

Abstract
The main purpose of this study was to determine whether the aging process in the mouse is associated with a pro-oxidizing shift in the redox state of glutathione and whether restriction of caloric intake, which results in the extension of life span, retards such a shift. Amounts of reduced and oxidized forms of glutathione (GSH and GSSG, respectively) and protein-glutathione mixed disulfides (protein-SSG) were measured in homogenates and mitochondria of liver, kidney, heart, brain, eye, and testis of 4, 10, 22, and 26 month old ad libitum-fed (AL) mice and 22 month old mice fed a diet containing 40% fewer calories than the AL group from the age of 4 months. The concentrations of GSH, GSSG, and protein-SSG vary greatly (∼10-, 30-, and 9-fold, respectively) from one tissue to another. During aging, the ratios of GSH:GSSG in mitochondria and tissue homogenates decreased, primarily due to elevations in GSSG content, while the protein-SSG content increased significantly. Glutathione redox potential in mitochondria became less negative, i.e., more pro-oxidizing, as the animal aged. Caloric restriction (CR) lowered the GSSG and protein-SSG content. Results suggest that the aging process in the mouse is associated with a gradual pro-oxidizing shift in the glutathione redox state and that CR attenuates this shift.
Keywords: Glutathione, Protein-mixed disulfides, S-thiolation, Mitochondria, Redox potential, Aging, Caloric restriction, Oxidative stress, Glutathiolation, Free radicals
Introduction
It is now widely accepted that the survival of aerobic cells depends on the presence of sufficient antioxidative defenses to modulate the concentrations of endogenously generated reactive oxygen species (ROS). Nevertheless, steady state amounts of oxidatively damaged macromolecules have been detected under normal physiological conditions, which has been inferred to suggest that a certain level of oxidative stress prevails even in cells of young, healthy animals [1–3]. Additionally, the amounts of oxidatively modified macromolecules have been found to increase with age in tissues of a variety of different species [2,4,5]. Such accumulation of oxidative damage has been hypothesized to be a primary causal factor in the senescence-associated attenuations of various physiological functions [6].
Notwithstanding these observations, the nature of the relationship between the accrual of oxidative damage and senescence-associated losses in cellular functions remains ambiguous. One major unanswered question is whether the level of oxidative stress, i.e., the imbalance between antioxidant capacity vs. oxidant load, remains steady or whether it increases with age. The resolution of this issue is important because an increase in the severity of oxidative stress with age would imply a relatively more rapid rate of accrual of oxidative damage and loss of functional capacity as the animal ages.
There is no single measure of intracellular antioxidative capacity or of the net oxidant load that may exist at a particular age. The redox state of cells, believed to be closely reflective of their antioxidant:pro-oxidant balance, is determined by measuring the amounts and the ratios of the interconvertible, reduced, and oxidized forms of various redox couples, such as (i) reduced: oxidized glutathione (GSH:GSSG), (ii) cysteine:cystine, (iii) reduced:oxidized thioredoxin, (iv) NAD(H):NAD+, and (v) NADP(H):NADP+ (reviewed in [7]). However, the glutathione redox couple is two to four orders of magnitude more abundant than any other redox couple; furthermore, it is metabolically linked to other redox couples by directly or indirectly donating the reducing equivalents for the reduction of their oxidized forms [8–10]. Glutathione (γ-L-glutamyl-L-cysteinylglycine) is, therefore, widely considered to be a representative indicator of the cellular redox state of tissues [7,11–13]. Oxidized glutathione (GSSG) can also interact with accessible cysteinyl thiols of many cellular proteins to form protein-glutathione mixed disulfides, a reaction also referred to as S-thiolation or glutathiolation [14–17]. Indeed, a variety of studies have indicated that S-thiolation of proteins is among the most sensitive and physiologically relevant cellular alteration during periods of upsurge in oxidative stress (reviewed in [15,17]). Accordingly, we used the glutathione redox state and protein-SSG content as surrogates for determining whether or not the imbalance between pro-oxidants and antioxidants increases during the aging process.
The present study reports a comparison of age-related changes in glutathione redox state and protein-SSG content in the homogenates and mitochondria of six different organs of C57BL/6Nnia mice. In addition, glutathione redox state and amounts of protein-SSG were compared between 22 month old ad libitum-fed (AL) mice and calorically restricted (CR) mice. The reason for examining the age-related patterns in the calorically restricted mice was that this regimen results in a significant extension of the life span of mice [18,19]. Thus, it can be employed for determining whether or not an age-related alteration is associated with the life expectancy or physiological age of an animal or with its chronological age alone. Age-related alterations that correlate with life expectancy are reasoned to be likely to play a causal role in the aging process. In addition to the tissue homogenates, measurements of glutathione redox state and protein-SSG content were also made in the mitochondrial fraction because mitochondria are widely believed to play a central role in the aging process, as both the main generators of ROS and the targets of oxidative damage [20–23]. Furthermore, the mitochondrial redox state is compartmentalized from the other regions of the cell and, thus, may exhibit a variant pattern [11,24,25]. Intramitochondrial GSSG content has been found to be correlated with oxidative damage to mitochondrial DNA [26] and lipids [27]. Our objective in this study was to extend the characterization of the redox state of glutathione to a variety of different tissues of the mouse and to compare glutathione redox state and protein-SSG content in tissue homogenates and their respective mitochondria at different ages and between AL and CR animals.
Our results demonstrate that there is a distinct pro-oxidizing shift in glutathione redox state in tissue homogenates and mitochondria of mice during aging, which is attenuated by caloric restriction.
Materials and Methods
Reagents
GSH and GSSG were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Acetonitrile, meta-phosphoric acid, and 1-octane sulfonic acid were acquired from EM Science (Gibbstown, NJ, USA). Deionized water was prepared using a Millipore Milli-Q system (Milli-Q Corp., Bedford, MA, USA). All other chemicals were HPLC grade or of the highest purity available.
Animals
Male C57BL/6Nnia mice (ages 4 to 26 months) were obtained from the National Institute on Aging (NIA) and fed ad libitum or a diet containing 40% fewer calories than the AL animals, as described previously [28].
