

Abstract
The highly conserved mismatch (MMR) repair system corrects post-replicative errors and modulates cellular responses to genotoxic agents. Here, we show that the MMR system strongly influences cellular sensitivity to curcumin. Compared to MMR-proficient cells, isogenically-matched MMR-deficient cells displayed enhanced sensitivity to curcumin. Similarly, cells suppressed for MLH1 or MSH2 expression by RNA-interference displayed increased curcumin sensitivity. Curcumin treatment generated comparable levels of reactive oxygen species (ROS) and the mutagenic adduct 8-oxo-G in MMR-proficient and deficient cells; however, accumulation of γH2AX foci, a marker for DNA double strand breaks, occurred only in MMR-positive cells in response to curcumin treatment. Additionally, MMR-positive cells showed activation of Chk1 and induction of G2/M cell-cycle checkpoint following curcumin treatment and inhibition of Chk1 by UCN-01 abrogated Chk1 activation and heightened apoptosis in MMR-proficient cells. These results indicate that curcumin triggers accumulation of DNA DSB and induction of a checkpoint response through a MMR-dependent mechanism. Conversely, in MMR-compromised cells, curcumin-induced DSB is significantly blunted, and as a result, cells fail to undergo cell-cycle arrest, enter mitosis and die via mitotic catastrophe. The results have potential therapeutic value, especially in the treatment of tumors with compromised MMR function.
Keywords: Curcumin, Apoptosis, Mismatch repair, cell cycle arrest, DNA damage
Introduction
Curcumin (diferuloylmethane), a dietary pigment derived from the rhizome Curcuma longa, is a promising anti-cancer drug that is currently in phase II clinical trial [1, 2]. Curcumin's anti-cancer property is attributed to its selective cell-death inducing ability in tumor cells [2-4]. In normal and primary cells, curcumin is either inactive or inhibits proliferation but does not elicit a cytotoxic response. For example, in primary untransformed mouse embryonic fibroblast line C3H/10T1/2, rat embryonic fibroblasts, and human foreskin fibroblasts, curcumin failed to initiate cell-death although it inhibited proliferation of both normal and transformed cells in a non-selective manner [2-4].
The principal mode of cell death induced by curcumin is apoptosis [5]. Failure to undergo growth arrest before or during mitosis in response to stress triggers aberrant chromosome segregation, which ultimately culminates in the activation of apoptosis through a mechanism termed mitotic catastrophe. Indeed, a recent study showed that curcumin disrupts mitotic spindle structure and induces micro-nucleation [6]. It has been proposed that mitotic catastrophe induced by some anti-cancer agents such as paclitaxel stems from cellular damage in combination with dysregulated cell-cycle checkpoint activation resulting in spindle and chromosome segregation abnormalities during mitosis [7-9]. More specifically, inactivation or dysfunction of the chromosomal passenger complex (CPC), which is necessary for coordinating chromosomal and cytoskeletal events during mitosis, often leads to mitotic cell death [10]. To that end, a recent report documented that, in BCR-Abl transformed cells, curcumin induces mitotic catastrophe and cell-death through disruption of CPC and down regulation of survivin, a modulator of cell division and apoptosis [11]. In the apoptosis-resistant HL-60 subline HCW-2, curcumin induces mitotic catastrophe by inhibiting the expression of survivin [12] and, in the breast cancer line MCF7, curcumin-induced apoptosis was associated with assembly of aberrant, mono-polar mitotic spindles that are impaired in their ability to segregate chromosomes [6, 13]. Other studies documented that curcumin-induced apoptosis and cell-cycle arrest in melanoma cells was associated with downregulation of NFκB activation, decreased iNOS and DNA-PK catalytic subunit expression, upregulation of p53, p21(Waf1/Cip1), p27(Kip1) and Chk2 [14, 15]. Curcumin-mediated inhibition of NF-κB via the NIK/IKK signaling complex results in excessive generation of reactive oxygen species (ROS) which ultimately trigger apoptosis through activation of c-Abl>JNK signaling [16, 17]. In sum, available evidence indicates that curcumin targets multiple signaling pathways to induce cell-cycle arrest and/or cell death.
The mismatch repair (MMR) system is an evolutionarily conserved DNA repair mechanism primarily responsible for resolving post-replicative mismatches in DNA [18,19]. Expectedly, deficiencies in MMR increase the rate of genomic mutations and susceptibility to multiple forms of cancer, including colorectal tumors [18,19]. In addition to its capacity as a repair mechanism, MMR is essential for the activation of signaling cascades activated in response to certain genotoxic insults [20, 21]. For example, MMR-deficient cells fail to trigger G2/M cell-cycle arrest following treatment with SN1 methylating agents such as N-methyl-N-nitro-N-nitrosoguanidine (MNNG), procarbazine, temozolomide, and the anti-metabolite 6-thioguanine [18-21]. MMR-deficient cell lines are also resistant to the cytotoxic effects of these agents, a phenotypic effect termed alkylation tolerance [20,21]. MMR complexes also recognize other types of damage such as cisplatin adducts [22] and oxidized bases such as 8-oxo-dG [23].
