and 10 μm NMDA reduced apoptosis from 45 to 25% (p < 0.05) (Fig. 6). In contrast, shNFATc4-N prevented the NMDA neuroprotection against LY294002 (65 or 54% apoptosis with 0 or 10 μm NMDA, respectively, p > 0.05) (Fig. 6). We also performed similar experiments to evaluate whether shNFATc3 affected NMDAR-dependent neuroprotection of PI3K-inhibited neurons. Unexpectedly, we observed a neuroprotective effect of shNFATc3 (A. Vashishta and M. Hetman, unpublished observation). Although analysis of NFATc3 contribution to NMDAR-dependent survival was not possible because of the shNFATc3-mediated rescue of LY294002-treated neurons, the latter activity suggests that, at least during PI3K inhibition, NFATc3 induces rather than prevents apoptosis. Although the proapoptotic activity of NFATc3 is under current investigation (Vashishta and Hetman, unpublished observation), our results indicate specific requirement of NFATc4 for moderate NMDAR activity to protect against neuronal apoptosis.
Figure 6.

Requirement of NFATc4 for the NMDA-induced neuroprotection. DIV4 cortical neurons were cotransfected with control shRNA (shGFP) or shNFATc4-N together with a β-gal expression plasmid pON260 (1.0 + 0.2 μg of plasmid DNA/5 × 105 cells, respectively). Forty-eight hours later, cells were exposed to 30 μm PI3K inhibitor LY294002 (LY) or vehicle (Veh, 0.2% DMSO). In addition, cells were treated with 10 μm NMDA as indicated. After 24 h, LY294002 increased apoptosis in shGFP- or shNFATc4-transfected neurons. In shGFP-transfected neurons, NMDA suppressed the apoptotic response to LY294002. After NFATc4 knockdown, NMDA-mediated neuroprotection was removed. Data represent four independent experiments ± SEM; *p < 0.05; nsp > 0.05.
Because the major function of NFATs is regulation of gene transcription, it is likely that NFATc4 provides antiapoptotic neuroprotection by mediating the NMDAR-induced increases in survival gene expression. Thus, we analyzed the 5′ regulatory regions of several previously identified NMDAR-regulated antiapoptotic survival genes for the presence of the NFAT consensus binding site core 5′WGGAAAW3′, with W being A or T (Hogan et al., 2003). The search included genomic sequences from human, mouse, and rat (if available) to verify the evolutionary conservation of the NFAT regulatory elements. Several putative NFAT binding sites were found in the 5′ regulatory regions of alivin, l1, and bdnf genes. Six of eight alternative bdnf promoters contain evolutionary conserved NFAT elements. Therefore, alivin, L1, and BDNF may be regulated by the NR2B-responsive NFATc4.
To further evaluate this possibility, we determined the effects of NFATc4 knockdown on the basal mRNA levels of alivin, L1, and BDNF. Four days after electroporating shNFATc4-N, mRNA levels of endogenous NFATc4 or BDNF declined to 20 or 60% of those in neurons receiving the control shRNA, respectively (p < 0.01) (Fig. 7). In contrast, mRNAs of alivin or L1 were unaffected by shNFATc4. Consistent with NFAT contribution to regulation of BDNF expression, a 24 h treatment with the 0.2 μg/ml PP2B inhibitor FK506 reduced BDNF mRNA levels to 58 ± 2.7% untreated controls (p < 0.01). These results indicate that, in cultured rat cortical neurons, NFATc4 regulates BDNF mRNA levels.
Figure 7.

NFATc4 as a regulator of BDNF expression. Freshly isolated cortical neurons were electroporated with control shRNA (shGFP) or shNFATc4-N together with DN-p53 that was added to reduce electroporation toxicity (2.5 + 0.5 μg of plasmid DNA/8 × 106 cells, respectively). Four days later, RNA was isolated and expression of NFATc4 as well as its NMDAR-responsive candidate target genes including bdnf, alivin, and l1 was determined using qRT-PCR. rRNA levels (18S) were used for normalizations. The shNFATc4-N significantly reduced NFATc4 and BDNF mRNA levels. Average of three independent experiments are presented. **p < 0.01; ***p < 0.001; nsp > 0.05. Error bars are SEM.