Isolation of mitochondria
Mice were killed by cervical dislocation and their tissues (liver, kidney, brain, testis, heart, and eyeballs) were quickly removed and placed in ice-cold antioxidant buffer (50 mM potassium phosphate buffer, pH 7.4, containing 2 mM EDTA and 0.1 mM butylated hydroxytoluene). On the day of the experiment, 20 mice (4 animals in each age group, including CR) were dissected within 1 h. All subsequent procedures were performed at 4°C. Tissues were homogenized in 10 vol (w/v) of the indicated tissue-specific isolation buffer: (i) for liver tissue, 0.25 M sucrose, 3 mM EDTA, 10 mM Tris buffer, pH 7.4; (ii) for kidney, 0.22 M mannitol, 70 mM sucrose, 10 mM EGTA, 2 mM Hepes, pH 7.4; (iii) for heart, 0.3 M sucrose, 0.03 M nicotinamide, 0.02 M EDTA, pH 7.4; and (iv) for brain, testis, and eye, 0.32 M sucrose, 1 mM EDTA, 10 mM Tris, pH 7.4. Tissue homogenization procedures and specific protocols for isolation of mitochondria by the standard method of differential centrifugation were used according to previously published procedures [29–31]. Mitochondria from all tissues were isolated within 1 to 2 h after tissue dissection, except brain mitochondria, which require longer isolation time (up to 3 h) due to additional steps using Percoll gradient centrifugation [32].
Sample preparation for glutathione analysis
The first step in sample preparation for glutathione analysis is precipitation of proteins by acidification. We tested three different acids: trichloroacetic acid (TCA), meta-phosphoric acid (MPA), and perchloric acid (PA). MPA at the final concentration of 5% (w/v) was found to be the best precipitant as only 1–3% of GSH was oxidized 24 h after the addition of MPA on ice. The presence of a metal chelator (EDTA) in the sample decreased the GSH oxidation to virtually undetectable levels (< 0.001%). In contrast to MPA, TCA and PA were considered unsuitable because 20–30% GSH was oxidized under the same conditions. It should be noted that MPA prepared from clear crystals (obtained from EM Science) is more desirable for this purpose than the white powdered substance (Sigma Chemical Co.). Concentrated solutions of MPA, to be used for deproteinization, were prepared fresh immediately prior to use. Just after homogenization of the tissues, 200 μl aliquots of the crude homogenates were mixed with 200 μl of ice-cold 10% (w/v) MPA, incubated for 30 min on ice, and centrifuged for 20 min at 14,000 × g at 4°C Supernatants were transferred to autosample vials and either injected immediately or stored at −80°C until analysis. Storage of deproteinized tissue samples in 5% (w/v) MPA at − 80°C for up to 4 months was found to have no effect on the concentrations of GSH and GSSG.
For measurements of mitochondrial glutathione content, aliquots (150–200 μl) of fresh mitochondrial preparations were washed once with a 100-fold excess of buffer and pelleted by centrifugation This washing step was found to be critical to remove excess GSH and GSSG from the cytosolic fraction. The mitochondria were then resuspended in 200 μl of 5% (w/v) MPA. After 30 min of incubation on ice, samples were centrifuged for 20 min at 18,000 × g at 4°C. Supernatants were transferred into autosample vials and used for injections immediately or stored at − 80°C until analysis.
For the analysis of protein-glutathione mixed disulfides (protein-SSG), protein pellets from the initial precipitation of homogenates and mitochondria were resuspended and subjected to extensive washing to remove the free (nonprotein-bound) GSH and GSSG. Washing included three cycles of resuspension of the precipitate by sonication for 15 s on ice in 1.5 ml of 5% (w/v) MPA containing 0.5 mM EDTA. After each wash, precipitated protein was recovered by centrifugation for 20 min at 18,000 × g at 4°C, and the supernatants were discarded. The release of protein-bound GSH (incubation of protein pellets in 100 mM phosphate buffer for 1.5 h at 37°C) was achieved according to the procedure of Rossi and coworkers [33].
The protein contents of homogenates and mitochondria were determined by the BCA protein assay, according to the manufacturer's instructions (Pierce, Rockford, IL, USA).
HPLC-coulometric EC detection
Aminothiols were separated by HPLC equipped with a Shimadzu Class VP solvent delivery system (Shimadzu Corp., Kyoto, Japan), using a reverse-phase C18 Luna (II) column (3 μm, 4.6 × 150 mm) obtained from Phenomenex (Torrance, CA, USA). The mobile phase for isocratic elution consisted of 25 mM monobasic sodium phosphate, 0.5 mM of the ion-pairing agent 1-octane sulfonic acid, 2% (v/v) acetonitrile, pH 2.7, adjusted with 85% phosphoric acid. The flow rate was 1.0 ml/min. Under these conditions, the separation of aminothiols was completed in 35 min; GSSG was the last eluting peak, with a retention time of 30 min. Deproteinated samples were injected directly onto the column using a Shimadzu autosampler (Shimadzu Corp.). Calibration standards of aminothiols were prepared, by the dilution of 2 mM stock solutions of GSH and GSSG in 5% (w/v) meta-phosphoric acid, and were injected at regular intervals to ensure uniform standardization. Each sample was injected twice, and the average of the peak areas was used for calculations of the aminothiol concentrations. Following HPLC separation, aminothiols were detected with a model 5600 CoulArray electrochemical detector (ESA, Inc., Chelmsford, MA, USA) equipped with a four-channel analytical cell. Increasing potentials of +400, +600, +750, and +875 mV were applied on channels 1–4, respectively. GSH and GSSG were detected on channel 3 (+ 750 mV). The low potentials on channels 1 (+ 400 mV) and 2 (+ 600 mV) were used as an oxidative screen to eliminate interfering compounds that oxidize at a lower voltage than the GSH and GSSG.
Determination of redox potential
Redox potentials of the glutathione redox couple (2GSH → GSSG + 2e− + 2H+) in mitochondria were calculated by using experimentally determined concentrations of GSH and GSSG in the Nernst equation [7,13] as follows:
Eo is the standard potential of GSH (−288 mV at pH 7.8 in mitochondria), R is the gas constant (8.31 J/deg mol), T is the absolute temperature (310 K), n is the number of electrons transferred (2), and F is the Faraday constant (96,406 J/V).
Statistics
Data are presented as mean ± SD for four separate preparations (n = 4) of homogenates and mitochondria from pooled organs of four different animals in each age group. Statistical significance was determined by paired t-tests; p < .05 was considered to be significant.