Recently, defects in G2/M checkpoint activation observed in MMR-deficient cells following SN1 methylator exposure have been attributed to dysregulation of MMR-dependent activation of the cell-cycle checkpoint kinases Chk1 and Chk2 [24, 25]. Biochemical studies revealed that components of the MMR system interact with the damage-responsive kinases ATM/ATR potentially facilitating phosphorylation and activation of Chk1/Chk2 kinases [24-27]. Taken together, absence of MMR confers resistance to SN1 methylators due to failure to recognize and respond to DNA lesions that normally activate a robust cellular response.
In this report, we demonstrate that cells compromised or deficient in MMR function (Msh2 and Mlh1) exhibit increased sensitivity to curcumin. Elucidation of the mechanism behind this enhanced sensitivity revealed that the levels of oxidative DNA damage induced by curcumin were independent of the MMR status. However, activation of Chk1 and Chk2 kinases and induction of G2/M arrest triggered by curcumin required MMR function. Compared to MMR-proficient cells, MMR-deficient counterparts displayed reduced DNA double strand breaks (DSBs) and ATM activation suggesting that DSB formation induced by curcumin is predominantly a MMR-dependent process. Together, the findings indicate that the MMR system protects cells from curcumin cytotoxicity, in part, by activating the G2/M checkpoint. In clear contrast, MMR-deficient cells fail to activate this response, and likely transit into mitosis with a damaged genome which subsequently activates a robust apoptotic response.
Materials and Methods
Materials
Curcumin (cat #C7727), NAC (cat #A9165), GSH (cat # G4251) was obtained from Sigma-Aldrich (St. Louis, MO) and dissolved in DMSO prior to use. 7-hydroxystaurosporine (UCN-01) was obtained from the National Cancer Institute (Bethesda, MD). Chk2 inhibitor II (2-[4-(4-Chlorophenoxy) phenyl]-1H-benzimidazole-5-carboxamide) was purchased from Calbiochem (La Jolla, CA). Antibodies specific for phospho-Chk1 (S345), phospho-Chk2 (T68), and total Chk1 and Chk2 were obtained from Cell Signaling (Danvers, MA). Antibodies specific for γH2AX, MLH1 and MSH2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). HRP-conjugated secondary antibodies were purchased from Novus Biologicals (Littleton, CO). Cleaved PARP (cat #ab2317) and caspae-3 (cat#ab2302) antibodies were obtained from AbCam (Cambridge, MA).
Cell lines
The MMR-proficient colorectal tumor line HCT116+ch3 was created by the stable transfer of a portion of human chromosome 3, bearing a wild-type copy of the hMlh1 gene, into MLH1-deficient line HCT116 [28]. The control HCT116+ch2 cell line is an MLH1-deficient derivative of HCT116 that has a portion of human chromosome 2 introduced by microcell fusion. HCT116+ch2 and HCT116+ch3 cells were maintained in DMEM containing 10% FBS supplemented with 400 μg/ml of geneticin (G418) as described [28]. The MSH2-proficient derivative of the human endometrial adenocarcinoma cell line Hec59 (Hec59+ch2) was maintained in RPMI containing 400 μg/ml geneticin [29]. The colorectal tumor line RKO was cultured in DMEM containing 5-azacytidine (5 μM) for 5 days to restore expression of the epigenetically silenced hMlh1 gene. The resultant RKO/Mlh1+ and the parental RKO cells were cultured as described [30].
Viability Assays
5-dimethylthiazol-2-yl-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay was performed using CellTiter 96 Aqueous One Solution Proliferation Assay System (Promega). This assay measures the bioreduction by intracellular dehydrogenases of the tetrazolium compound MTS in the presence of the electron-coupling reagent phenazine methosulfate. MTS and phenazine methosulfate were added to the culture wells, and the mixture was incubated at 37°C for 3 h. Absorbance was measured at 490 nm using a microplate reader and is directly proportional to the number of viable cells in the cultures. The relative toxicity was calculated by comparing with untreated cells.
RNA interference
Overlapping synthetic oligonucleotides corresponding to sequences specific for the human Chk1 (5′–GAAGCAGTCGCAGTGAAGAT-3′), Chk2 (5′-GAACCTGAGG-ACCAAGAACC-3′), MSH2 (5′- GTTCGTCAGTATAG AGTTGAA-3′), and MLH1 (5′-GGTTCACTACTAGTAAACTG-3′) transcripts were hybridized and cloned into pSIREN-RETRO-Q (Clontech, La Jolla, CA). The recombinant pSiren plasmid was co-transfected with pCL-ampho plasmid encoding the packaging viral DNA into 293T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The supernatant containing the viral DNA was collected, filtered and used to infect HCT116+ch3 cells. Cells were selected by incubation with puromycin (1 μg/ml) for 4 days and downregulation of target gene expression was confirmed by immunoblotting.