At least in rodents, transcriptional regulation of BDNF promoter IV provides a major coupling between BDNF expression and neuronal activity/NMDAR (Hong et al., 2008). Therefore, we tested a possibility that NFATc4 regulates promoter IV. Compared with its wild-type counterpart, the mutant promoter IV with disruption of the conserved NFAT/MEF2 composite site at position 140–156 (relative to the major transcription start site of human bdnf exon IV) had greatly reduced basal transcriptional activity and diminished responsiveness to NMDAR stimulation (Fig. 8A). We also evaluated effects of manipulating NFATc4 on the NMDAR-mediated activation of the promoter IV. In neurons that were stimulated with 10 μm NMDA for 20 h, overexpression of NFATc4 increased promoter IV activation, whereas shNFATc4-N-mediated knockdown of NFATc4 reduced promoter IV activation (Fig. 8B). Analysis of five independent experiments revealed that the NMDAR-mediated activation of promoter IV was enhanced by 40 ± 9.5% or reduced by 25 ± 3.65% in NFATc4- or shNFATc4-transfected neurons, respectively (p < 0.001). These results indicate that NFATc4 contributes to activation of BDNF promoter IV.
Figure 8.

NFATc4 regulation of the NMDA-responsive BDNF promoter IV. A, DIV4 cortical neurons were cotransfected with the β-gal expression plasmid EF1αLacZ and either the wild-type (WT) or mutant (MT) form of human BDNF promoter IV–luciferase reporter construct (0.125 + 0.2 μg of plasmid DNAs/5 × 105 cells, respectively). The mutation consisted of four substitutions inactivating the composite binding sites for NFAT/MEF2 that is fully conserved between primates and rodents (positions 140–156 from the major transcription start site of exon 4). Two days after transfection, cells were stimulated with 10 μm NMDA for 20 h. Disruption of the NFAT/MEF site inhibited NMDA-induced activation of the BDNF promoter IV. Data ± SD are averages of triplicate sister cultures; similar trends were observed in three independent experiments. B, DIV4 cortical neurons were cotransfected with expression plasmids for NFATc4 or shNFATc4-N together with the rat BDNF promoter IV–CAT reporter construct and the β-gal expression plasmid EF1αLacZ (0.2 or 1.1 + 0.2 + 0.125 μg of plasmid DNAs/5 × 105 cells, respectively). The control plasmids included the empty NFATc4 expression vector pBJ5 (vector) and the shNFATc4-N control shGFP. NMDA stimulation was as in A. In B, data from a representative experiment are shown. Analysis of five independent experiments revealed that the wtNFATc4 enhanced NMDA-mediated promoter IV activation by 40 ± 9.5% and the shNFATc4 reduced the activation by 25 ± 3.65% (p < 0.001).
The NR2B NMDAR appears as a major NMDAR subtype that, during moderate stimulation, activates NFATc4 (Figs. 1, 3). Therefore, we tested the effects of selective NR2B inhibition on expression of candidate NFATc4-regulated survival genes in cortices of P7 rat pups or in cultured cortical neurons. At P7, single systemic injections of NMDAR antagonists induced apoptotic cell death of cortical neurons, indicating their critical dependence on NMDAR activity (Ikonomidou et al., 1999). Three hours after administering the nonselective MK801 (10 mg/kg, s.c.) or the NR2B-selective ifenprodil (20 mg/kg, s.c.), cortical levels of BDNF mRNA reached 22 or 30% of saline-treated controls (p < 0.001) (Fig. 9A). In contrast, L1 or alivin mRNA levels were significantly reduced in response to MK801 but not ifenprodil (42%, p < 0.05 or 34%, p < 0.001, respectively) (Fig. 9A). Also, in DIV7 cultured cortical neurons, either NR2B-selective or nonselective NMDAR blockade reduced BDNF mRNA levels (Fig. 9B). Although, the nonselective MK801 appeared more effective than the NR2B-selective ifenprodil or Ro-25-6981, these differences were not significant (MK801 vs ifenprodil or Ro-25-6981, p > 0.05). Thus, in P7 cortex or in cultured cortical neurons, moderate activity of NR2B NMDAR contributes to the regulation of BDNF mRNA levels. This is consistent with the role of NR2B in driving NFAT-mediated transcription and the contribution of the latter to BDNF expression.