Results
Comparison of distributions of GSH, GSSG, and protein-SSG in different tissues of 4 month old mice
The amounts of GSH, GSSG, and protein-SSG were determined in homogenates (Table 1 and Fig. 1) and mitochondrial fractions (Table 2 and Fig. 1) of liver, kidney, heart, brain, eye, and testis of mice at 4, 10, 22, and 26 months of age. In the 4 month old mice, GSH content varied up to ∼ 10-fold in various tissue homogenates, with the following rank order: liver = testis > brain = heart > eye > kidney. The GSSG amount in the 4 month old mice also varied up to ∼ 10-fold among homogenates of different tissues, and displayed the following rank order: liver = heart > testis > eye > brain > kidney. The GSH:GSSH ratio in homogenates ranged from 230:1 to 36:1, with the following rank order: brain > testis > eye > liver = kidney > heart.
Table 1.
Concentrations of Reduced Glutathione (GSH) and Glutathione Disulfide (GSSG) in Homogenates of Different Tissues of Ad Libitum–Fed (AL) Mice and Calorically Restricted (CR) Mice at 4, 10, 22, and 26 Months of Age
| Tissue/age (month) | GSH (nmol/mg protein) | GSSG (nmol/mg protein) | GSH:GSSG ratio |
|---|---|---|---|
| Liver | |||
| 4 | 27 ± 2 | 0.46 ± 0.07 | 61 ± 11 |
| 10 | 29 ± 2 | 0.41 ± 0.08 | 71 ± 16 |
| 22 (AL) | 27 ± 1 | 0.49 ± 0.10 | 56 ± 12 |
| 26 | 25 ± 2 | 0.68 ± 0.14 | 38 ± 9* |
| 22 (CR) | 25 ± 2 | 0.53 ± 0.08 | 47 ± 4 |
| Kidney | |||
| 4 | 2.8 ± 0.2 | 0.05 ± 0.01 | 59 ± 11 |
| 10 | 2.7 ± 0.1 | 0.05 ± 0.01 | 51 ± 8 |
| 22 (AL) | 2.6 ± 0.1 | 0.08 ± 0.01 | 33 ± 3 |
| 26 | 2.6 ± 0.1 | 0.07 ± 0.01* | 25 ± 3* |
| 22 (CR) | 2.5 ± 0.2 | 0.08 ± 0.01 | 34 ± 5 |
| Heart | |||
| 4 | 16.1 ± 1.6 | 0.44 ± 0.04 | 36 ± 5 |
| 10 | 15.4 ± 0.2 | 0.47 ± 0.01 | 32 ± 1 |
| 22 (AL) | 15.9 ± 1.1 | 0.49 ± 0.05 | 33 ± 3 |
| 26 | 14.9 ± 0.7 | 0.61 ± 0.04* | 25 ± 3* |
| 22 (CR) | 14.9 ± 1.2 | 0.44 ± 0.05 | 34 ± 5 |
| Brain | |||
| 4 | 17.0 ± 1.6 | 0.08 ± 0.01 | 230 ± 31 |
| 10 | 15.0 ± 0.5 | 0.06 ± 0.01 | 242 ± 36 |
| 22 (AL) | 14.1 ± 1.4 | 0.11 ± 0.02 | 135 ± 28 |
| 26 | 13.0 ± 1.6* | 0.14 ± 0.02* | 92 ± 17* |
| 22 (CR) | 14.3 ± 1.2 | 0.06 ± 0.01† | 230 ± 20† |
| Eye | |||
| 4 | 11.8 ± 0.3 | 0.11 ± 0.01 | 103 ± 5 |
| 10 | 12.9 ± 0.6 | 0.13 ± 0.02 | 98 ± 15 |
| 22 (AL) | 11.2 ± 0.3 | 0.18 ± 0.01 | 61 ± 5 |
| 26 | 10.6 ± 0.3* | 0.25 ± 0.02* | 41 ± 3* |
| 22 (CR) | 11.7 ± 0.2 | 0.20 ± 0.01 | 58 ± 3 |
| Testis | |||
| 4 | 26 ± 2 | 0.16 ± 0.03 | 165 ± 29 |
| 10 | 26 ± 2 | 0.24 ± 0.01 | 110 ± 5 |
| 22 (AL) | 25 ± 2 | 0.24 ± 0.02 | 104 ± 12 |
| 26 | 23 ± 0.3* | 0.24 ± 0.04* | 97 ± 14* |
| 22 (CR) | 24 ± 1 | 0.15 ± 0.04† | 161 ± 27† |
Data are presented as mean ± standard deviation of measurements in tissue homogenates from four different groups with four animals in each group.
Significant statistical difference (p < .05) compared to 4 month old (young) mice.
Values from CR mice are significantly different (p < .05) from the AL controls.
Fig. 1.
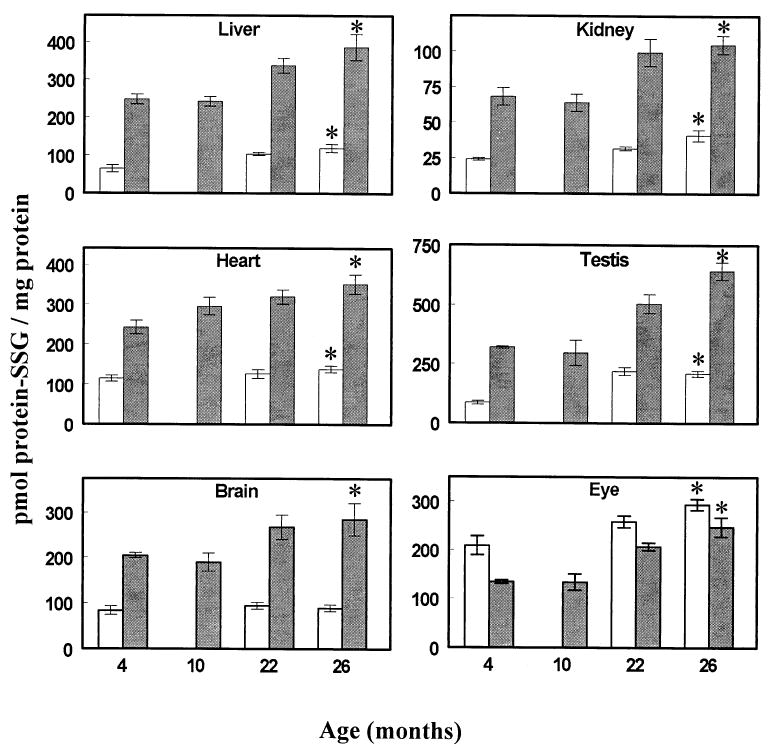
Protein-glutathione mixed disulfides (protein-SSG) in homogenates (open bars) and mitochondria (closed bars) of different tissues of mice at various ages. All values represent the mean ± standard deviation of n = 4 groups, with four animals pooled in each group. * p < .05 for old (26 month old) vs. young (4 month old) values, based on paired t-test.
