

Autophagy (xenophagy) degrades intracellular bacteria. The cargoes are degraded after the fusion of xenophagosomes with lysosomes. However, the molecular mechanism underlying the fusion remains unclear. Here we show that combinational SNARE proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes.
Abstract
Autophagy plays a crucial role in host defense, termed antimicrobial autophagy (xenophagy), as it functions to degrade intracellular foreign microbial invaders such as group A Streptococcus (GAS). Xenophagosomes undergo a stepwise maturation process consisting of a fusion event with lysosomes, after which the cargoes are degraded. However, the molecular mechanism underlying xenophagosome/lysosome fusion remains unclear. We examined the involvement of endocytic soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) in xenophagosome/lysosome fusion. Confocal microscopic analysis showed that SNAREs, including vesicle-associated membrane protein (VAMP)7, VAMP8, and vesicle transport through interaction with t-SNAREs homologue 1B (Vti1b), colocalized with green fluorescent protein-LC3 in xenophagosomes. Knockdown of Vti1b and VAMP8 with small interfering RNAs disturbed the colocalization of LC3 with lysosomal membrane protein (LAMP)1. The invasive efficiency of GAS into cells was not altered by knockdown of VAMP8 or Vti1b, whereas cellular bactericidal efficiency was significantly diminished, indicating that antimicrobial autophagy was functionally impaired. Knockdown of Vti1b and VAMP8 also disturbed colocalization of LC3 with LAMP1 in canonical autophagy, in which LC3-II proteins were negligibly degraded. In contrast, knockdown of Syntaxin 7 and Syntaxin 8 showed little effect on the autophagic fusion event. These findings strongly suggest that the combinational SNARE proteins VAMP8 and Vti1b mediate the fusion of antimicrobial and canonical autophagosomes with lysosomes, an essential event for autophagic degradation.
INTRODUCTION
Autophagy is an intracellular degradation system for cytoplasmic materials, such as proteins and organelles, which are directed to lysosomes by a membrane-mediated process (Seglen and Bohley, 1992; Yoshimori, 2004). This system functions as a survival mechanism during short-term starvation by degrading some nonessential components to obtain nutrients for biosynthetic reactions in mammalian cells. In addition, autophagy is essential for cellular survival, differentiation, development, homeostasis, and host defense (Ravikumar et al., 2002; Komatsu et al., 2006; Hara et al., 2006; Kamimoto et al., 2006). After signaling for autophagy, a flat membrane sac structure, called the isolation membrane, elongates to surround a portion of the cytoplasm and eventually forms a closed double-membrane structured vacuole, which is termed an autophagosome (Yoshimori, 2004). Autophagosomes then undergo a stepwise maturation process that consists of a fusion event with lysosomes, which allows the autophagic vacuole to acquire lysosomal proteases and the vacuolar-type proton ATPase. Then, the interior of the autophagosome becomes acidified and the cytoplasmic materials are subjected to degradation. At this final stage after fusion with lysosomes, autophagosomes are referred to as autolysosomes (Yoshimori, 2004). This fusion event, essential for degradation, has been studied by morphological analysis using electron microscopy, whereas such molecular analyses of the underlying mechanism have progressed more slowly (Eskelinen, 2005; Eskelinen and Saftig, 2009).
Autophagy is emerging as a central component of various immunological functions, such as innate and adaptive immune activation, as well as antimicrobial host defense against diverse viral, bacterial, and parasitic infections (Deretic, 2009). Among them, its most principal manifestation is capturing and digesting intracellular foreign microbial invaders. Group A Streptococcus (GAS) is an extracellular pathogen that causes human diseases based on its ability to bind to extracellular matrix proteins and produce a wide range of toxins. GAS can invade nonphagocytic cells (e.g., epithelial cells, keratinocytes), although once inside a cell it is unable to proliferate and degraded by mechanisms that until recently were unidentified (Cunningham, 2000). We showed previously that the bacterium invades nonphagocytic human cells via endocytosis, then escapes from endosomes to the cytoplasm (Nakagawa et al., 2004), after which the pathogen is entrapped within antimicrobial autophagosomes, termed GAS-containing autophagosome-like vacuoles (GcAVs). Initially, the structures of GcAVs do not include lysosomal membrane protein (LAMP)1 similar to autophagosomes, although they are subsequently associated with and colocalize with that protein. Finally, GAS is degraded by GcAVs possessing lysosomal degradation enzymes (Nakagawa et al., 2004). This antimicrobial autophagy, which has been likened to “eating the enemy,” was recently classified as xenophagy selective for degradation of intracellular bacteria and viruses (Levine, 2005). Although antimicrobial autophagosomes (xenophagosomes) were morphologically shown to be formed by the fusion of multiple small precursor structures of xenophagosomes (Nakagawa et al., 2004, Yamaguchi et al., 2009), their mechanism of fusion with lysosomes remains far from clear and is a major topic of investigation.
Soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) are generally accepted as major players in the final stage of docking and subsequent fusion of diverse vesicle-mediated transport events (Hong, 2005). SNAREs are functionally classified into v-SNAREs, which are associated with the vesicle/container, and t-SNAREs, which are associated with the target compartment (Hong, 2005). SNAREs are also structurally divided into Q-SNAREs (those having a Gln/Q residue) and R-SNAREs (those having an Arg/R residue), and Q-SNAREs are further subdivided into Qa-, Qb-, and Qc-SNAREs based on the amino acid sequence of the SNARE domain. It is unknown whether SNAREs are involved in xenophagosome-lysosome or canonical autophagosome-lysosome fusion events.
In the present study, we investigated the involvement of Vti1b in combinations with the v-SNAREs VAMP7 and VAMP8 in the fusion of GcAVs (xenophagosomes) with lysosomes. The results of our experiments using small interfering RNA (siRNA)-knockdown strongly indicate that the SNARE proteins Vti1b and VAMP8 mediate xenophagosome-lysosome fusion. Furthermore, those SNARE proteins were also found to mediate the fusion of canonical autophagosomes with lysosomes.