Immunoblotting
Cells were harvested by scraping, washed with ice-cold PBS, and lysed in cold 1× lysis buffer containing 10 mM Tris HCl pH 8.0, 240 mM NaCl, 5 mM EDTA, 1 mM DTT, 0.1 mM PMSF, 1% Triton X-100, 1 mM sodium vanadate, and 1 μg/ml of leupeptin, pepstatin, and aprotinin by incubation on ice for 20 min. Lysates were cleared by centrifugation and protein concentration was determined using Bradford assay. For immunoblotting, proteins were resolved on 4-12% SDS-PAGE, electro-transferred onto Immobilon-P (Millipore, Billerica, MA) membrane. The membrane was then probed with indicated primary antibody, subsequently with HRP-conjugated secondary antibody, and developed using chemiluminescence. Where indicated, the membrane was stripped by incubation in stripping buffer containing 65 mM Tris-HCl pH 6.7, 100 mM β-mercaptoethanol (BME) and 2% SDS for 30 min at 40° C. The membrane was then re-probed with the indicated antibody.
Microscopy
Cells were grown on pre-sterilized glass cover slips and treated with DMSO (Mock) or curcumin (30 μM) in the presence or absence of NAC (5 mM) for 1 h. Cells were then washed three times with Hank's Balanced Salt Solution (HBSS) containing 10 mM Hepes, 2 mM CaCl2, and 4 mg/ml BSA. Cells were fixed in 4% paraformaldehyde in HBBS for 5 min and then permeabilized in 100% methanol for 5 min. Cells were then stained for γH2AX foci by incubation with anti-H2AX antibody for 1 hr at 37°C. Cells were subsequently stained with Texas-Red conjugated secondary antibody. DNA was counterstained with DAPI. γH2AX foci in each nucleus were counted using a Nikon fluorescence microscope (TE S2000) equipped with CCD camera. At least 100 cells were scored for each time point.
Flow cytometry
Mock (DMSO) and curcumin-treated cells were washed twice with 1X PBS and fixed in ice-cold 70% ethanol for 30 min on ice and stored at 4° C prior to analysis. For staining, cells were incubated in PBS containing 1 mg/ml RNase A and 40 μg/ml propidium iodide for 30 min in the dark at 37°C and then analyzed by flow cytometry. Approximately 3 × 104 cells were evaluated in each sample and DNA histograms were analyzed using ModFit (Verity Software, Topsham, ME) software. All flow cytometry experiments were performed in triplicate and Student's t-test was conducted to determine statistical significance.
Measurement of 8-oxo-guanine
8-oxo-G was measured using the 8-OxyDNA assay Kit from Calbiochem. Following treatment with DMSO (mock) or curcumin, cells were fixed, permeabilized and the FITC-antibody conjugate, that binds 8-oxoguanine moiety, is added. The presence of oxidized DNA is indicated by a green/yellow fluorescence and measured using flow cytometry.
Clonogenic Survival Assay
Logarithmically growing cells were plated at a density of 3 × 103 cells per well in a 10 cm dish and treated the next day with varying concentrations of curcumin (0, 10, 20, 30, 40 and 50 μM) for 5 hrs. Cells were washed and replaced with fresh media and grown for 14 days. Colonies were stained using 0.25% crystal violet and 10% formalin in 80% methanol for 30 min, washed with water, and counted.
Results
MMR-deficient cells display heightened sensitivity to curcumin
In addition to resolving post-replicative base-pairing errors, the MMR system plays a critical role in activation of key cellular responses induced by genotoxins such as SN1 methylators [21]. To evaluate a potential role for MMR in curcumin-induced cellular responses, we exposed matched MMR-proficient (HCT116+ch3, HEC59+ch2 and RKO/MLH+) and MMR-deficient (HCT116+ch2, HEC59 and RKO) cells to 30 μM curcumin and then assessed cytotoxic response by Trypan Blue exclusion (Fig 1A). Curcumin was mildly cytotoxic in the MMR-proficient cell lines at 48 hr after treatment, with each line showing <10% Trypan Blue staining. In contrast, MMR-deficient lines displayed >30% Trypan Blue positive cells in response to curcumin at this time point. Statistical analysis showed significant difference (p < 0.01) in Trypan Blue positive cells between matched MMR-proficient/deficient cell lines.
Fig. 1. MMR-deficient cells display heightened sensitivity to curcumin.