Table 2.
Concentrations of Reduced Glutathione (GSH) and Glutathione Disulfide (GSSG) in Mitochondria of Different Tissues of Ad Libitum–fed (AL) Mice and Calorically Restricted (CR) Mice at 4, 10, 22, and 26 Months of Age
| Tissue/age (months) | GSH (nmol/mg protein) | GSSG (nmol/mg protein) | GSH:GSSG ratio |
|---|---|---|---|
| Liver | |||
| 4 | 11.1 ± 1.6 | 0.07 ± 0.02 | 162 ± 51 |
| 10 | 10.3 ± 2.3 | 0.09 ± 0.01 | 116 ± 35 |
| 22 (AL) | 11.1 ± 0.9 | 0.16 ± 0.01 | 68 ± 6 |
| 26 | 12.1 ± 1.6 | 0.21 ± 0.02* | 57 ± 10* |
| 22 (CR) | 11.5 ± 2.2 | 0.10 ± 0.02† | 114 ± 32† |
| Kidney | |||
| 4 | 2.1 ± 0.2 | 0.007 ± 0.001 | 323 ± 32 |
| 10 | 2.1 ± 0.1 | 0.007 ± 0.001 | 287 ± 25 |
| 22 (AL) | 2.0 ± 0.1 | 0.008 ± 0.001 | 233 ± 31 |
| 26 | 1.7 ± 0.1* | 0.009 ± 0.001* | 186 ± 17* |
| 22 (CR) | 1.9 ± 0.1 | 0.006 ± 0.001† | 343 ± 37† |
| Heart | |||
| 4 | 5.4 ± 0.2 | 0.22 ± 0.01 | 25 ± 2 |
| 10 | 5.5 ± 0.2 | 0.24 ± 0.02 | 23 ± 2 |
| 22 (AL) | 4.2 ± 0.1 | 0.35 ± 0.03 | 12 ± 1 |
| 26 | 4.9 ± 0.2* | 0.29 ± 0.01* | 17 ± 1* |
| 22 (CR) | 5.2 ± 0.4† | 0.25 ± 0.03† | 21 ± 2† |
| Brain | |||
| 4 | 7.0 ± 0.5 | 0.013 ± 0.001 | 564 ± 87 |
| 10 | 7.4 ± 0.1 | 0.012 ± 0.002 | 642 ± 102 |
| 22 (AL) | 6.9 ± 0.3 | 0.017 ± 0.003 | 423 ± 57 |
| 26 | 6.6 ± 0.2 | 0.020 ± 0.001* | 512 ± 122 |
| 22 (CR) | 7.5 ± 0.4 | 0.012 ± 0.003 | 560 ± 74† |
| Eye | |||
| 4 | 6.9 ± 0.08 | 0.022 ± 0.001 | 314 ± 19 |
| 10 | 6.8 ± 0.2 | 0.025 ± 0.002 | 277 ± 21 |
| 22 (AL) | 5.2 ± 0.1 | 0.029 ± 0.001 | 181 ± 9 |
| 26 | 5.9 ± 0.2* | 0.031 ± 0.004* | 187 ± 19* |
| 22 (CR) | 5.7 ± 0.3† | 0.021 ± 0.003† | 285 ± 51† |
| Testis | |||
| 4 | 7.1 ± 0.1 | 0.022 ± 0.003 | 319 ± 39 |
| 10 | 7.3 ± 0.5 | 0.031 ± 0.005 | 242 ± 44 |
| 22 (AL) | 7.0 ± 0.4 | 0.041 ± 0.004 | 172 ± 13 |
| 26 | 6.2 ± 0.1* | 0.041 ± 0.002* | 152 ± 7* |
| 22 (CR) | 6.6 ± 0.4 | 0.023 ± 0.003† | 286 ± 34† |
Data are presented as mean ± standard deviation of measurements in tissue homogenates from four different groups with four animals in each group.
Significant statistical difference (p < .05) compared to 4 month old (young) mice.
Values from CR mice are significantly different (p < .05) from the AL controls.
Generally, in mitochondria of 4 month old mice, the amount of GSH per mg protein was considerably lower than that in the respective tissue homogenate, i.e., 40% as much in liver, 74% in kidney, 31% in heart, 41% in brain, 58% in eye, and 26% in testis. The GSSG content in mitochondria was also relatively lower than in the corresponding tissue homogenate. However, the GSH: GSSG ratio was much higher in mitochondria than in homogenates of tissues other than the heart, exhibiting the following rank order: brain > testis = eye = kidney > liver > heart. The protein-SSG content in homogenates of 4 month old mice varied 8.7-fold in different tissues of the 4 month old mice, with the following rank order: eye > heart > testis = brain > liver > kidney (Fig. 1). The amounts of protein-SSG were considerably higher in mitochondria than in the respective homogenates, except in the eye; i.e., being higher by 3.7-fold in liver, 2.8-fold in kidney, 2-fold in brain, 1.6-fold in eye, and 3.7-fold in testis. The rank order of mitochondrial protein-SSG content among different tissues was as follows: testis > liver = heart > brain > eye > kidney, which does not match the rank order observed among tissue homogenates.
Overall, the data suggested that there were large variations in GSH, GSSG, and protein-SSG content among different tissues as well as between mitochondria and their respective tissue homogenates. These results indicate the existence of unique glutathione redox states in each tissue.