MATERIALS AND METHODS
Cell Culture and Transfection
All cell lines were cultured in DMEM (Wako Pure Chemicals, Osaka, Japan) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA). Human breast adenocarcinoma MCF7 cells stably expressing enhanced green fluorescent protein (EGFP)-LC3 were a kind gift from Dr. Koichi Matsunaga (Yoshimori laboratory). Human cervical epithelial HeLa cells stably expressing EGFP-LC3 were constructed as described previously (Mizushima et al., 2001). Human lung adenocarcinoma epithelial A549 cells stably expressing EGFP-LC3 were constructed as described previously (Matsunaga et al., 2009). Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For amino acid starvation, cells were cultured in Earle's balanced aalt (EBS) solution (Sigma-Aldrich, St. Louis, MO) without amino acids and fetal bovine serum. Stable transformants were selected in complete medium containing 500 μg/ml G418 (Sigma-Aldrich).
Antibodies
The following antibodies were used: mouse monoclonal anti-LAMP1 (clone H4A3; Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal anti-VAMP7 (clone 158.2; Covalab, Villeurbanne, France), mouse monoclonal anti-Vti1b (clone 7; BD Biosciences, Sparks, MD), mouse monoclonal anti-Syntaxin 8 (clone 48; BD Biosciences), mouse monoclonal anti-β-actin (clone AC-74; Sigma-Aldrich), rabbit polyclonal anti-LAMP1 (Abcam, Cambridge, MA), rabbit polyclonal anti-VAMP8 (Covalab), mouse polyclonal anti-Syntaxin 7 (Abnova, Taipei, Taiwan), and rabbit polyclonal anti-LC3 (MBL, Nagoya, Japan). Alexa Fluor-conjugated secondary antibodies (goat anti-mouse immunoglobulin [Ig]G and goat anti-rabbit IgG) were purchased from Invitrogen and used for fluorescence microscopy.
Western blotting was done as described previously (Kamimoto et al., 2006). To determine the intensities of the blotting bands, each was selected using the selection tool in ImageJ software (http://rsb.info.nih.gov/ij/), and each band intensity was quantified using the Plot lane command.
siRNAs and Plasmids
Two sets of siRNA duplexes for each SNARE were used to knock down VAMP7 (siRNA duplex sets: SYBL1-HSS110395 and SYBL1-HSS110396), VAMP8 (VAMP8-HSS112730 and VAMP8-HSS112731), Vti1b (Vti1b-HSS145663 and Vti1b-HSS145664), Syntaxin 7 (STX7-HSS112238 and STX7-HSS112239), Syntaxin 8 (STX8-HSS114122 and STX8-HSS114123), and a siRNA-negative control (Stealth RNAi; all from Invitrogen). siRNA-SYBL1-HSS110395 corresponded to nucleotides 240-264 of human vamp7 (GenBank accession NM_005638) located at the NH2-terminal domain. siRNA-SYBL1-HSS110396 corresponded to nucleotides 517-541, which are located in the coiled-coil domain (R-SNARE motif). siRNA-VAMP8-HSS112730 corresponded to nucleotides 194-218 of human vamp8 (GenBank accession NM_003761), which are located in the coiled-coil domain (R-SNARE motif). siRNA-VAMP8-HSS112731 corresponded to nucleotides 23-47 located in the NH2-terminal domain. siRNA-Vti1b-HSS145663 and iRNA-Vti1b-HSS145664 corresponded to nucleotides 318-342 and 387-411 of human vti1b (GenBank accession NM_006370), respectively. These nucleotides are located in the NH2-terminal domain. siRNA-STX7-HSS112238 corresponded to nucleotides 666-690 of human syntaxin7 (GenBank accession NM_003569), which are located in the coiled-coil domain (Q-SNARE motif). siRNA-STX7-HSS112239 corresponded to nucleotides 742-766 located in the transmembrane domain. siRNA-STX8-HSS114122 corresponded to nucleotides 462-486 of human syntaxin8 (GenBank accession NM_004853), which are located in the coiled-coil domain (Q-SNARE motif). siRNA-STX8-HSS114123 corresponded to nucleotides 663-687 located in the transmembrane domain. A plasmid encoding monomeric Cherry (mCherry) protein was a generous gift from Dr. Roger Y. Tsien (University of California, San Diego, San Diego, CA). To construct an mCherry-C1 plasmid, polymerase chain reaction (PCR) was used to generate mCherry cDNA with the exogenous restriction sites of NheI and BglII at the 5′ and 3′ ends, respectively, lacking the termination codon. The PCR fragment obtained after digestion with each restriction enzyme was used to replace EGFP cDNA of pEGFP-C1. To construct mCherry-Sec20, Slt1, Syntaxin 6, VAMP7, and Vti1b plasmids, cDNA was cloned from genomic DNA isolated from HeLa cells and then inserted into pmCherry C1 using engineered SalI and BamHI sites. To construct an mCherry-VAMP8 plasmid, cDNA was cloned from genomic DNA isolated from HeLa cells and inserted into pmCherry C1 using engineered SalI and KpnI sites. The expression vectors for mRFP-GFP-LC3 (tf-LC3) and mRFP-LC3 plasmids have been described previously (Kimura et al., 2007).
Fluorescence Microscopy
For immunostaining, the cells were washed with phosphate-buffered saline (PBS), fixed with 3% paraformaldehyde in PBS for 15 min, and permeabilized with 50 μg/ml digitonin in blocking solution (0.1% gelatin in PBS) for 10 min. After being washed twice with PBS, the cells were incubated in blocking solution for 20 min and subsequently with primary antibodies diluted with blocking solution at room temperature for 1 h. After being washed twice with PBS, the cells were probed with secondary antibodies conjugated with Alexa 405, 488, 594, or 633 (Invitrogen). Samples were examined under a fluorescence laser scanning confocal microscope (model LSM510; Carl Zeiss, Thornwood, NY) using Zeiss LSM Image Browser software (Carl Zeiss).