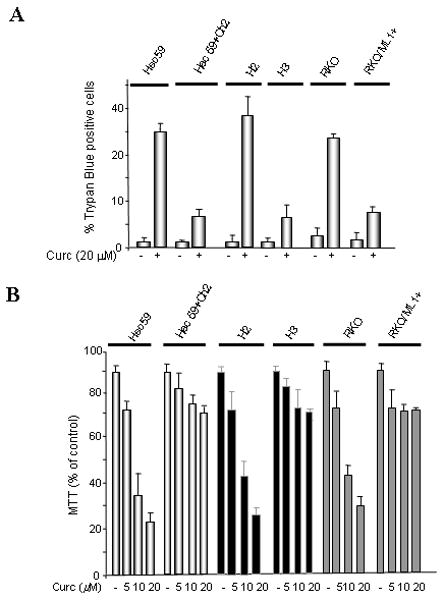
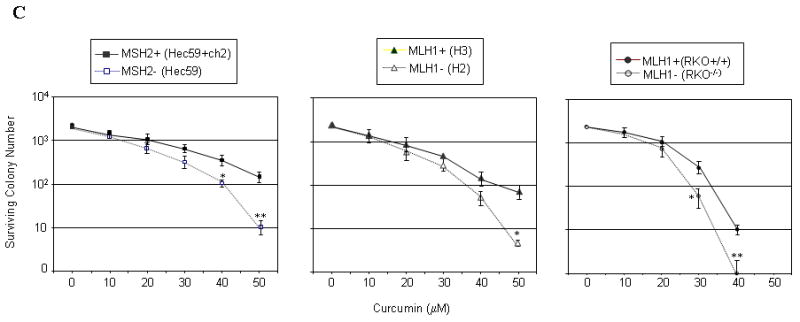
A. MLH1-deficient (HCT116+ch2, RKO), MSH2-deficient (Hec59) and matched MLH1-proficient (HCT116+ch3, RKO+MLH1+), MSH2–proficient (Hec59+ch2) cells were either mock (DMSO) treated or exposed to curcumin (Curc; 30 μM) and cell viability was assessed 48 h later by Trypan Blue exclusion. B. Cell lines outlined in panel A were treated with 0, 5, 10 and 20 μM of curcumin and viability was measured by MTT assay. C. Indicated cell lines were exposed to various doses of curcumin (0, 10, 20, 30, 40 and 50 μM), colonies counted 14 days after drug, and subsequently plotted against curcumin concentration. Each point represents the mean value obtained from at least three independent experiments, error bars = 1 SD.
As an independent approach, we used MTT assay to examine the effect of MMR on curcumin sensitivity. In this set of experiments, we not only scored the cytotoxic effects of curcumin in our MMR-proficient/deficient cells but also examined the dose-dependent nature of this response. In close agreement with the results obtained using Trypan Blue exclusion, we observed a modest cytotoxic effect in MMR-proficient cells at each curcumin dose tested (5, 10, and 20 μM) (Fig 1B). Again, we observed a heightened cytotoxic response in MMR-deficient cells and measured a statistically significant (p < 0.01) difference between matched MMR-proficient and deficient cells treated with 10 and 20 μM curcumin.
Next, we examined the effect of MMR status on cell survival in response to curcumin using clonogenic assays on our panel of matched MMR-proficient/deficient cells. Colony forming ability in each cell line was measured in response to 10, 20, 30, 40, and 50 μM curcumin. This analysis revealed that survival in the MSH2-deficient HEC59 line was significantly reduced (p< 0.05, Student's t-test) compared to matched MSH2-proficient HEC59+ch2 cells treated with 40 and 50 μM curcumin (Fig 1C). Consistent with this result, we measured statistically significant (p< 0.01) reduced survival at curcumin doses of ≥ 40 μM in MLH1-deficient HCT116+ch2 cells compared to matched MLH1-proficient HCT116+ch3 cells. MLH1-deficient RKO showed significantly reduced (p<0.05) survival following treatment with ≥ 30 μM curcumin when compared to MLH1-proficient RKO/MLH1+ cells.