The effect of age on GSH, GSSG, and protein-SSG concentrations in homogenates of different tissues
Comparisons among 4, 10, 22, and 26 month old mice indicated that GSH content declined significantly only in the brain, eye, and testis (Table 1). In contrast, GSSG content in tissue homogenates exhibited a significant increase between 4 and 26 months of age, which ranged from 121% in the eye to 89% in the brain, 48% in the liver and testis, 43% in the kidney, and 35% in the heart. In all the tissues examined, GSH:GSSG ratios decreased significantly, ranging from 60% overall decline in the brain and eye to 52% in the kidney, 43% in the testis, 38% in the liver, and 31% in the heart. The protein-SSG content of tissue homogenates, measured at 4, 22, and 26 months of age, showed significant age-related increases in all the tissues except brain (Fig. 1). Thus, aging seemed to be associated with a uniform decline in the GSH:GSSG ratios and a selective increase in protein-SSG content in the tissue homogenates.
The effect of age on mitochondrial GSH, GSSG, and protein-SSG content
Mitochondrial GSH content declined (p < .05) gradually between 4 and 26 months of age in the eye, testis, kidney, and heart, which showed 14%, 12%, 19%, and 9% declines, respectively (Table 2). In contrast, the amount of intramitochondrial GSSG increased significantly with age in all of the tissues examined, with an increase of ∼200% in liver, 82% in testis, 63% in brain, 60% in heart, and 40% in kidney and eye. The GSH: GSSG ratio in mitochondria decreased significantly with age in all the tissues examined except the brain. The magnitude of the decline was 40% in eye, 42% in kidney, 52% in testis, 65% in liver, and 31% in heart. This pro-oxidizing shift in the GSH:GSSG ratio resulted primarily from a relative increase in GSSG concentration and, to a lesser extent, a decrease in GSH content.
During the interval from 4 to 26 months of age, mitochondrial protein-SSG content increased (p < .05) by 56% in the liver, 54% in the kidney, 45% in the heart, 39% in the brain, 85% in the eye, and 102% in the testis (Fig. 1). The relative as well as the absolute magnitudes of these increases were greater than in the corresponding tissue homogenates.
The effect of age on mitochondrial glutathione redox potential
Using the Nernst equation [7,13], the glutathione redox potentials were calculated in the mitochondria at pH 7.8 and 37°C. The redox potentials were not calculated in the tissue homogenates, however, because the cells are known to contain several distinct redox compartments, and the relative volume of a tissue occupied by different cellular and extracellular compartments could not be readily discerned.
To calculate the glutathione redox potential, experimentally determined amounts of intramitochondrial GSH and GSSG were converted into molar concentrations, assuming that 1 mg mitochondrial protein occupied an average volume of 1 μl [34]. In 4 month old mice, the redox potential of glutathione in mitochondria was found to be most positive, i.e., most pro-oxidizing, in the kidney and heart (−282 and −262 mV, respectively) and most negative or pro-reducing (ranging between −295 and −305 mV) in the liver, testis, eye, and brain (Fig. 2).
Fig. 2.
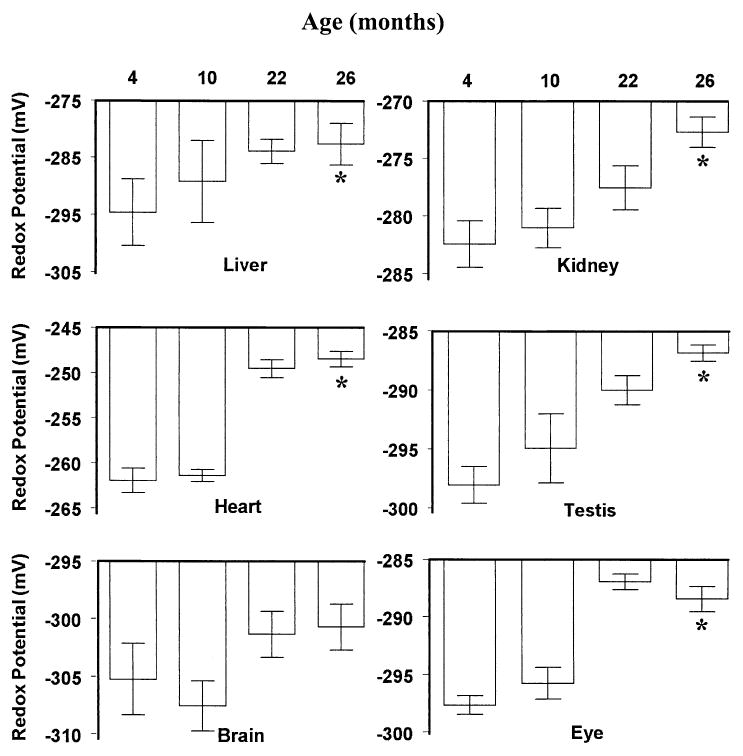
Glutathione redox potentials in mitochondria isolated from liver, kidney, heart, brain, eye, and testis of mice at 4, 10, 22, and 26 months of age. Redox values were calculated using the Nernst equation and the experimentally determined mitochondrial GSH and GSSG concentrations. Relatively more negative values indicate a more reducing state, whereas more positive values indicate the converse. All values represent the mean ± standard deviation of n = 4 groups, with four animals pooled in each group. * p < .05 for old (26 month old) vs. young (4 month old) values, based on paired t-test.
The glutathione redox potential of the mitochondria from the various tissues studied, except those from the brain, tended to become significantly more pro-oxidizing with age, starting from 4 or 10 months of age. The most severe age-associated loss in mitochondrial redox potential (∼15 mV) occurred in the heart, whereas the smallest decline (4.5 mV) was in the brain. The decreases in other tissues ranged between 9 and 12 mV.
The effect of caloric restriction on GSH, GSSG, and protein-SSG content and mitochondrial glutathione redox potential
AL mice and those fed 60% of the food eaten by the AL mice, starting from the age of 4 months, were compared at 22 months of age. In tissue homogenates, CR had no effect on GSH concentration (Table 1), whereas GSSG concentration was significantly lowered in the brain (42%) and in the testis (37%). In mitochondria (Table 2), CR had no effect on GSH concentration except in the heart and eye, where it was increased by 24% and 9%, respectively. However, the GSSG concentration was significantly decreased in all the tissues except the brain, and the GSH:GSSH ratio was significantly increased (by 32–75%) in all tissues. The protein-SSG contents in homogenates of kidney, testis, and eye were significantly lower in CR mice than in the AL mice (Fig. 4). The differences between the CR and AL mice in homogenates of the other tissues were not significant.
Fig. 4.