Figure 9.

Role of the NR2B in maintenance of BDNF mRNA expression. A, P7 rat pups received single subcutaneous injections of 0.9% NaCl (vehicle control), 10 mg/kg MK801, or 20 mg/kg ifenprodil (IFN). After 3 h, expression of the candidate NMDAR-regulated NFAT target genes was analyzed by qRT-PCR. MK801 or IFN reduced BDNF expression, whereas alivin and l1 declined only in response to MK801. Hence, NR2B appears critical for maintenance of BDNF expression in developing rat cortex. B, In DIV7 cortical neurons, 24 h treatments with 10 μm MK801 (MK), 10 μm ifenprodil (IFN), or 0.5 μm Ro-25-6981 (RO) lowered BDNF mRNA levels. Veh, Vehicle. In A, three animals were treated for each data point; in B, data represent three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars are SEM.
If BDNF is among the survival targets of NFATc4, one could expect that its supplementation could protect against apoptotic toxicity of the shNFATc4. Thus, cortical neurons were transfected with shNFATc4-N or shGFP used as a control. Forty-eight hours after transfection, cells were treated with 0 or 10 ng/ml BDNF. After the next 24 h, apoptosis analysis revealed 15 or 28% apoptotic neurons that received shGFP or shNFATc4, respectively (p < 0.01) (Fig. 10A). BDNF lowered apoptosis of shNFATc4-N-transfected neurons from 28 to 12% (p < 0.001) (Fig. 10A). In contrast, 10 μm NMDA, which similarly to BDNF protected against PI3K inhibition (Hetman et al., 2002), failed to suppress shNFATc4-induced apoptosis (data not shown). These results are consistent with the notion that BDNF maps downstream of the NMDAR-regulated antiapoptotic transcription factor NFATc4 (Fig. 10B).
Figure 10.

Rescue of shNFATc4-transfected neurons by exogenous BDNF. A, Cortical neurons were transfected as in Figure 5. After 48 h, cells were treated with BDNF for the next 24 h. The shNFATc4-induced apoptosis was suppressed by BDNF. Data from three independent experiments ± SEM are depicted. **p < 0.01; ***p < 0.001. B, Our results support a model of NR2B-activated survival networks, in which BDNF maps downstream of NFATc4.
Discussion
In this study, we demonstrated that, in cortical neurons, NFATc4 contributes to the NR2B NMDAR-activated antiapoptotic gene expression program. We also identified BDNF as one of the NFATc4-regulated neuroprotective genes.
NFATc4 was expressed in both cultured rat cortical neurons and developing rat cortex in which its expression overlapped with the period of survival dependence on NMDAR activity. Although NFATc1 and NFATc2 were also expressed in developing cortex, only NFATc3 demonstrated an NFATc4-like temporal overlap with NMDAR-dependent cortical neuron survival. Therefore, the NMDAR-NFATc3/c4 signaling may serve development-specific roles, including regulation of neuronal survival and/or differentiation.
The NMDAR-mediated regulation of NFATc3/c4 involved calcineurin/PP2B-dependent nuclear translocation, ERK1/2 activity, and interactions with a partner transcription factor of yet unknown identity. Requirement of PP2B and ERK1/2 is consistent with the activation mechanism that regulates all calcium-dependent isoforms of NFAT in various cell types responding to diverse stimuli (Crabtree and Olson, 2002; Hogan et al., 2003). Conversely, cell type determines the nature of the NFAT partner(s), including representatives of such transcription factor families as MEF2, AP1, Foxo, or inducible cAMP early repressor (ICER) (Crabtree and Olson, 2002; Hogan et al., 2003). Although identification of the NFATc3/c4 partner in NR2B-stimulated cortical neurons will be a subject of our future research, it is tempting to speculate that NFATc4 may synergize with the antiapoptotic MEF2s or be antagonized by the proapoptotic ICER or Foxo family members (Brunet et al., 1999; Mao et al., 1999; Gaudilliere et al., 2002; Jaworski et al., 2003).