Curcumin-induced cell death is suppressed in MMR-proficient cells
To further evaluate the role of MMR in activating the cell death response to curcumin, we knocked down MSH2 or MLH1 expression in MMR-proficient cells (HCT116+ch3) by RNA interference; specifically, using retroviruses encoding gene-specific short hairpin RNA (shRNA) sequences. Immunoblotting of the lysates transduced with MLH1 and MSH2 shRNA-encoding retroviruses confirmed marked suppression of MLH1 and MSH2 expression compared to cells transduced with control luciferase shRNA (shLuc) encoding virus (Fig. 2A). This panel of cell lines was treated with 30 μM curcumin and the sub-G1 cell population was measured 48 h post-treatment by flow cytometry following staining with propidium iodide (Fig 2B). Results showed notable accumulation of sub-G1 population in curcumin-treated cells compared to mock-treated cells. However, a comparison of cell death induction within the treated panel of cell lines revealed notably less cell death in shLuc cells (8.6 % sub-G1) than in shMSH2 or shMLH1 cells (27.2% and 17.9% sub-G1, respectively). When the results of three independent experiments were combined, shMLH1 and shMSH2 cells showed a statistically significant (p<0.01, p<0.05, respectively) increase in sub-G1 than in shLuc cells in response to curcumin treatment (Fig 2C). Immunoblotting of lysates formed from curcumin-treated cells with anti-caspase-3 and anti-PARP antibody showed increased cleavage in treated shMSH2 and shMLH1 cells than in treated shLuc cells (Fig 2D) consistent with activation of the intrinsic apoptotic pathway. Parallel findings were also observed with matched MMR-proficient/deficient cells (suppl. Figs S1, S2panels A & B). Here again, at 24 and 48 h after curcumin treatment, MMR-negative cells (H2; Hec59) displayed increased cell death compared to matched MMR-proficient cells. At the 24 hr time point, MMR-positive cells (H3; Hec59+Ch2) showed accumulation of cells with 4N DNA content (G2/M arrest) but at a later time point (48 hr), a decrease in the G2/M population and an apparent increase in G1 population was observed. These results indicate that the cell cycle arrest induced by curcumin in MMR+ cells is transient in nature, presumably allowing these cells enhanced survival against curcumin. Immunoblotting of lysates obtained from these cells with anti-caspase-3 and anti-PARP antibodies confirmed reduced apoptotic response in MMR+ cells compared to MMR- cells (Figs S1, S2 panel C). Together, these findings reinforced that loss of either MSH2 or MLH1 enhances curcumin sensitivity.
Fig. 2. ShRNA-mediated suppression of MSH2 or MLH1 expression enhances curcumin sensitivity.
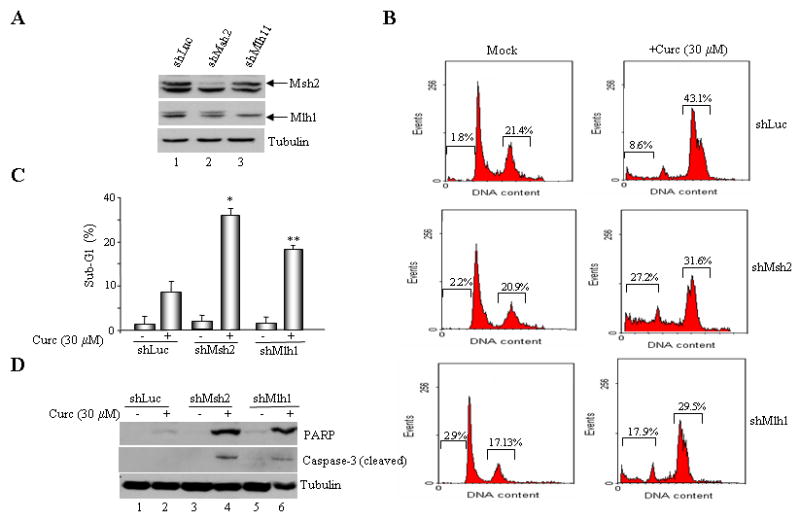
A. HCT116+ch3 (H3) cells were stably transduced with retrovirus encoding luciferase (Luc), MSH2 or MLH1-specific RNAi sequences. Following puromycin selection, lysates were formed and immunoblotted with anti-MSH2 (top), MLH1 (middle) or tubulin (bottom) antibody. A cross-reactive protein band was observed with both anti-MLH1 and MSH2 antibodies. B. ShLuc, shMSH2 and shMLH1 cells were mock (DMSO) treated or exposed to curcumin (30 μM), and 36 h later, stained with PI and assayed for DNA content. Percentage of cells with 4N (G2/M) and sub-G1 DNA content is indicated. C. The percent sub-G1 cell population in mock- and curcumin-treated shLuc, shMSH2 and shMLH1 cells, measured at 36 h post-treatment, is graphed. Displayed is the mean value obtained from three independent experiments, error bar = +1 SD. *denotes P<0.01, **denotes P<0.05. D. Lysates prepared from mock- and curcumin-treated shLuc, shMLH1 and shMSH2 cells were subjected to immunoblotting with anti-PARP and anti-caspase-3 antibody that specifically detects the cleaved form of PARP (top) and caspase-3 (middle). Anti-tubulin (bottom) confirmed equal protein loading.
Following our observation that MMR-deficient cells display enhanced sensitivity to curcumin, we determined if curcumin-induced cell stress is dependent upon a functional MMR system. Since curcumin has been characterized as raising cellular levels of reactive oxygen species (ROS) [17,18], we measured ROS production using 2′,7′-dichlorofluorescein diacetate. Result showed no significant difference in DCF fluorescence in the matched sets of MMR-proficient/deficient cells (see Suppl. Fig S3 A, B). We also measured 8-oxo-G, a prominent mutagenic lesion formed by increased levels of ROS, in shLuc, shMSH2 and shMLH1 cells. Again, no significant difference in curcumin-induced accumulation of 8-oxo-G was observed in these lines (see Suppl. Fig S3 C, D). Clearly, the disparity in curcumin sensitivity observed in MMR-deficient and proficient cells is not attributable to quantitative differences in the stress itself.