Identification of GAS and GcAV was performed as described previously (Nakagawa et al., 2004; Yamaguchi et al., 2009). In brief, green fluorescent protein (GFP)-LC3 and DNA of GAS were observed in GFP and 4,6-diamidino-2-phenylindole (DAPI) channels, respectively. Obtained images were merged to compare the two signal patterns. GFP-LC3 puncta closely surrounding and containing a GAS chain were identified as GcAV. To quantify the colocalization among different compartments, GFP-LC3, SNAREs, and LAMP1 were observed in the GFP, red fluorescent protein (RFP), and cyanine (Cy)5 channels, respectively. Obtained images were merged, and then the overlapped areas of the images were measured using the overlay tool of LSM Image Browser software (Carl Zeiss). Merged compartments with >95% overlap were determined to be colocalized. Colocalization of monomeric (m)RFP with GFP pixels of tfLC3, LAMP1, or dextran was also determined using the profile tool of LSM Image Browser software (Carl Zeiss), as described previously (Kimura et al., 2007). Obtained images were merged to compare the two signal patterns, and plot profiles derived from the merged images were quantified. Colocalization of RFP-LC3, GFP-LC3, and each SNARE was observed in the GFP, RFP, and Cy5 channels, respectively. Obtained puncta images were merged to compare the three signal patterns, and colocalization of these three signals was determined by manual counting.
Alexa Fluor 488-conjugated dextran (Mr 10 000) was purchased from Invitrogen. The reagent was added to the medium of HeLa cells to a final concentration of 0.5 mg/ml, followed by incubation at 37°C with 5% CO2 overnight. The next day, the cells were transferred to fresh medium, incubated for 4 h to chase, and transfected with siRNA for the control, VAMP7, VAMP8, and Vti1b. At 24 h after siRNA transfection, the cells were transfected with plasmids expressing mRFP-LC3. At 24 h after plasmid transfection, the cells were starved in EBS solution for 3 h and then fixed and analyzed with confocal microscopy. Detection of cathepsin L activity by fluorescent microscopy was performed using a Magic Red Cathepsin L detection kit (Immunochemistry Technologies, Bloomington, MN) according to the manufacturer's protocol.
Proteinase Inhibitor
For Western blotting of LC3, E64d and pepstatin A were purchased from the Peptide Institute (Osaka, Japan). Cells were transfected with siRNA for the control, VAMP7, VAMP8, and Vti1b. After 48 h of incubation, the cells were cultured in EBS solution with E64d (10 μg/ml) and pepstatin A (30 μg/ml), or without proteinase inhibitors (control; dimethyl sulfoxide [DMSO] 1.1 μg/ml) for appropriate times. Lysate samples were examined by Western blotting using the anti-LC3 and actin antibodies.
GAS Infection
Infection with GAS (strain JRS4) was performed as described previously (Nakagawa et al., 2004). In brief, bacterial cells were added to cell cultures without antibiotics for 1 h, and then infected cells were washed with PBS and antibiotics (100 μg/ml gentamicin and 100 U/ml penicillin G) were added for an appropriate period to kill extracellular bacteria.
Gas Invasion Assays
GAS cells were incubated separately with 0.1 mCi of [methyl-3H]thymidine (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) for 12 h, after which they were harvested and washed with PBS. 3H-Labeled GAS (multiplicity of infection [MOI]), 100] was added to HeLa cells for 1 h and then washed three times with PBS to remove external nonadherent bacteria cells. Next, GAS-infected HeLa cells were incubated for 2 h in medium containing antibiotics, after which the cells were disrupted by addition of 100 μl of distilled water with incubation at 37°C for 10 min. The numbers of invading organisms were determined using a liquid scintillation counter (model LSC-5100; Aloka, Tokyo, Japan) and from the amounts of 3H recovered from infected cells, with the results expressed as percentages of the total numbers of GAS organisms added.
Bacterial Viability Assay
A colony-forming units viability assay was performed as described previously (Nakagawa et al., 2004). GAS (MOI, 100) was added to HeLa cells for 1 h and then washed three times with PBS to remove external nonadherent bacteria cells. Next, GAS infected HeLa cells were incubated for 2 h in medium containing antibiotics, after which the cells were disrupted by addition of 100 μl of distilled water with incubation at 37°C for 10 min. Serial dilutions of the lysates were plated on THY agar plates, and colony counting was performed.
Statistics
All values shown in figures are presented as the mean ± SD. Statistical significance (p value) was determined using one-way analysis of variance (ANOVA) with Scheffe posttest (SPSS, Chicago, IL).
RESULTS
Localization of GFP-LC3 with SNAREs in GcAVs (Xenophagosomes)
To determine whether SNARE proteins become localized in GcAVs of GAS-infected HeLa cells, endosome SNAREs (VAMP7, VAMP8, and Vti1b), endoplasmic reticulum (ER)-residing SNAREs (Sec20 and Slt1), and trans-Golgi network (TGN) residing Syntaxin 6 were examined. As shown in Figure 1, GFP-LC3 puncta associated with GcAVs were found to colocalize with those of VAMP7, VAMP8, and Vti1b, whereas the other SNAREs were not markedly overlapped with GFP-LC3.
Figure 1.

Colocalization of GFP-LC3 with SNAREs in GcAVs. (A) HeLa cells stably expressing GFP-LC3 were transfected with plasmids to express mCherry-Sec20, Slt1, Syntaxin 6, Vti1b, VAMP7, and VAMP8. At 24 h after transfection, the cells were infected with GAS for 180 min at an MOI of 100, as described in Materials and Methods. Cellular and bacterial DNAs were stained with DAPI. The boxed regions in the top panels are enlarged in the bottom panels. Bars, 10 μm (top) and 5 μm (bottom). (B) Colocalization frequencies of GcAVs with several mCherry signals were manually determined as the percentage of the total number of GcAVs. Data shown represent results of >60 cells and each ratio (percentage) represents the mean value ± SD from three independent experiments.
To determine whether these localizations occurred before or after lysosomal fusion, we next examined the colocalization of VAMP7, VAMP8, and Vti1b with LAMP1 and GFP-LC3 (Figures 2 and 3). Immediately after infection, VAMP7 and VAMP8 puncta were negligibly merged with GFP-LC3 and LAMP1, whereas they gradually became colocalized with them over time, indicating that VAMP7 and VAMP8 become localized in GcAVs after fusion with lysosomes. In contrast, GcAVs were found to possess Vti1b from an early stage, and LAMP1 merged with Vti1b and GFP-LC3 in LAMP1-acquired GcAVs. These results suggest that VAMP7 and VAMP8 originate from lysosomes, whereas Vti1b is derived from xenophagosomes before fusion with lysosomes.
