

Abstract
Background:
Rett syndrome (RTT) is a neurodevelopmental disorder primarily seen in females, most with a mutation in MECP2. Epilepsy has been reported in 50%–80%. Previous reports were based on small sample sizes or parent-completed questionnaires, or failed to consider the impact of specific MECP2 mutations.
Methods:
The Rare Disease Consortium Research Network for RTT is an NIH-funded project to characterize the clinical spectrum and natural history of RTT in advance of clinical trials. Evaluations include clinical status (classic vs atypical RTT), MECP2 mutations, clinical severity, and presence, frequency, and treatment of seizures.
Results:
Enrollment as of June 2008 is 602; 528 (88%) meet clinical criteria for classic RTT. Of these, 493 (93%) have MECP2 mutations. Age range was 8 months to 64 years. A total of 360 (60%) were reported to have seizures, including 315 (60%) classic and 45 (61%) atypical RTT. Physician assessment of the 602 indicated that 48% had seizures. There was no significant difference in seizure occurrence by race/ethnicity. A significant age impact for seizures was seen and seizures were infrequent before age 2 years. MECP2 mutations most frequently associated with epilepsy were T158M (74%) and R106W (78%), and less frequently R255X and R306C (both 49%). Individuals with seizures had greater overall clinical severity, and greater impairment of ambulation, hand use, and communication.
Discussion:
Seizures are common in Rett syndrome, have an age-related onset and occurrence, vary by mutation, and are associated with greater clinical severity. This information represents a key consideration for designing clinical trials.
GLOSSARY
- AED
= antiepileptic drug;
- CSS
= Clinical Severity Scale;
- MBA
= Motor-Behavioral Assessment;
- RTT
= Rett syndrome;
- VNS
= vagus nerve stimulator.
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder which occurs in 1.09/10,000 females. Typical RTT is diagnosed based on a set of clinical criteria; atypical RTT is diagnosed when some, but not all, of the typical RTT clinical criteria are present.1 Mutations in the gene encoding methyl-CpG-binding protein 2 (MECP2) cause the majority of cases of typical RTT and are identified in 90%–95% of cases.2 The NIH-funded Rare Disease Network for Rett syndrome (clinicaltrials.gov, Identifier NCT00296764) is characterizing the clinical spectrum and natural history of RTT preparatory to the initiation of anticipated clinical trials. The RTT consortium consists of 3 primary RTT centers (Baylor College of Medicine, Greenwood Genetic Center, and University of Alabama at Birmingham) and 4 traveling sites across the United States. In advance of clinical trials, understanding the comorbidities such as seizures is critical. A feature of RTT is the wide-ranging neurologic manifestations. Among these, seizures are reported to occur frequently. However, previous studies have been flawed because of small sample size, failure to use established clinical criteria to define RTT, and lack of correlation with mutational status. The Rare Disease Research Network for RTT is designed to address these concerns and to define the natural history of RTT. This report concerns the initial effort and provides information regarding the occurrence of seizures. This information may impact on the design of clinical trials in RTT.
METHODS
As part of the Rare Disease Clinical Research Network, the natural history study of RTT was organized in 2003. Institutional review board applications from each of the three sites were submitted, and have been approved for the use of human subjects for this study. At present, more than 750 participants with RTT (typical and atypical) or with a mutation in the methyl-CpG-binding protein 2 gene (MECP2) have been enrolled. From this cohort, complete data are available for 602 participants, 528 of whom meet the consensus criteria for classic RTT. Each participant in this analysis was evaluated by members of the RTT consortium based at the Baylor College of Medicine, the Greenwood Genetic Center, or the University of Alabama at Birmingham utilizing a common set of data collection instruments and the current consensus diagnostic criteria.1 Evaluations occurred twice annually through age 12 years or annually beginning at age 13 years. At each study visit, a detailed history and physical examination and a comprehensive set of anthropometric measurements were obtained. In addition, 2 quantitative measures of clinical status, the Clinical Severity Scale (CSS) and the Motor-Behavioral Assessment (MBA), were completed.2,3 All data were then exported to the Data Technology Coordinating Center at the University of South Florida. This data repository was utilized for the present analyses. With respect to seizures, parent report of seizures, description of seizures, age at onset of seizures, frequency of occurrence of seizures, and current and past treatments of seizures was recorded. Physician assessment of the clinical description, determination of whether this represented seizures, and clinical type of seizure was recorded.
Standard protocol approvals, registrations, and patient consent.