The MMR system is required for curcumin-induced checkpoint signaling
Recently, our group and others determined that the dysregulated checkpoint response exhibited by MMR-deficient cells in response to SN1 methylators stems, in part, from defective Chk1 and Chk2 activation [24, 25, 31]. In addition to the increased cell death observed in MMR-deficient cells, we also observed a decreased G2/M arrest when compared to MMR-proficient cells similarly treated with curcumin (Fig 2C). Based on this rationale, we examined MMR+ and MMR- cells for activation of Chk1 and Chk2 by immunoblotting with phospho-specific Chk1 (phospho-S317) and Chk2 (phospho-T68) antibodies. Whereas MMR-positive (HCT116+ch3; H3) cells showed clearly detectable levels of phosphorylated Chk1 and Chk2, matched MMR-negative (HCT116+ch2; H2) cells showed a blunted response at 2, 4 and 6 h after curcumin treatment (Fig. 3A). Similarly, curcumin-induced phosphorylation of Chk1 and Chk2 was observed in treated MSH2-proficient (Hec59+ch2) cells but not in MSH2-deficient (Hec59) cells (Fig. 3B). Blunted Chk1 and Chk2 phosphorylation were also observed in shMLH1 and shMSH2 cells in response to curcumin treatment (Fig. 3C). Together, the results showed that curcumin activates Chk1/Chk2 kinase through a mechanism that is dependent on intact MMR system.
Fig. 3. Curcumin activates Chk1/Chk2 kinases in a MMR-dependent manner.
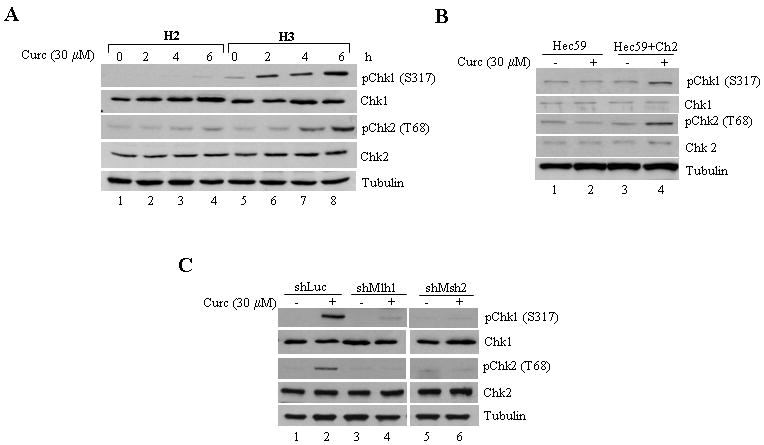
A. MLH1-deficient (HCT116+ch2; H2) and proficient (HCT116+ch3; H3) cells were treated with curcumin (30 μM) and collected 0, 2, 4 and 6 h post-treatment. Lysates were formed, and subjected to immunoblotting with phospho-specific Chk1 (S317), total Chk1, phospho-specific Chk2 (T68), total Chk2, and tubulin antibodies. B. MSH2-deficient (Hec59) and proficient (Hec59+ch2) cells were either mock-treated or exposed to curcumin (30 μM) and Chk1 and Chk2 phosphorylation and protein levels were assessed 4 h post-drug by immunoblotting. C. Curcumin (30 μM) treated shLuc, shMLH1, and shMSH2 cells were assessed for Chk1 and Chk2 activation by immunoblotting with phospho-specific Chk1 and Chk2 antibodies. Total Chk1 and Chk2 levels were assessed by immunoblotting with anti-Chk1 and Chk2 antibodies and anti-tubulin to ensure equal loading.
Inhibition of Chk1 abrogates G2/M arrest and enhances curcumin sensitivity
To determine whether the MMR-dependent phosphorylation of Chk1 and Chk2 mediates G2/M arrest and protects cells from curcumin-induced cytotoxicity we inhibited Chk1 activity with the drug UCN-01 [32]. As expected, treatment of MMR-proficient (HCT116+ch3 cells) with 30 μM curcumin induced robust accumulation of cells in the G2/M phase of the cell cycle (44.1%) accompanied by minimal (5.5%) levels of cell death (sub-G1 cells) (Fig 4A). However, incubation of HCT116+ch3 cells with 0.3 μM UCN-01 resulted in 24.3% of the cells displaying sub-G1 DNA content representing a statistically-significant (p<0.05) increase in curcumin-induced cell death compared to control cells. Chk2 inhibitor II, a compound that specifically inhibits this kinase caused a modest increase in the percentage of apoptotic cells in response to curcumin treatment (Fig. 4 A, B).