Figure 2.

Colocalization of GFP-LC3 with VAMP7, VAMP8, and Vti1b in GcAVs. HeLa cells stably expressing GFP-LC3 were infected with GAS for 60 min at an MOI of 100. Infected cells were washed and incubated with antibiotics for the indicated time periods and then fixed and incubated with anti-LAMP1, VAMP7, VAMP8, and Vti1b antibodies. Cellular and bacterial DNAs were stained with DAPI. The boxed regions in the left panels are enlarged in the right panels. Bars, 10 μm (left) and 5 μm (right).
Figure 3.

Colocalization of GcAVs with SNAREs and LAMP1. The colocalization frequencies of GcAVs with VAMP7, VAMP8, and Vti1b in Figure 2 were determined. SNARE- and LAMP1-negative cells are shown in green, SNARE-positive and LAMP1-negative cells in white, SNARE-negative and LAMP1-positive cells in red, and SNARE- and LAMP1-positive cells in blue. The numbers of those signals were manually counted and are presented as a percentage of the total number of GcAVs. Data shown represent results of >60 cells.
Knockdown of VAMP8 and Vti1b Disturbs Bactericidal Effects of GcAVs
Next, we examined the effects of knockdown of VAMP7, VAMP8, and Vti1b with siRNAs on GcAVs. The expression of these SNARE proteins was significantly suppressed in HeLa cells (Figure 4A). After infecting HeLa cells with GAS, GFP-LC3–positive GcAVs were clearly merged with LAMP1 in the control and VAMP7-depleted cells (Figure 4, B and C), whereas siRNA knockdown of VAMP8 and Vti1b apparently inhibited the colocalization of GFP-LC3 with LAMP1. However, GcAV formation efficiency was not altered by knockdown of these SNAREs (Figure 4D). These results indicate that VAMP8 and Vti1b are directly involved with the fusion of xenophagosomes with lysosomes. The effects of knockdown of these SNAREs were also analyzed in regard to their cellular bactericidal effects. Although the invasive efficiency of GAS into cells was not altered by knockdown (Figure 4E), bactericidal efficiency was significantly diminished in VAMP8- and Vti1b-depleted cells (Figure 4F).
Figure 4.

Knockdown of VAMP8 and Vti1b disturbed the antimicrobial effects of GcAVs. (A) HeLa cells were transfected with siRNA for the control, VAMP7, VAMP8, and Vti1b. At 48 h after transfection, the cells were lysed and then examined by Western blotting using anti-Vti1b, -VAMP7, -VAMP8, and actin antibodies. (B) HeLa cells expressing GFP-LC3 were treated with siRNA in the same manner as described in A. After 48 h, cells were infected with GAS for 60 min at an MOI of 100. After an additional 120 min of incubation with antibiotics for bacterial killing, the cells were fixed and incubated with anti-LAMP1 antibodies and observed with a confocal microscope. Cellular and bacterial DNAs were stained with DAPI. The boxed regions in the left panels are shown enlarged in the right panels. Bars, 10 μm (left) and 5 μm (right). (C) Colocalization frequencies of GcAVs with LAMP1 signals were manually determined and are presented as the percentage of total number of GcAVs. Data shown represent results of >60 cells, and each ratio (percentage) represents the mean value ± SD from three independent experiments. *p < 0.01; **p < 0.05 by one-way ANOVA and Scheffé's posttest. (D) Numbers of cells containing GcAVs were counted and are presented as the percentage of the total number of GAS infected cells. HeLa cells stably expressing GFP-LC3 were transfected with siRNA and infected with GAS in the same manner as described in B. Data shown represent results of >30 cells and each ratio (percentage) represents the mean value ± SD from three independent experiments. (E) Efficiency of GAS invasion was measured as described in Materials and Methods. Data are shown as the mean ± SD from three independent experiments. (F) Viability of invaded GAS in HeLa cells was evaluated as described in Materials and Methods. Data are shown as the mean ± SD from three independent experiments. *p < 0.01; **p < 0.05 by one-way ANOVA and Scheffé's posttest.
Knockdown of VAMP8 and Vti1b Disturbs Maturation of Canonical Autophagosomes
We also analyzed the effects of knockdown of SNARE proteins on canonical autophagy using HeLa cells. It is known that LC3 contains two forms: LC3-I that resides in cytosol as a soluble protein and LC3-II that is associated with autophagosomes (Eskelinen, 2005). Under starvation, LC3-I is converted to LC3-II, and autophagosomes are visualized as GFP-LC3 puncta. In the control cells, punctate GFP-LC3 structures were clearly observed at 120 min and then disappeared, with subsequent longer starvation of 240 min (Figure 5, A and B), due to the degradation of LC3-II in autolysosomes as well as detachment from autolysosomes (Eskelinen, 2005). Although knockdown of VAMP8 or Vti1b negligibly altered the number of GFP-LC3 dots in both starved and fed conditions at 120 min, it apparently inhibited the disappearance of GFP-LC3 puncta at 240 min. Knockdown of VAMP7 resulted in little effects in regard to LC-II protein level. In contrast, considerable amounts of LC3-II protein remained in the VAMP8- and Vti1b-depleted cells (Figure 5, C and D). In a previous study, after cells were treated with lysosomal protease inhibitors such as E64d and pepstatin A, degradation of LC3-II was inhibited, whereas that of LC3-I was not affected (Mizushima and Yoshimori, 2007). That report also demonstrated that the amount of LC3-II at a certain time point does not indicate the total amount of autophagic flux, rather that flux is more accurately represented by differences in the amounts of LC3-II between samples in the presence and absence of lysosomal protease inhibitors. We examined autophagic flux using E64d and pepstatin A, and we observed vastly different densities between LC3-II bands in the presence and absence of protease inhibitors in both control and VAMP7-depleted cells, indicating the induction of normal autophagic flux (Figure 6, A and B). In contrast, there were only slight differences between band densities with and without the inhibitors in the VAMP8- and Vti1b-depleted cells. Furthermore, knockdown of VAMP8 and Vti1b clearly disturbed the maturation of canonical autophagosomes in other cell types, including MCF7 and A549 cells (Supplemental Figures 1–4). These results indicate that knockdown of VAMP8 and Vti1b prevents LC3-II degradation in autolysosomes and suggest that VAMP8 and Vti1b are also involved in maturation of canonical autophagosomes.