The shRNA-mediated knockdown of NFATc4 but not NFATc3 induced cortical neuron apoptosis. Also, shNFATc4 antagonized NR2B NMDAR-mediated neuroprotection. Therefore, NFATc4 is required for the NR2B-dependent cortical neuron survival. Although survival promoting links between glutamate signaling and the NFATc4 have not been demonstrated before this study, proapoptotic effects of NFATc4 inhibition have been reported in cultured CGNs (Benedito et al., 2005). Conversely, overexpression of active NFATc2/NFAT1 protected CGNs against trophic/neuronal activity deprivation (Benedito et al., 2005). Thus, NFATc4 may underlie neuronal activity/neurotransmitter-mediated survival in various neuronal populations throughout the CNS. However, as exemplified by the joint recruitment of NFATc3/c4 during metamphetamine-induced adult neuronal death (Jayanthi et al., 2005), other NFAT isoforms may cooperate with NFATc4 to support survival of developing neurons.
Although detailed analysis of forebrain development in NFATc4 knock-out mice has not been reported, these animals appeared normal likely attributable to functional compensation by other NFAT isoforms (Graef et al., 2001). Indeed, only double inactivation of NFATc3 and NFATc4 revealed NFAT requirement for axonal growth (Graef et al., 2003). Unfortunately, these double knock-out mice cannot be used to study glutamate-dependent survival in developing forebrain as they die in utero before forebrain appears (Graef et al., 2003). In concert with our results indicating that the NFATc4 mediates NMDA-dependent survival but is dispensable for the antiapoptotic effects of BDNF (A. Habas and M. Hetman, unpublished observations), neurotrophin survival response of cultured sensory neurons was not compromised by triple knock-out of NFATc4/c3/c2 (Graef et al., 2003). Therefore, the prosurvival recruitment of NFATc4 may require neuronal activity/neurotransmission and, as such, occur during synaptogenesis but not at the earlier stages of development.
In addition to its role as an antiapoptotic transducer of physiological survival signals in developing neurons, NFAT-driven transcription may also contribute to injury-induced neuronal death (Jayanthi et al., 2005; Luoma and Zirpel, 2008; Sama et al., 2008). Therefore, NFAT-driven transcription may either support or antagonize neuronal survival. The cell type and/or the character of the NFAT activating stimulus and/or its signaling context likely determine the opposite survival outcomes of NFAT activation. Finally, different NFATc isoforms may engage in contradictory regulations of neuronal survival.
Although antiapoptotic effects of NFATc4 may involve regulation of multiple antiapoptotic and proapoptotic genes, we have identified bdnf as one of the survival-promoting targets of NFAT. Thus, inhibition of NFATc4 or NR2B NMDAR reduced BDNF expression. Moreover, disruption of the composite NFAT/MEF2 site in the NMDAR-regulated BDNF promoter IV reduced its basal activity as well as responsiveness to NMDA. Conversely, enhancement or reduction of NFATc4 levels increased or decreased NMDA activation of promoter IV, respectively. Finally, exogenous BDNF protected against NFATc4 knockdown (Fig. 10), which by itself did not affect BDNF neuroprotection against several proapoptotic stimuli (Habas and Hetman, unpublished observations). These results are in agreement with previous studies that demonstrated requirement of BDNF for neuroprotective effects of the NMDAR (Marini and Paul, 1992; Bhave et al., 1999; Chen et al., 1999). In addition, they add NFATc4 to the list of BDNF transcriptional regulators in NMDAR-stimulated cortical neurons.