Fig. 4. Pharmacological inhibition or shRNA-mediated suppression of Chk1 abrogates G2/M arrest following exposure to curcumin.
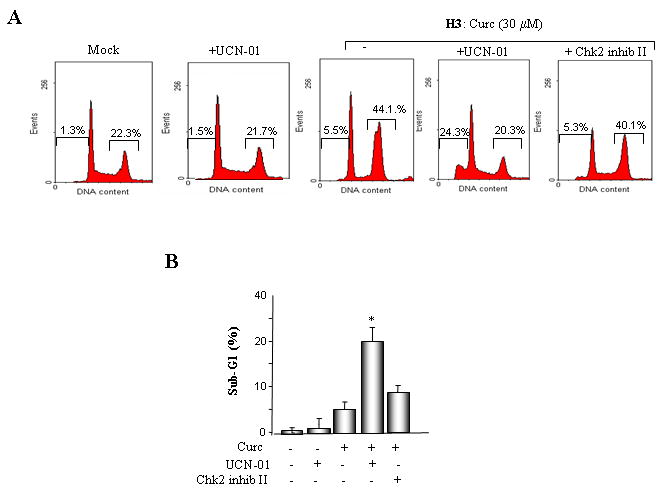
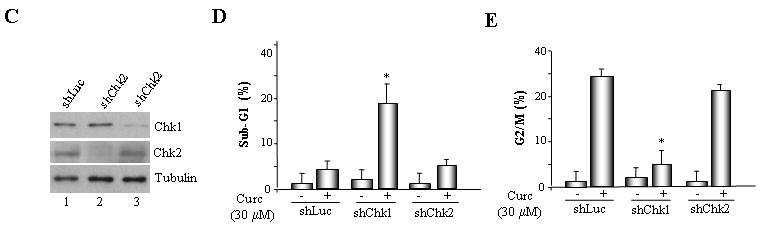
A. MLH1-proficient (HCT116+ch3; H3) cells were pre-incubated with UCN-01 or Chk2 inhibitor II and subsequently mock treated or exposed to curcumin (30 μM). Additionally, HCT116+ch3 cells were either untreated or treated with UCN-01 only. DNA content was measured 36 h post-treatment by PI staining/flow cytometry. Percentage of cells with 4N (G2/M) and sub-G1 DNA content is indicated. B. Graphed is the percentage of cells displaying sub-G1 DNA content in curcumin-treated H3 cells in the presence and absence of UCN-01 or Chk2 inhibitor II. The mean of three independent experiments is indicated, error bars= +1 SD. *denotes P<0.01. C. ShLuc, shChk1 or shChk2 cell lysates were immunoblotted with Chk1 (top), Chk2 (middle) or tubulin (bottom) antibodies. D. ShLuc, shChk1 and shChk2 cells were either mock (DMSO) treated or exposed to curcumin (30 μM) and 36 h later DNA content in the cell population was quantified by PI staining/flow cytometry. Graphed is the mean percentage of sub-G1 cell population from three independent experiments, error bar = +1 SD. *denotes P<0.01. E. ShLuc, shChk1 and shChk2 cells were either mock (DMSO) treated or exposed to curcumin (30 μM) and 36 h later DNA content in the cell population was quantified by PI staining/flow cytometry. Graphed is the mean percentage of G2/M cell population from three independent experiments, error bar = +1 SD. *denotes P<0.01.
To independently assess the roles of Chk1 and Chk2 in curcumin-induced cytotoxicity we suppressed Chk1, Chk2 expression in HCT116+ch3 (H3) using gene-specific shRNAs. After confirming reduced Chk1, Chk2 expression in shChk1 and shChk2 cells by immunoblotting (Fig 4C), these cells, along with control shLuc cells, were treated with 30 μM curcumin and analyzed at 36 h by flow cytometry. As anticipated, in response to curcumin treatment, shChk1 cells displayed a significant increase (p<0.01) in the sub-G1 population in contrast to shLuc cells (20.3% vs 6.3%, respectively) (Fig. 4D). Conversely, curcumin-induced G2/M arrest was decreased significantly (p<0.01) in shChk1 cells compared to curcumin-treated shLuc cells (8.4% vs 51.3%; Fig. 4E). Curcumin-treated shChk2 cells displayed a modest decrease in G2/M cells than treated shLuc cells. In sum, the data demonstrated that, in these colorectal cancer cells, Chk1 confers protection against curcumin toxicity by activating G2/M checkpoint response.
Curcumin-generated ROS activates Chk1/Chk2 and induces DNA damage in a MMR-dependent manner
Curcumin's anti-proliferative effects are associated with elevated levels of intracellular ROS. Thus, we sought to test if curcumin generated ROS activates Chk1 and Chk2. To address this issue, we examined Chk1 and Chk2 activation by curcumin in HCT116+ch3 (H3) cells in the presence of the radical scavenger, N-acetyl-L-cysteine (NAC) or glutathione (GSH). Results showed that both NAC and GSH efficiently abrogated Chk1 and Chk2 phosphorylation by curcumin (Fig. 5A). Additionally, flow cytometric analysis of these cells indicated a significant decrease (p< 0.01) in G2/M population when HCT116+ch3 cells were pretreated with NAC prior to the addition of curcumin (Fig. 5B). These results established that curcumin-induced Chk1/Chk2 activation and G2/M checkpoint induction is mediated by ROS.