Figure 5.

Knockdown of VAMP8 and Vti1b disturbed the maturation of canonical autophagosomes. (A) HeLa cells stably expressing GFP-LC3 were treated with siRNA in the same manner as described in Figure 4A. The cells were further cultured in growth medium (Fed) or EBS solution (Starved) for the indicated periods and then fixed and observed with a confocal microscope. Cellular DNA was stained with DAPI (blue). Bars, 10 μm. (B) Quantitative analysis of the number of GFP-LC3 puncta per cell shown in A was performed using ImageJ software. More than 100 cells were examined. Data are shown as the mean ± SD (C) siRNA-treated HeLa cells were cultured in Fed and Starved conditions for 240 min. The cellular lysates were subjected to western blotting using anti-LC3 and actin antibodies. (D) Quantitative analysis of Western blot bands shown in C was performed using ImageJ software. Data show the ratios of LC3-II band intensities to the actin bands. Values are shown as the mean ± SD from three independent experiments.
Figure 6.
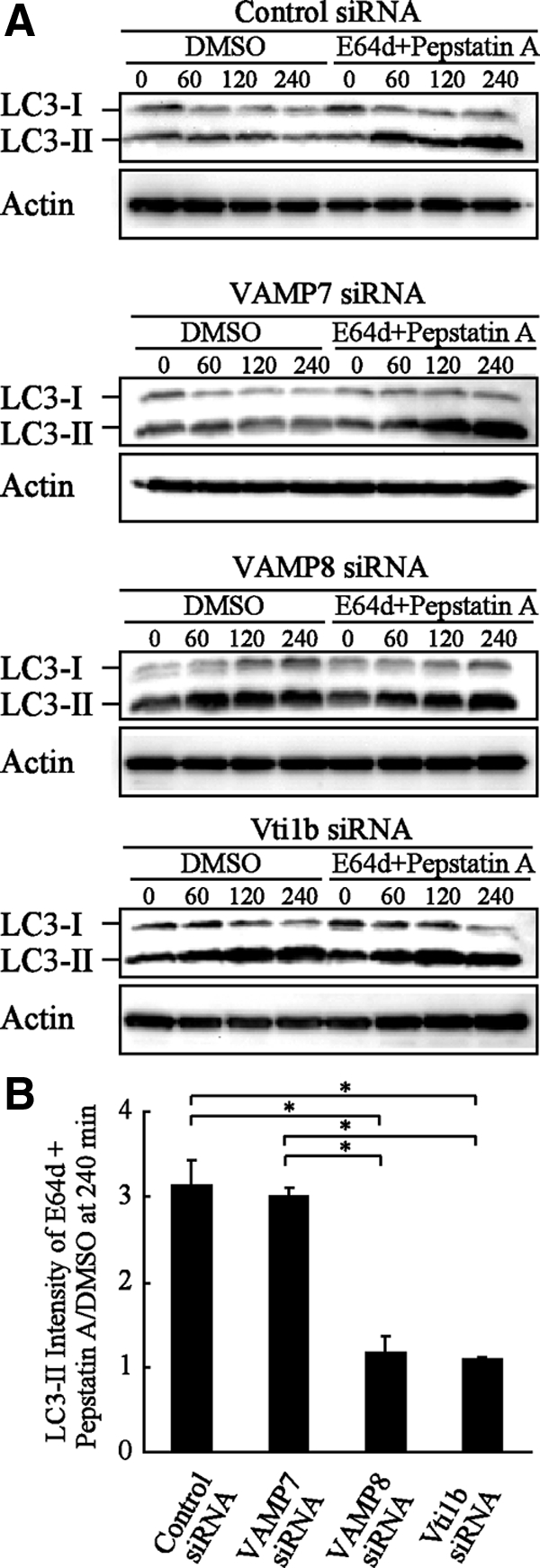
LC3-II proteins accumulated without degradation in VAMP8- and Vti1b-depleted HeLa cells. (A) HeLa cells were treated with siRNA in the same manner as described in Figure 4A. At 48 h after transfection, the cells were cultured in starved solution (EBS) with or without proteinase inhibitors for the indicated times. The cellular lysates were examined to measure the amounts of LC3-II proteins with Western blotting using anti-LC3 and actin antibodies. DMSO was used as the control. (B) Quantitative analysis of the relative intensities of the LC3-II bands (inhibitor treated/control) after 240 min in A was performed using ImageJ software. The mean values ± SD are shown from three independent experiments. *p < 0.01 by one-way ANOVA and Scheffé's posttest.
Impaired Degradation of LC3 Proteins in Autophagosomes of VAMP8- and Vti1b-depleted Cells
Fusion of autophagosomes with lysosomes provides an acidic environment to digest the interior. A recent study showed that GFP loses its fluorescence in acidic conditions and is subsequently degraded by lysosomal hydrolases, whereas mRFP is stable under degradation conditions (Kimura et al., 2007). In contrast, the life span of xenophagosomes (GcAVs) is significantly longer than that of canonical autophagosomes, and GFP-LC3 notably remains on GcAVs for up to several hours or even days (Nakagawa et al., 2004). Therefore, mRFP-LC3 as well as mRFP-GFP in tandem with fluorescently tagged LC3 (tfLC3) have been devised for dissecting the maturation process of autophagosomes to autolysosomes (Kimura et al., 2007). Using tfLC3, we analyzed the distributions of mRFP and GFP signals in LC3 proteins of canonical autophagosomes in VAMP8- and Vti1b-depleted cells. mRFP puncta were inconsistently merged with those of GFP in the control as well as VAMP7-depleted cells at 180 min after starvation, indicating that GFP signals were attenuated by the acidic condition in the autolysosomes, whereas mRFP remained (Figure 7A). In contrast, the puncta of these markers were clearly colocalized in VAMP8- and Vti1b-depleted cells, indicating negligible attenuation of GFP, because few LC3 proteins were sorted to autolysosomes. Quantitative analysis also indicated that a majority of GFP signals remained with mRFP in autophagosomes in VAMP8- and Vbti1b-depleted cells (Figure 7B), indicating prevention of fusion between autophagosomes and lysosomes in those cells.