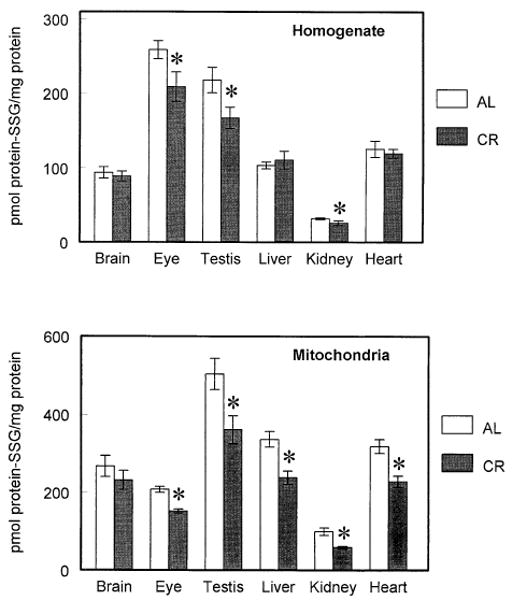
Protein-glutathione mixed disulfides (protein-SSG) in homogenates (top) and mitochondria (bottom) of different tissues of ad libitumfed (AL) mice and calorically restricted (CR) mice at 22 months of age. All values represent the mean ± standard deviation of n = 4 groups, with four animals pooled in each group. * p < .05 based on paired t-test.
The mitochondrial glutathione redox potential was significantly more pro-reducing in the kidney, heart, eye, and testis of CR than AL mice (Fig. 3). The liver and brain exhibited no significant differences. The mitochondrial protein-SSG concentration was significantly lowered by CR in all the tissues except the brain (Fig. 4).
Fig. 3.
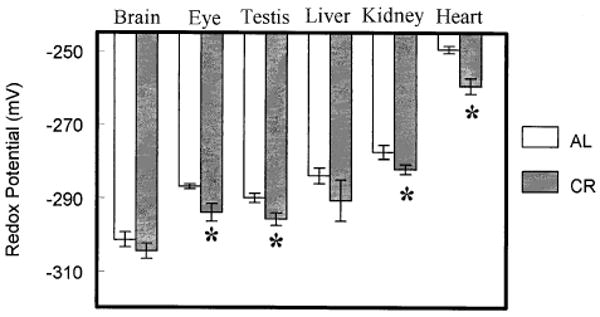
Glutathione redox potentials in mitochondria isolated from liver, kidney, heart, brain, eye, and testis of ad libitum-fed (AL) mice and calorically restricted (CR) mice at 22 months of age. Redox values were calculated using the Nernst equation and experimentally determined mitochondrial GSH and GSSG concentrations. All values represent the mean ± standard deviation of n = 4 groups, with four animals pooled in each group. * p < .05 based on paired t-test.
Discussion
The main findings of this study were as follows: (i) the GSH, GSSG, and protein-SSG contents and GSH: GSSG ratios varied in different tissues and also exhibited disparity between mitochondria and their respective tissue homogenates; (ii) the GSH:GSSG ratios in mitochondria and tissue homogenates decreased during aging, primarily due to a relative increase in GSSG concentration; (iii) the mitochondrial redox potential became less negative during aging, reflecting a pro-oxidizing shift; (iv) protein-SSG content increased with age in homogenates of some tissues and mitochondria of all the tissues examined; and (v) caloric restriction diminished the magnitude of the age-related alterations in glutathione redox state and protein-SSG content in the tissue homogenates and the mitochondria.
It is well recognized in the literature that measurements of GSH and GSSG in biological specimens can be affected during the isolation procedures due to the artificial oxidation of GSH [13,15,16]. Analytical procedures involving enzymatic, fluorimetric, and colorimetric assays were found in the earlier studies to have unsatisfactory sensitivity and/or inadequate specificity and/or low reproducibility. Later, procedures employing high-performance liquid chromatography, in combination with UV-absorbance or fluorescence detection, were also found to be complicated and time-consuming, with insufficient sensitivity for small samples and, importantly, an inability to simultaneously measure GSH and GSSG. Moreover, multiple derivatization steps involving acidification, neutralization, and thiol blocking could lead to over- or underestimation of GSH/GSSG content. In comparison, ion-pairing HPLC in combination with coulometric electrochemical detection, the procedure used in the present work, offers significant advantages. It is simple (no sample manipulation required), fast (analysis completed in 35 min), and specific (selective oxidation of GSH and GSSG at high electrode potential and elimination of interfering compounds at low electrode potential). Also, as indicated in Materials and Methods, this procedure resulted in minimal GSH oxidation. Indeed, the values of GSH:GSSG ratios reported here are among the highest published values, including those from samples of tissues that were perfused and rapidly frozen in liquid nitrogen [11,13,15,26].
The glutathione redox status of a tissue is dependent upon two factors: the amount of GSH and the ratio between GSH and GSSG. Results of this study demonstrate that different tissues of the mouse differ vastly, not only in their total contents of GSH and GSSG but also in GSH:GSSG ratios. For instance, GSH content varied ∼10-fold in tissue homogenates and ∼5-fold in mitochondria of different tissues, whereas GSSG showed up to ∼10-fold differences in homogenates and ∼33-fold differences in mitochondria from various tissues. Similarly, GSH:GSSG ratios exhibited ∼6-fold variation among tissue homogenates and ∼22-fold among mitochondria. The magnitude of intertissue variation in protein-SSG content was up to 8.7-fold in the homogenates and 4.7-fold in the mitochondria. Such remarkable differences suggest the existence of unique redox milieus in different tissues. These intertissue variations in the redox state may stem from variations in parameters such as the rates of ROS generation, activities of antioxidative enzymes, and the concentrations of various low molecular weight antioxidants [7,11,12]. Indeed, rates of mitochondrial O2− and H2O2 generation vary several-fold among different tissues of the mouse [35]. Similarly, the activities of the main antioxidative enzymes, superoxide dismutase, catalase, and glutathione peroxidase, differ greatly among various tissues of the mouse [35]. Why different tissues maintain such vastly different redox states and why mitochondria, which are the main sites of O2−/H2O2 generation, have relatively lower amounts of GSH than the homogenates are important but presently unresolved issues.