Consistent with BDNF importance for brain development and adult brain plasticity, the bdnf gene structure analysis revealed complex regulation of its expression (Aid et al., 2007; Pruunsild et al., 2007). Transcription of rodent BDNF is controlled by at least eight alternative promoters (Aid et al., 2007). In rodent cerebral cortex or in cultured rat or mouse cortical neurons, BDNF promoter IV (formerly known as promoter III) is the major target for neuronal activity modulation of BDNF transcription (Shieh et al., 1998; Tao et al., 1998; Aid et al., 2007; Hong et al., 2008). Also, its activity increases postnatally overlapping with the period of neuronal survival dependence on NMDAR (Aid et al., 2007). In the BDNF promoter IV region, we identified at least one potential NFAT binding site that is conserved between rodents and primates. NFAT may also bind to NFκB sites, and one of them has been suggested to activate promoter IV in NMDA-stimulated CGNs (Lipsky et al., 2001; Hogan et al., 2003). Thus, NFATc4 effects on BDNF promoter IV activity may be through direct interactions with DNA and the transcription machinery. CREB binding to the promoter IV has been shown recently to underlie the major component of the NMDAR-mediated BDNF upregulation in cortical neurons (Hong et al., 2008). Although these results identified CREB as a necessary component for NMDA-induced BDNF transcription, they did not rule out significant contributions by other transcription factors. In fact, it has been shown that CREB may be instrumental for promoter IV recruitment of such calcium-regulated transcriptional regulators as CREB binding protein or the NFAT partner MEF2D (Hong et al., 2008). Because the consensus NFAT site at the position 140 of promoter IV partially overlaps with a consensus site for a potential NFAT partner, MEF2, it is tempting to speculate that NFATc4 and MEF2D interact to regulate BDNF transcription. The non-CREB regulators of promoter IV may be either corequired for its activation by NMDAR and/or determine the maximum levels of the activation.
Besides NMDAR and neuronal activity, BDNF expression is stimulated by the BDNF itself (Groth and Mermelstein, 2003). Because BDNF induction by BDNF was sensitive to PP2B inhibition and because NFATc4 overexpression increased BDNF mRNA levels, it has been proposed that NFATc4 regulates BDNF (Groth and Mermelstein, 2003). By using the NFAT-specific loss-of-function approach, our study adds a piece of critical evidence to support that notion.
Our results indicate that stimulation of BDNF expression plays a role in NR2B NMDAR-mediated cortical neuron survival. Similar observations have been reported for the neuronal activity- and/or NMDAR-dependent survival of cultured rat cortical neurons or CGNs (Marini and Paul, 1992; Ghosh et al., 1994; Bhave et al., 1999; Chen et al., 1999). In addition to promoting survival, BDNF contributes to other key events in the developing nervous system, including morphogenesis or induction of apoptosis (Huang and Reichardt, 2001; Miller and Kaplan, 2001). It remains to be tested whether the NMDAR–NFATc4–BDNF pathway plays a role in these processes. Although inhibition of NMDAR/BDNF cascade is implicated in the pathogenesis of fetal alcohol syndrome, NFATc4 hypoactivity plays a role in pathogenesis of Down syndrome (Bhave et al., 1999; Ikonomidou et al., 2000; Arron et al., 2006). Thus, disruption of the NMDAR– NFATc4 signaling may contribute to genetically or environmentally induced developmental brain disorders that produce mental retardation.
Interestingly, of the three NMDAR-regulated survival genes investigated in this study, only BDNF responded to both NR2B-selective NMDAR blockade and NFATc4 knockdown. Conversely, antiapoptotic cell adhesion molecules alivin and L1 were sensitive to nonselective NMDAR inhibition but not to NR2B NMDAR blocker ifenprodil or shNFATc4. These results suggest specificity of NFATc4 involvement in mediating the effects of NR2B NMDAR stimulation. Likewise, CREB-mediated transcription has been proposed to be preferentially activated by the NR2A receptors (Hardingham et al., 2002). Therefore, NMDAR isoform-specific recruitment of nonoverlapping transcription factors may underlie the differences in gene expression programs that were reported after activation of different NMDAR pools. Alternatively, regulation of NFATc4 may engage various subtypes of NMDAR dependent on the period of nervous system development. After the maturation-associated switch of the synaptic NMDAR from NR2B to NR2A (Cull-Candy and Leszkiewicz, 2004), the synaptic activity-regulated NFATc4 may also alter its NMDAR subtype dependence.
In summary, we identified a novel survival signaling pathway that suppresses apoptosis of developing cortical neurons. This pathway consists of the NR2B NMDAR, the transcription factor NFATc4, and the neurotrophin BDNF. Given the broad developmental impact of both NMDAR and BDNF, it is possible that NMDAR–NFATc4–BDNF signaling affects not only neuronal survival but also other key steps of neuronal differentiation, including synaptogenesis.