Figure 7.

Impaired degradation of LC3 proteins in autophagosomes from VAMP8- and Vti1b-depleted cells. (A) HeLa cells were transfected with siRNA for the control, VAMP7, VAMP8, and Vti1b. At 24 h after transfection, the cells were further transfected with plasmids expressing tf-LC3. After 24 h of incubation, the cells were subjected to a starved condition for 180 min and then fixed and observed with a confocal microscope. The boxed regions in the left panels are enlarged in the right panels. Bars, 10 μm (left) and 5 μm (right). (B) The colocalization frequencies of mRFP with GFP signals shown as tf-LC3 pixels in A were determined using LSM Image Browser software (Carl Zeiss) and are presented as the percentage of total number of mRFP pixels. Values are shown as the mean ± SD of >60 cell images. *p < 0.01 by one-way ANOVA and Scheffé's posttest.
To determine whether lysosomal enzyme failure in VAMP8- and Vti1b-depleted cells is the cause of this phenotype, the distribution of mRFP-LC3 in lytic compartments was further examined. RFP signals were clearly merged with LAMP1 in the control and VAMP7-depleted cells after 180 min of starvation (Figure 8, A and C), whereas their coexistence was merely observed in VAMP8- and Vti1b-depleted cells. Next, we used another lysosome marker, dextran (Bright et al., 2005), and preloaded HeLa cells with Alexa 488 dextran for marking lysosomes via endocytosis and then starved them for 180 min, after which the colocalization of dextran with mRFP-LC3 was examined as described previously (Kimura et al., 2007). This assay also indicated that colocalization of the markers for autophagosomes and lysosomes was significantly inhibited by siRNA for VAMP8- and Vti1b, respectively (Figure 8, B and D).
Figure 8.

Colocalization of mRFP-LC3 with LAMP1 in VAMP8- and Vti1b-depleted cells. (A) HeLa cells were transfected with siRNA for the control, VAMP7, VAMP8, and Vti1b. At 24 h after transfection, the cells were further transfected with plasmids expressing mRFP-LC3. After 24 h of incubation, the cells were subjected to a starved condition for 180 min, followed by fixation and incubation with anti-LAMP1 antibodies and then observed with a confocal microscope. Cellular DNA was stained with DAPI. Bars, 10 μm. (B) HeLa cells were preloaded with Alexa 488 dextran for marking lysosomes, as described in Materials and Methods. Cellular DNA was stained with DAPI. Bars, 10 μm. (C) The colocalization frequencies of mRFP shown as LAMP1 pixels were determined using LSM Image Browser software (Carl Zeiss) and are presented as the percentage of total number of mRFP pixels. Values are shown as the mean ± SD of >30 images. *p < 0.01 by one-way ANOVA and Scheffé's posttest. (D) The colocalization frequency of mRFP-LC3 shown as Alexa 488 dextran pixels was determined using LSM Image Browser (Carl Zeiss) and presented as the percentage of total number of mRFP pixels. The mean value ± SD of >30 cell images is shown. *p < 0.01 by one-way ANOVA and Scheffé's posttest.
We also examined whether knockdown of Vti1b or VAMP8 had a negative influence on lysosomal maturation and proteolytic activation. The lysosomal cysteine peptidase cathepsin L significantly contributes to terminal degradation of proteins in lysosomes (Mohamed and Sloane, 2006). To validate the effects on lysosomal maturation and proteolytic activation, we examined cathepsin L activities in HeLa and MCF7 cells after SNARE knockdown by using a Magic Red Cathepsin L detection kit, which enables determination of cathepsin L activity in whole cells with fluorescent microscopy (Droga Mazovec et al., 2008; Razi et al., 2009). As shown in Supplemental Figure 5, fluorescent signals were clearly observed around the nuclei in control cells under both fed and starved conditions, whereas they were apparently attenuated by the addition of lysosomal protease inhibitors (E64d and pepstatin A). In contrast, fluorescence was clearly observed in VAMP7-, VAMP8-, and Vti1b-depleted cells in a manner similar to the control cells. These results indicate that lysosomal maturation and proteolytic activation were not diminished by knockdown of VAMP8 and Vti1b.
Vti1b interacts with Syntaxin 7 and Syntaxin 8 to form the t-SNARE complex, which mediates the formation of both late endosomes and lysosomes (Antonin et al., 2000). Thus, we examined whether Syntaxin 7 and Syntaxin 8 are also involved in autophagic fusion with lysosomes. It was observed that GFP-LC3-positive GcAVs were clearly merged with LAMP1 in Syntaxin 7- and Syntaxin 8-depleted cells in a manner similar to the control cells (Figure 9). In addition, knockdown of Syntaxin 7 and Syntaxin 8 did not interfere maturation of canonical autophagosome (Figure 10). These results indicate that Syntaxin 7 and Syntaxin 8 are not involved in the formation of GcAVs or canonical autophagy.
Figure 9.

Antimicrobial effects of GcAVs on Syntaxin 7 and Syntaxin 8-depleted cells. (A) HeLa cells were transfected with siRNA for the control, Syntaxin 7, and Syntaxin 8. At 48 h after transfection, the cells were lysed and examined by Western blotting using anti-Syntaxin 7, -Syntaxin 8, and actin antibodies. (B) HeLa cells stably expressing GFP-LC3 were transfected with siRNA for the control, Syntaxin 7, and Syntaxin 8. After 48 h, the cells were infected with GAS for 180 min at an MOI of 100 as described in Materials and Methods. After fixation, the cells were incubated with anti-LAMP1 antibodies and observed with a confocal microscope. Cellular and bacterial DNA were stained with DAPI. The boxed regions in the top panels are enlarged in the bottom panels. Bars, 10 μm (top) and 5 μm (bottom). (C) Colocalization frequencies of GcAVs with LAMP1 signals were manually determined and are presented as the percentage of total number of GcAVs. Data shown represent results of >30 cells.