The observed age-associated changes in the amounts of GSH, GSSG, and protein-SSG and GSH:GSSG ratios demonstrate unequivocally that aging is associated with a significant pro-oxidizing shift in all of the organs of the mouse examined here. The most proximate reason for the shift in GSH:GSSG ratios seems to be the increase in GSSG concentration rather than major losses in GSH content. The age-related increases in GSSG content can potentially arise from corresponding elevations in rates of mitochondrial generation of O2−/H2O2 and/or declines in antioxidative defenses. However, there is no consistent age-related decline in GSH content of tissues, in activities of enzymes involved in GSH regeneration and synthesis, or in activities of major antioxidative enzymes [36–42]. In contrast, previous studies in this and other laboratories have shown that the rates of mitochondrial O2−/H2O2 generation tend to increase during aging [35,36,40].
Thus, the age-related changes in GSH:GSSG ratios can be linked more plausibly to increased oxidant production than to altered antioxidant levels. Irrespective of the underlying causes, the pro-oxidizing shifts in the GSH:GSSG ratios and in GSSG concentration can potentially have multiple physiological effects. For instance, the reaction between GSSG and cysteinyl residues in proteins leads to glutathiolation or the formation protein-mixed disulfides, which can inactivate a variety of proteins, such as thioredoxin, enolase, aldolase, and pyrophosphatases [10,43,44]. Alternatively, glutathiolation can also activate certain proteins [43]. In either case, glutathiolation leads to a cascade of physiological perturbations associated with the enhancement of oxidative stress. Also, there can be a plethora of effects on cellular signaling molecules whose activities are modulated by the redox state of the cells [15,17,45,46]. Indeed, perturbations of the redox state, induced by either depletion or augmentation of reductants such as the GSH precursor N-acetylcysteine, can have deleterious effects, which illustrates the physiological importance of the maintenance of an optimal redox state [47–49].
The absolute values of intramitochondrial redox potential of the GSH/GSSG couple depend on factors such as the concentrations of GSH and GSSG, pH of the mitochondrial matrix, and the volume of mitochondria [13]. pH affects the redox potential (E) due to its effect on Eo (see Nernst equation in Materials and Methods); for instance, an increase of 0.1 pH unit would decrease redox potential by ∼6 mV and vice versa [7,13]. Similarly, a change in mitochondrial volume due to swelling or shrinkage would affect redox potential by altering GSH and GSSG concentrations. For example, a 2-fold increase in liver mitochondrial volume would decrease the redox potential by 9 mV, assuming E = −295 mV, based on concentrations of 11 mM GSH and 0.07 mM GSSG and a pH of 7.8 at 37°C. In the present study, the intramitochondrial redox potential of the GSH/GSSG couple in different tissues declined with age, by amounts ranging from 4.5 to 15 mV, which is equivalent to a decrease of 0.1 to 0.2 units of pH. Since age-related changes in the mitochondrial volume and pH were not determined, the present values of redox potential should be considered as tentative. The actual values in organs from older animals may indeed be higher because mitochondrial electrochemical potential tends to decline and mitochondrial volume to increase during aging [50].
A notable finding of this study was that the age-related increases in GSSG concentrations, decreases in GSH:GSSG ratios, and increases in mitochondrial glutathione redox potentials and glutathiolation of proteins were all attenuated by caloric restriction, a regimen that is known to extend the life span of mice. Although the nature of the mechanisms by which CR affects life span remains controversial, there is mounting evidence that attenuation of oxidative stress may be at least one of the underlying mechanisms. CR has been shown to retard the age-associated increase in the rate of mitochondrial generation of O2−/H2O2 and accrual of oxidative damage to DNA and proteins in mice [35,51]. The present study shows that the attenuation of oxidative damage by CR is also correlated with retardation of the pro-oxidizing shift in the redox state of glutathione.
To conclude, the observed age-associated, pro-oxidizing shift of glutathione redox state in various organs of the mouse can be interpreted to reflect a widening of the imbalance between antioxidants and pro-oxidants during the aging process. Such a shift most likely entails a relatively more rapid rate of augmentation of oxidatively modified macromolecules as an organism ages. This view is consistent with the findings that the amounts of oxidatively modified macromolecules in several different animal models increase exponentially, rather than linearly, with age [52]. One major implication of the age-related increase in the level of oxidative stress/damage may be that if such stress were indeed a causal factor in the aging process, then the rate of aging could also be speculated to increase exponentially as the organism ages.
Acknowledgments
This research was supported by the grant RO1 AG13563 from the National Institutes of Health, National Institute on Aging. We are grateful to Dr. R. J. Mockett for critical reading of the manuscript.