This study was approved by the institutional review boards of the participating centers. Written, informed consent was obtained from all guardians of patients participating in the study (clinicaltrials.gov, Identifier NCT00296764).
Statistical analysis.
We summarized the overall prevalence of seizures and prevalence of seizure for MECP2 mutation type and clinical features collected at the enrollment of 602 patients. The proportions were reported for categorized outcomes and the means and standard deviations were reported with minimum and maximum values for continuously measured outcomes. The association between the prevalence of seizures and other clinical features was investigated employing logistic regression models for P (having seizures) and adjusting for age in years at the enrollment. Each clinical feature was examined in a univariate model, and the multiple model was set up by backward variable selection from the univariate analysis results. We also investigated the association between the prevalence of seizures and common mutation types from logistic regression models for P (having seizures), adjusting for age in years at the enrollment. A p value less than 0.05 was considered significant. SAS 9.1.3 (SAS Institute, Cary, NC) was used for all statistical analyses.
RESULTS
Overall, 360 (60%) study participants had a history of seizures. There were 528 participants with classic RTT and 315 had seizures (60%). Of the 74 with atypical RTT, 45 had seizures (61%) (figure 1). There was no significant difference in the occurrence of seizures in typical vs atypical RTT (p value 0.984). However, evaluation of the clinical description of the reported seizures by the RTT specialist physicians indicated only 48% (n = 291) of the whole group were considered to have epileptic seizures.

Figure 1 Occurrence of epilepsy at first visit
Eighty-five percent of participants reported ethnicity/race as white (n = 512), of which 63% (323) noted seizures. There were smaller numbers of other ethnic/race groups; however, no significant difference in the occurrence of seizures was found for any of these groups. For example, there were 78 Hispanic patients, of which 47 (59%) were reported to have seizures.
The age range of the participants was 8 months to 64 years. There was a significant age-related occurrence of seizures. Of the 9 participants under age 2 years at the initial visit, none was reported to have seizures; for those 3–<5 years (n = 99), 33% were reported to have seizures; for those 5–<10 years (n = 175), 62% were reported to have seizures; for those 10–<15 years (n = 94), 80% were reported to have seizures; for those 15–<20 years (n = 64), 86% were reported to have seizures; for those 20–<30 years (n = 70), 84% were reported to have seizures (figure 2).

Figure 2 Frequency of epilepsy in Rett syndrome by age
MECP2 mutations were identified in 540 participants (90%), including 493 (93%) of the classic RTT. Seizures were reported in 317 (59%) of those with mutations and 41 (73%) of those without a mutation. Among those with mutations, 326 (60%) had 1 of the 8 most common mutations (R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C), 51 (9%) had c-terminal deletions, and 46 (8.5%) had large deletions. When adjusted for age, seizures were reported more frequently for those with a T158M mutation (74%, p < 0.05). No significant differences were found for other mutations, although those with R255X and R306C tended to have a lower occurrence of seizures (49% each) (figure 3).
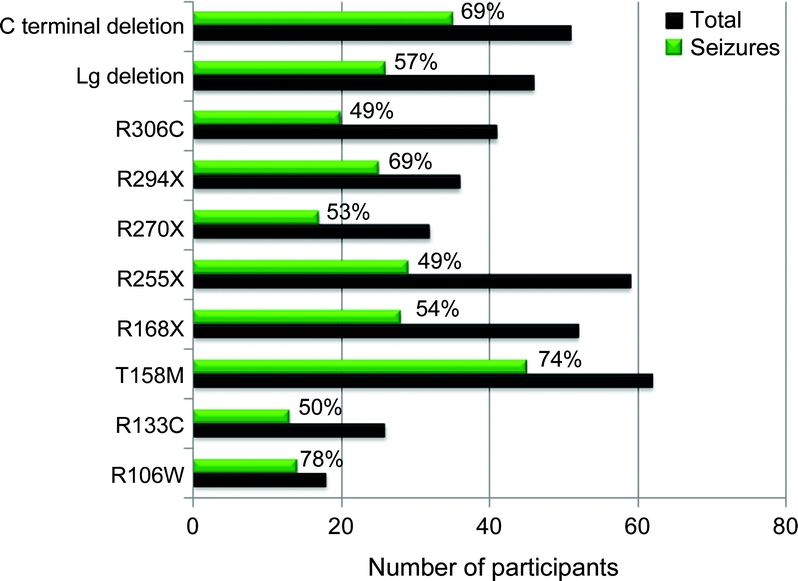
Figure 3 Frequency of epilepsy in Rett syndrome by mutation type
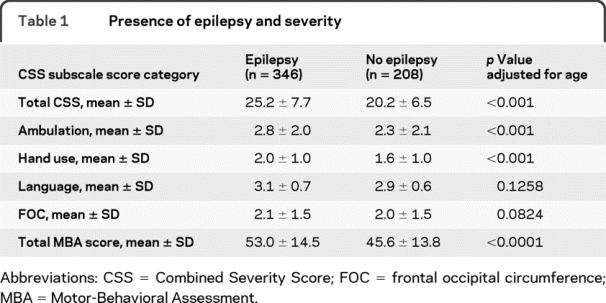
We examined overall clinical severity scores on both the CSS and MBA and on specific features, absent or lost ability to walk, hand use, communication skills, and head growth. Since age could confound the relationship between seizures and each clinical feature, a model adjusting for age was used to investigate the association. As shown in table 1, when adjusted for age, the occurrence of seizures was associated with higher overall clinical severity scores on both the CSS (p < 0.001) and MBA (p < 0.001), as well as with walking and hand use. While greater severity score for communication was observed, when adjusted for age the difference in this feature was not significant. Head growth was also not significantly different between the 2 groups (table 1).