Figure 10.

Maturation of canonical autophagosomes in Syntaxin 7 and Syntaxin 8-depleted cells. (A) MCF7 cells were transfected with siRNA for the control, Syntaxin 7, and Syntaxin 8. At 48 h after transfection, the cells were lysed and then examined by Western blotting using anti-Syntaxin 7, -Syntaxin 8, and actin antibodies. (B) MCF7 cells stably expressing GFP-LC3 were treated with siRNA in the same manner as described in A. The cells were further cultured in growth medium (Fed) or EBS solution (Starved) for the indicated times, then fixed and observed with a confocal microscope. Cellular DNA was stained with DAPI (blue). Bars, 10 μm. (C) siRNA-treated MCF7 cells were cultured in Fed and Starved conditions for 240 min, and then the cellular lysates were subjected to Western blotting using anti-LC3 and actin antibodies.
Collectively, these findings strongly suggest that the combinational SNARE proteins VAMP8 and Vti1b also mediate the fusion of autophagosomes with lysosomes.
Localization of VAMP8 and Vti1b
We also examined the localization of VAMP8 and Vti1b in terms of canonical autophagy by using tfLC3. At 60 min after beginning starvation, mRFP-LC3 (autolysosome marker) accounted for >15% of the LC3 puncta, whereas the residual portion was positive for mRFP-GFP-LC3 (autophagosome marker; Supplemental Figure 6A). mRFP-GFP-LC3 was negligibly colocalized with VAMP8 at 60 min after beginning starvation, and mRFP-LC3 puncta were clearly merged with those of VAMP8, indicating that VAMP8 resides in autolysosomes (Supplemental Figure 6, B and C). In contrast, mRFP-GFP-LC3 was apparently merged with Vti1b, whereas a few mRFP-LC3 puncta were colocalized with Vti1b (Supplemental Figure 6, B and C), showing that Vti1b resides in autophagosomes. These results also indicate that autophagosome-lysosome fusion is likely mediated by a combination of autophagosome-derived Vti1b and lysosomal VAMP8.
DISCUSSION
Our results showed that the fusion events of antimicrobial and canonical autophagosomes with lysosomes are mediated by the combinational SNARE proteins VAMP8 and Vti1b. In contrast, Syntaxin 7 and Syntaxin 8, which also function as t-SNAREs, were not shown to be involved in that fusion. This is the first report of the involvement of SNAREs in the autophagic process in mammals and several of our findings are worthy of special mention.
First, it is of interest that the late stages of autophagic maturation interconnect with the endocytic pathway by sharing the SNARE machinery, which is known to be involved in homotypic fusion within late endosomes. In this study, a unique combination of endocytic SNAREs was shown to function in autophagy, whereas previous studies have suggested that autophagosomal maturation has several other features similar to the progression of endosomes to lysosomes. For example, it was revealed that autophagosome–endosome fusion depends on Vps4/SKD1 (Nara et al., 2002), Rab11 (Fader et al., 2008), homotypic fusion and protein sorting (HOPS) complex (Liang et al., 2008), Hrs (Tamai et al., 2007), and the ESCRT III complex (Rusten et al., 2007; Lee et al., 2007), whereas autophagosome-lysosome fusion was shown to be mediated by Rab7 (Gutierrez et al., 2004; Jager et al., 2004; Yamaguchi et al., 2009), UV radiation resistance-associated gene (Liang et al., 2008), the HOPS complex (Lindmo et al., 2006), presenilin (Esselens et al., 2004), and LAMP2 (Eskelinen et al., 2004). It is also interesting that Vti1b was found to be recruited to autophagosomes before lysosomal fusion. Although the mechanism of Vti1b delivery to autophagosomes remains to be determined, it is an important issue, as it is possible that it is acquired by fusion with endosomes. We also found that xenophagosomes and autophagosomes share the same SNARE machinery to fuse with lysosomes, even though antimicrobial autophagy against GAS differs from canonical autophagy in several other aspects, such as size, morphology, and the conditions required for initiation (Yoshimori and Amano, 2009). We recently reported that GcAVs are formed through the fusion of multiple isolation membrane like-structures (Yamaguchi et al., 2009). In spite of these discrepancies, our results indicate that GcAVs and autophagosomes share the same set of SNARE machinery to fuse with lysosomes.
The present study also revealed the involvement of SNAREs in autophagosome–lysosome fusion in mammalian cells. Phenotypical alteration by deletion of Vti1b was examined previously using knockout (KO) mice (Atlashkin et al., 2003). Contrary to expectation, most of the vti1b-KO mice in that study behaved normally and were indistinguishable from wild-type mice, with no defects in transport to the lysosomes. Furthermore, only a limited percentage (20%) of the mice was physically smaller, whereas multivesicular bodies and autophagic vacuoles were accumulated in hepatocytes. Compensatory activation/hyperfunction of other related genes/molecules might have prevented the phenotypical alteration in those KO mice. In contrast, the present knockdown assay clearly showed that an alteration was caused by the deficiency. In addition, recent studies with yeast have implicated the SNAREs Vam3 (possible mammalian orthologue for syntaxin7) and Vti1 (mammalian homologue for Vti1a) in the fusion of autophagosomes with vacuoles of Saccharomyces cerevisiae (Darsow et al., 1997; Fischer von Mollard and Stevens, 1999; Ishihara et al., 2001). Mammalian Vti1a and Vti1b share 30% of their amino acid residues with each other (Advani et al., 1998). Therefore, we examined the effect of Vti1b depletion on expression of Vti1a protein and confirmed that there were no differences in regard to Vti1a protein level between the control and Vti1b knockdown cells (Supplemental Figure 7). Similarly to Syntaxin 7 and Syntaxin 8, Vti1a is not likely to be involved in autophagic fusion with lysosomes.