Footnotes
This work was supported by National Institutes of Health Grants NS047341-01 and RR015576-06 (M.H.), Kentucky Spinal Cord and Head Injury Research Trust Grant 3-5 (M.H.), The Commonwealth of Kentucky Challenge for Excellence (M.H.), and Norton Healthcare (M.H.). T.T. and P.P. were supported by Estonian Ministry of Education and Research Grant 0140143, Estonian Enterprise Grant EU27553, and Estonian Science Foundation Grant 7257. Drs. Gerald Crabtree, Neil Clipstone, Yuryi Usachev, and Chi-Wing Chow provided reagents used in this study.
References
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KC, Groth RD, Mermelstein PG. Immunolocalization of NFATc4 in the adult mouse brain. J Neurosci Res. 2005;82:762–770. doi: 10.1002/jnr.20695. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Chen S, Mantei N, Dong L, Schachner M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J Neurobiol. 1999;38:428–439. doi: 10.1002/(sici)1097-4695(19990215)38:3<428::aid-neu10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Cherrington JM, Mocarski ES. Human cytomegalovirus iel transactivates the α promoter-enhancer via an 18-base-pair repeat element. J Virol. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD. A model for neuronal competition during development. Science. 2008;320:369–373. doi: 10.1126/science.1152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Groth RD, Mermelstein PG. Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci. 2003;23:8125–8134. doi: 10.1523/JNEUROSCI.23-22-08125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habas A, Kharebava G, Szatmari E, Hetman M. NMDA neuroprotection against a phosphatidylinositol-3 kinase inhibitor, LY294002 by NR2B-mediated suppression of glycogen synthase kinase-3beta-induced apoptosis. J Neurochem. 2006;96:335–348. doi: 10.1111/j.1471-4159.2005.03543.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–938. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem. 2006;6:787–799. doi: 10.2174/156802606777057553. [DOI] [PubMed] [Google Scholar]
- Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z. ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J Biol Chem. 2002;277:49577–49584. doi: 10.1074/jbc.M111227200. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Mioduszewska B, Sánchez-Capelo A, Figiel I, Habas A, Gozdz A, Proszynski T, Hetman M, Mallet J, Kaczmarek L. Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J Neurosci. 2003;23:4519–4526. doi: 10.1523/JNEUROSCI.23-11-04519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, Cadet JL. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 2005;102:868–873. doi: 10.1073/pnas.0404990102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita K, Kharebava G, Zheng JJ, Hetman M. Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci. 2006;26:10020–10032. doi: 10.1523/JNEUROSCI.2644-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky RH, Xu K, Zhu D, Kelly C, Terhakopian A, Novelli A, Marini AM. Nuclear factor kappaB is a critical determinant in N-methyl-d-aspartate receptor-mediated neuroprotection. J Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- Marini AM, Paul SM. N-methyl-d-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci U S A. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci. 2001;58:1045–1053. doi: 10.1007/PL00000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Lindner R, Tedeschi A, Forsberg K, Green A, Wuttke A, Gaub P, Di Giovanni S. NFAT-3 is a transcriptional repressor of the growth-associated protein 43 during neuronal maturation. J Biol Chem. 2009;284:18816–18823. doi: 10.1074/jbc.M109.015719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007;90:397–406. doi: 10.1016/j.ygeno.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybold VS, Coicou LG, Groth RD, Mermelstein PG. Substance P initiates NFAT-dependent gene expression in spinal neurons. J Neurochem. 2006;97:397–407. doi: 10.1111/j.1471-4159.2006.03744.x. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Vihma H, Pruunsild P, Timmusk T. Alternative splicing and expression of human and mouse NFAT genes. Genomics. 2008;92:279–291. doi: 10.1016/j.ygeno.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- Yang TT, Chow CW. Transcription cooperation by NFAT.C/EBP composite enhancer complex. J Biol Chem. 2003;278:15874–15885. doi: 10.1074/jbc.M211560200. [DOI] [PubMed] [Google Scholar]