Table 1 Presence of epilepsy and severity
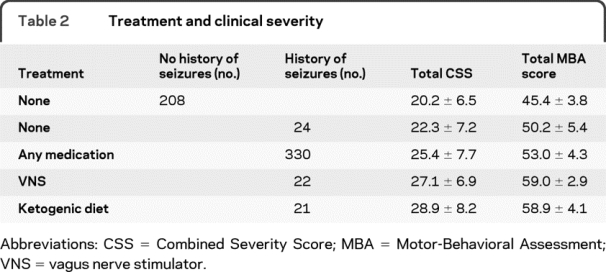
For participants with reported seizures, 36% experienced no seizures during the 6 months prior to their initial evaluation; 27% had monthly seizures; 20% had weekly seizures; and 11% had daily seizures. With respect to treatment of seizures, 20% received no treatment, 37% received 1 antiepileptic drug (AED), and 8% received 3 or more AEDs. Alternative therapies included the vagus nerve stimulator (VNS), used by 13%, and the ketogenic diet, used by 12%. The most commonly used AEDs were carbamazepine (37%), lamotrigine (33%), and levetiracetam (26%). Overall clinical severity scores on the CSS and MBA increased (greater severity) with increasing complexity of treatment from those receiving no treatment, to those on any medication (higher severity scores), to those managed with a VNS or ketogenic diet (highest severity scores) (table 2).
Table 2 Treatment and clinical severity
DISCUSSION
Seizures represent a significant clinical problem in RTT and negatively impact the quality of life of parents and caregivers of patients with RTT.4 Seizures occur frequently in RTT and begin at age 2 to 3 years with increases in occurrence with age through puberty. In the present study, at initial evaluation 60% of participants were reported to have seizures. This number is lower than that reported in studies from Europe and Australia.5,6 These studies reported rates as high as 80%–94% compared to 60% of our whole cohort. We observed no significant difference in reported seizures between those with typical and atypical RTT or with ethnicity/race. However, most of our participants had typical RTT (88%). Two factors may contribute to these differences. First, there was a significant impact of age on the reported occurrence of seizures. In this study, 86% of RTT individuals were reported to have seizures by age 20 years. This is similar to the occurrence reported in other studies. Our study and the other 2 studies suggest that early onset, less than 3 years of age, of seizures in RTT is unlikely, and onset after age 20 years is rare. Secondly, we previously reported the use of video-EEG to verify epileptic seizures in RTT.7 This study found that only one-third of parent-reported seizure behaviors were associated with an EEG seizure discharge. Epileptic seizures in RTT may be overreported. Indeed, in our current study careful review of reported seizure behaviors by clinicians determined that only 48% of the RTT participants were having epilepsy in contrast to the parent report of 60%.
Most individuals with typical RTT had MECP2 mutations (93%). Seizures were reported less frequently in those individuals meeting the clinical criteria for RTT with MECP2 mutations (52%) and more frequently in those participants meeting the clinical criteria for RTT without MECP2 mutations (73%). Individual MECP2 mutations appeared to confer different susceptibility to seizures. A significantly higher occurrence of seizures (74%) was reported for those participants with a T158M mutation, while 2 mutations, R255X and R306C, tended to have lower occurrence (49%) of seizures. A similar finding was reported from the Australian cohort for R255X and for R294X, but not for the other mutations observed in our larger cohort.