The involvement of VAMP8 and Vti1b in bacterial invasion of host cells remains unclear. In this study, knockdown of VAMP8 and Vti1b showed negligible effects on the invasive efficiency of GAS, suggesting the nonparticipation of these SNAREs in the bacterial invasive event. However, it was reported previously that VAMP8 is recruited to bacteria-induced membrane ruffles, which facilitates invasion of HeLa cells by Salmonella enterica serovar Typhimurium (Dai et al., 2007). Furthermore, knockdown of VAMP8 by siRNA reduced the invasion level of Salmonella, indicating that Salmonella exploits host SNARE proteins and vesicle trafficking to promote bacterial entry. GAS uses a different strategy to enter cells as compared with Salmonella (Cossart and Sansonetti, 2004) and their diverse underlying mechanisms might be the cause of these discrepant findings. The involvement of VAMP8 in bacterial invasion requires additional investigation.
In summary, our results present several new aspects to help unravel the mechanism underlying regulation of autophagic maturation events for the degradation of cargos. Autophagy is a versatile cellular machinery that has various physiological roles and its striking evolution has been shown to exploit, at least in part, interconnections with household pathway(s) by sharing ordinary machinery such as SNAREs. Additional investigations will help to elucidate the mechanism of not only canonical autophagy but also xenophagy formation.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grants-in-aid for scientific research (B) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Abbreviations used:
- GcAV
GAS-containing autophagosome-like vacuoles
- SNAREs
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- GAS
group A Streptococcus
- mRFP
monomeric red fluorescent protein
- LAMP
lysosomal membrane protein
- MOI
multiplicity of infection.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-08-0693) on January 20, 2010.
REFERENCES
- Advani R. J., Bae H. R., Bock J. B., Chao D. S., Doung Y. C., Prekeris R., Yoo J. S., Scheller R. H. Seven novel mammalian SNARE proteins localize to distinct membrane compartments. J. Biol. Chem. 1998;273:10317–10324. doi: 10.1074/jbc.273.17.10317. [DOI] [PubMed] [Google Scholar]
- Antonin W., Holroyd C., Fasshauer D., Pabst S., Von Mollard G. F., Jahn R. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J. 2000;19:6453–6464. doi: 10.1093/emboj/19.23.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlashkin V., Kreykenbohm V., Eskelinen E. L., Wenzel D., Fayyazi A., Fischer von Mollard G. Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol. Cell. Biol. 2003;23:5198–5207. doi: 10.1128/MCB.23.15.5198-5207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright N. A., Gratian M. J., Luzio J. P. Endocytic delivery to lysosomes mediated by concurrent fusion and kissing events in living cells. Curr. Biol. 2005;15:360–365. doi: 10.1016/j.cub.2005.01.049. [DOI] [PubMed] [Google Scholar]
- Cossart P., Sansonetti P. J. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Cunningham M. W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S., Zhang Y., Weimbs T., Yaffe M. B., Zhou D. Bacteria-generated PtdIns(3)P recruits VAMP8 to facilitate phagocytosis. Traffic. 2007;8:1365–1374. doi: 10.1111/j.1600-0854.2007.00613.x. [DOI] [PubMed] [Google Scholar]
- Darsow T., Rieder S. E., Emr S. D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Curr. Opin. Immunol. 2009;21:53–62. doi: 10.1016/j.coi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droga Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U., Salvesen G. S., Stoka V., Turk V., Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008;283:19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- Eskelinen E. L., et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol. Biol. Cell. 2004;15:3132–3145. doi: 10.1091/mbc.E04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen E. L. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Eskelinen E. L., Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Esselens C., et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004;166:1041–1054. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader C. M., Sanchez D., Furlan M., Colombo M. I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G., Stevens T. H. The Saccharomyces cerevisiae v-SNARE Vti1p is required for multiple membrane transport pathways to the vacuole. Mol. Biol. Cell. 1999;10:1719–1732. doi: 10.1091/mbc.10.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez M. G., Munafo D. B., Beron W., Colombo M. I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Hara T., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hong W. SNAREs and traffic. Biochim. Biophys. Acta. 2005;1744:493–517. [PubMed] [Google Scholar]
- Ishihara N., Hamasaki M., Yokota S., Suzuki K., Kamada Y., Kihara A., Yoshimori T., Noda T., Ohsumi Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol. Biol. Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., Eskelinen E. L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- Kamimoto T., Shoji S., Hidvegi T., Mizushima N., Umebayashi K., Perlmutter D. H., Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Komatsu M., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Lee J. A., Beigneux A., Ahmad S. T., Young S. G., Gao F. B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Liang C., Lee J. S., Inn K. S., Gack M. U., Li Q., Roberts E. A., Vergne I., Deretic V., Feng P., Akazawa C., Jung J. U. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindmo K., Simonsen A., Brech A., Finley K., Rusten T. E., Stenmark H. A dual function for Deep orange in programmed autophagy in the Drosophila melanogaster fat body. Exp. Cell Res. 2006;312:2018–2027. doi: 10.1016/j.yexcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Matsunaga K., et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mohamed M. M., Sloane B. F. Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Nakagawa I., et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Nara A., Mizushima N., Yamamoto A., Kabeya Y., Ohsumi Y., Yoshimori T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 2002;27:29–37. doi: 10.1247/csf.27.29. [DOI] [PubMed] [Google Scholar]
- Razi M., Chan E. Y., Tooze S. A. Early endosomes and endosomal coatomer are required for autophagy. J. Cell Biol. 2009;185:305–321. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Duden R., Rubinsztein D. C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Rusten T. E., et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 2007;17:1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Seglen P. O., Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–172. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- Tamai K., Tanaka N., Nara A., Yamamoto A., Nakagawa I., Yoshimori T., Ueno Y., Shimosegawa T., Sugamura K. Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem. Biophys. Res. Commun. 2007;360:721–727. doi: 10.1016/j.bbrc.2007.06.105. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H., Nakagawa I., Yamamoto A., Amano A., Noda T., Yoshimori T. Dynamic formation GAS-containing autophagosome-like vacuoles is dependent upon on Rab7. PLoS Pathog. 2009;5:e1000670. doi: 10.1371/journal.ppat.1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Amano A. Group A Streptococcus, a loser in the battle with autophagy. Curr. Top. Microbiol. Immunol. 2009;335:217–226. doi: 10.1007/978-3-642-00302-8_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.