Abbreviations
- AL
ad libitum
- CR
caloric restriction
- GSH
reduced glutathione
- GSSG
oxidized glutatione or glutathione disulfide
- protein-SSG
protein-glutathione mixed disulfides
- ROS
reactive oxygen species
References
- 1.Sies H. Biochemistry of oxidative stress. Angewandte Chemie. 1986;25:1058–1071. [Google Scholar]
- 2.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orr WC, Sohal RS. Oxidative stress as a governing factor in physiological aging. In: Sen CK, Sies H, Baeuerle PA, editors. Antioxidants and redox regulation of genes. San Diego, CA: Academic Press; 2000. pp. 517–530. [Google Scholar]
- 4.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 5.Grune T, Davies KJA. Oxidative processes in aging. In: Masoro EJ, Austad SN, editors. Handbook of the biology of aging. San Diego, CA: Academic Press; 2001. pp. 25–58. [Google Scholar]
- 6.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 7.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 8.Reed DJ. Regulation of reductive processes by glutathione. Biochem Pharmacol. 1986;35:7–13. doi: 10.1016/0006-2952(86)90545-9. [DOI] [PubMed] [Google Scholar]
- 9.Das KC, White CW. Redox systems of the cell: possible links and implications. Proc Natl Acad Sci USA. 2002;99:9617–9618. doi: 10.1073/pnas.162369199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci USA. 2002;99:9645–9649. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 12.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 13.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 14.Collison MW, Thomas JA. S-thiolation of cytoplasmic cardiac creatine kinase in vivo: inhibition in heart cells treated with diamide. Biochim Biophys Acta. 1987;928:121–129. doi: 10.1016/0167-4889(87)90112-1. [DOI] [PubMed] [Google Scholar]
- 15.Ziegler DM. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu Rev Biochem. 1985;54:305–329. doi: 10.1146/annurev.bi.54.070185.001513. [DOI] [PubMed] [Google Scholar]
- 16.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 17.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry. Glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 18.Weindruch R, Walford RL. Dietary restriction in mice beginning at 1 year of age: effects on life span and spontaneous cancer incidence. Science. 1982;215:1415–1418. doi: 10.1126/science.7063854. [DOI] [PubMed] [Google Scholar]
- 19.Weindruch R, Sohal RS. Caloric intake and aging. N Engl J Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shigenaga MK, Ames BN. Oxidants and mitogenesis as causes of mutation and cancer: the influence of diet. Basic Life Sci. 1993;61:419–436. doi: 10.1007/978-1-4615-2984-2_37. [DOI] [PubMed] [Google Scholar]
- 21.Nichols DG. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int J Biochem Cell Biol. 2002;34:1372–1381. doi: 10.1016/s1357-2725(02)00077-8. [DOI] [PubMed] [Google Scholar]
- 22.Miquel J, Economos AC, Fleming JA, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Campo R, Lopez-Torres M, Rojas C, Cadenas S, Barja G. Longevity and antioxidant enzymes, nonenzymatic antioxidants, and oxidative stress in the vertebrate lung: a comparative study. J Comp Physiol. 1994;163B:682–689. doi: 10.1007/BF00369520. [DOI] [PubMed] [Google Scholar]
- 24.Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci USA. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith CV, Jones DP, Guenthiner TM, Lash LH, Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol. 1996;140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 26.de la Asuncion GJ, Millan A, Pla R, Bruseghini L, Esteras A, Pallardo FV, Sastre J, Vina J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- 27.Martinez M, Ferrandiz ML, De Juan E, Miquel J. Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: relationship to cytochrome c oxidase decline. Neurosci Lett. 1994;170:121–124. doi: 10.1016/0304-3940(94)90254-2. [DOI] [PubMed] [Google Scholar]
- 28.Dubey A, Forster MJ, Lal H, Sohal RS. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch Biochem Biophys. 1996;333:189–197. doi: 10.1006/abbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- 29.Sohal RS, Svensson I, Sohal BH, Brunk UT. Superoxide anion radical production in different animal species. Mech Ageing Dev. 1989;49:129–135. doi: 10.1016/0047-6374(89)90096-1. [DOI] [PubMed] [Google Scholar]
- 30.Lash LH, Sall JM. Mitochondrial isolation from liver and kidney: strategy, techniques, and criteria for purity. In: Lash LH, Jones DP, editors. Mitochondrial dysfunction. San Diego, CA: Academic Press; 1993. pp. 8–29. [Google Scholar]
- 31.Arcos JC, Sohal RS, Sun SC, Argus MF, Burch GE. Changes in ultrastructure and respiratory control in mitochondria of rat heart hypertrophied by exercise. Exp Mol Pathol. 1968;8:49–65. doi: 10.1016/0014-4800(68)90005-1. [DOI] [PubMed] [Google Scholar]
- 32.Sims NR. Mitochondrial isolation from brain: strategy, techniques, and criteria for purity. In: Lash LH, Jones DP, editors. Mitochondrial dysfunction. San Diego, CA: Academic Press; 1993. pp. 29–41. [Google Scholar]
- 33.Rossi R, Cardaioli E, Scaloni A, Amiconi G, Di Simplicio P. Thiol groups in proteins as endogenous reductants to determine glutathione-protein mixed disulphides in biological systems. Biochim Biophys Acta. 1995;1243:230–238. doi: 10.1016/0304-4165(94)00133-i. [DOI] [PubMed] [Google Scholar]
- 34.Halestrap AP, Quinlan PT. The intramitochondrial volume measured using sucrose as an extramitochondrial marker overestimates the true matrix volume determined with mannitol. Biochem J. 1983;214:387–393. doi: 10.1042/bj2140387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation, and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 36.Barja G. Mitochondrial free radical production and aging in mammals and birds. Ann NY Acad Sci. 2000;854:224–238. doi: 10.1111/j.1749-6632.1998.tb09905.x. [DOI] [PubMed] [Google Scholar]
- 37.Sohal RS, Arnold LA, Sohal BH. Age-related changes in antioxidant enzymes and prooxidant generation in tissues of the rat with special reference to parameters in two insect species. Free Radic Biol Med. 1990;9:495–500. doi: 10.1016/0891-5849(90)90127-5. [DOI] [PubMed] [Google Scholar]
- 38.Rikans LE, Moore DR. Sex-dependent differences in the effects of aging on antioxidant defense mechanism of rat liver. Biochim Biophys Acta. 1991;1074:195–200. doi: 10.1016/0304-4165(91)90061-k. [DOI] [PubMed] [Google Scholar]
- 39.Sohal RS, Sohal BH. Hydrogen peroxide release by mitochondria increases during aging. Mech Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- 40.Matsuo M. Age-related alterations in antioxidant defenses. In: Yu BP, editor. Free radicals in aging. Boca Raton, FL: CRC Press; 1994. [Google Scholar]
- 41.Nohl H, Hegner D. Do mitochondria produce oxygen radicals in vivo? Eur J Biochem. 1978;82:563–567. doi: 10.1111/j.1432-1033.1978.tb12051.x. [DOI] [PubMed] [Google Scholar]
- 42.Sohal RS, Agarwal S, Sohal BH. Oxidative stress and aging in the Mongolian gerbil (Meriones unguiculatus) Mech Ageing Dev. 1995;81:15–25. doi: 10.1016/0047-6374(94)01578-a. [DOI] [PubMed] [Google Scholar]
- 43.Gilbert HF. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol. 1984;107:330–351. doi: 10.1016/0076-6879(84)07022-1. [DOI] [PubMed] [Google Scholar]
- 44.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 45.Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem Pharmacol. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- 46.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 47.Reed DJ. Glutathione: toxicological implications. Ann Rev Pharmacol Toxicol. 1990;30:603–631. doi: 10.1146/annurev.pa.30.040190.003131. [DOI] [PubMed] [Google Scholar]
- 48.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 49.Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- 50.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sohal RS, Agarwal S, Candas M, Forster MJ, Lal H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech Ageing Dev. 1994;76:215–224. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- 52.Stadtman ER, Levine RL. Protein oxidation. Ann NY Acad Sci. 2002;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]