Greater overall clinical severity, as well as impairment of ambulation and hand use, was observed in the participants who were reported to have seizures. This finding is similar to that reported in the Australian cohort. However, we did not find a significant relation between head growth and seizures, in contrast to that reported for the smaller Swedish cohort.6
The seizures in RTT have been described as benign.6 We did observe that the onset and occurrence of seizures appeared to diminish after puberty. A significant number of the participants (36%) were reported to be seizure-free for the previous 6 months. However, the occurrence of seizures in RTT negatively impacts family quality of life4 and is associated with greater clinical severity. Treatment may be an indication of seizure severity with respect to frequency of occurrence. Clinical severity was greater for those receiving AEDs and increased for those requiring alternative nonpharmacologic therapies such as VNS or ketogenic diet.
DISCLOSURE
Dr. Glaze serves as President of the Epilepsy Foundation of Southeast Texas and on the Medical Advisory Board of the International Rett Syndrome Foundation; serves as an Associate Editor of Sleep and on the Board of Directors of the American Academy of Sleep Medicine; and receives research support from the NIH (1 U54 RR019478 [Site PI]) the Blue Bird Circle, and Autism Speaks. Dr. Percy receives research support from the NIH (NCRR U54 RR019478 [Co-PI] and NICHD 38985 [PI]). Dr. Skinner receives research support from the NIH (U54 RR019478 [Site PI]). Dr. Motil serves on scientific advisory boards for the International Rett Syndrome Foundation and the National Foundation for Ectodermal Dysplasias; serves as Section Editor–Pediatric Nutrition and receives royalties from UpToDate, Inc.; and receives research support from the NIH (U54 RR019478 [Investigator]), and from the International Rett Syndrome Foundation. Dr. Neul receives research support from the NIH/NINDS (K08NS052240 [PI]) and from the International Rett Syndrome Foundation. Ms. Barrish receives research support from the NIH (U54 RR019478 [Project Manager]), the International Rett Syndrome Foundation, and from the Blue Bird Circle. Ms. Lane receives research support from the NIH (U54 RR019478 [Project Manager]), and from the International Rett Syndrome Foundation. Ms. Geerts receives research support from the NIH (U54 RR019478 [Investigator]), the Civitan/Sparks Clinics, University of Alabama in Birmingham, the Maternal and Child Health Leadership Education in Neurodevelopment Disabilities, and from the Alabama Center for Development Disabilities Education, Research and Service. Ms. Annese receives research support from the NIH (U54 RR019478 [Investigator]). Ms. Graham receives research support from the NIH (U54 RR019478, [Investigator]). Ms. McNair receives research support from the NIH (U54 RR019478 [Investigator]). Dr. Lee reports no disclosures.
Address correspondence and reprint requests to Dr. Daniel G. Glaze, The Blue Bird Circle Rett Center, Baylor College of Medicine, 6621 Fannin Street, CC1250.44, Houston, TX 77030 dglaze@bcm.edu
Study funding: This study was supported by the NIH (U54 RR019478).
Disclosure: Author disclosures are provided at the end of the article.
Received August 6, 2009. Accepted in final form January 12, 2010.
REFERENCES
- 1.Hagberg B, Hanefield F, Percy A, Skjeldel O. An update on clinically applicable diagnostic criteria in Rett syndrome. Eur J Pediatr Neurol 2002;6:293–297. [DOI] [PubMed] [Google Scholar]
- 2.Neul J, Fang J, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70:1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzgerald PM, Jankovic J, Glaze DG, Schultz RJ, Percy AK. Extrapyramidal involvement in Rett syndrome. Neurology 1990;40:293–295. [DOI] [PubMed] [Google Scholar]
- 4.Nadia BB, Isabelle G, Rima N, et al. Parental view of epilepsy in Rett syndrome. Brain Dev 2008;30:126–130. [DOI] [PubMed] [Google Scholar]
- 5.Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Paediatr Neurol 2007;11:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steffenburg U, Hagberg G, Hagberg B. Epilepsy in a representative series of Rett syndrome. Acta Paediatr 2001;90:34–39. [DOI] [PubMed] [Google Scholar]
- 7.Glaze DG, Schultz RJ, Frost JD. Rett syndrome: characterization of seizures versus non-seizures. Electroencephalogr Clin Neurophysiol 1998;106:79–83. [DOI] [PubMed] [Google Scholar]