Fig. 5. Curcumin-generated ROS activates Chk1 and Chk2 kinases.
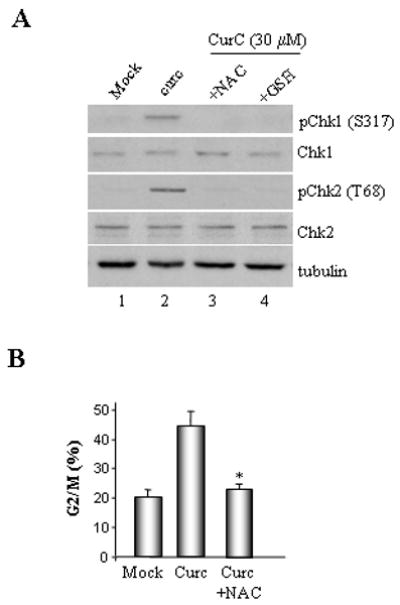
A. Lysates were prepared from HCT116+ch3 (H3) that were either mock-treated or exposed to 30 μM curcumin in the presence or absence of 5 mM NAC or 1 mM GSH. Lysates were then subjected to immunoblotting with phospho-specific Chk1 (S317) and Chk2 (T68) as well as total Chk1, Chk2 and tubulin antibodies. B. Mock and curcumin (30 μM) treated H3 cells were incubated in the presence or absence of 5 mM NAC as indicated. Cells were subsequently harvested, stained with PI and analyzed by flow cytometry. Mean percentage of G2/M cells measured in three independent experiments is graphed, error bars= +1 SD. *denotes P<0.01.
Elevated intracellular levels of ROS oxidize nucleotides resulting in replication-dependent induction of DNA double strand breaks (DSBs) [33]. Thus, we examined the formation of γH2AX foci, a well-known marker for DSB, in curcumin-treated shLuc, shMSH2 and shMLH1 cells. Staining of cells with phospho-specific (S139) H2AX antibody revealed γH2AX foci within 60 min after curcumin (30 μM) treatment, while very few mock-treated cells stained positive for γH2AX foci (Fig. 6A). Immunoblotting of the lysates prepared at various time points (1, 2 and 4 hr) after treatment showed a time-dependent increase in H2AX phosphorylation (Fig. 6B). Consistent with the role of ROS in DSB formation, we also observed that H2AX phosphorylation was blocked by treatment of cells with NAC (Fig. 6B). To address MMR-dependency of DSB formation following curcumin exposure, we examined H2AX phosphorylation in curcumin-treated shMLH1 and shMSH2 cells by immunoblotting. Results indicated substantial reduction in H2AX foci and phosphorylation in HCT116+ch3 with reduced expression of either MSH2 or MLH1 when compared to curcumin-treated control (shLuc) cells (Fig. 6C, D). Collectively, these results indicated that curcumin generated ROS induces DSB largely through a MMR-dependent mechanism.
Fig. 6. Curcumin-induced H2AX phosphorylation and γH2AX foci are blunted in MMR-deficient cells.

A. H3 cells were cultured on pre-sterilized glass cover slips and then either mock (DMSO) or curcumin (30 μM)-treated for 1 hr. Cells were fixed, stained with Ser139 phospho-H2AX antibody (red), and counterstained with DAPI (blue). Also shown is the merged red/blue image. B. H3 cells were either mock (DMSO) treated or exposed to curcumin (30 μM) in the presence or absence of NAC (5 mM) as indicated. At the indicated time point, lysates were formed and immunoblotted with phospho-H2AX (S139) (top), phospho-ATM (S1981) (middle), or tubulin (bottom) antibody. C. ShLuc, shMSH2 and shMLH1 cells were either mock-treated or exposed to 30 μM curcumin and collected 4 hr after. Lysates were formed and immunoblotted with anti-phospho H2AX (S139) (top) or anti-tubulin (bottom) antibody. D. Proposed model for curcumin-induced checkpoint/apoptotic response.
Discussion
Numerous groups have investigated the effects of curcumin on cultures of human tumor cell lines and determined that curcumin exhibits strong anti-proliferative activity, activates the G2/M checkpoint and, depending upon the cell type under investigation, triggers apoptosis [3, 5, 6, 12]. In addition, it has been determined that curcumin's anti-proliferative effect is attributable to sharp rise in superoxide anion concentrations in treated cells [16, 17]. Similarly, we observed that curcumin exposure results in increased ROS levels and the oxidized purine adduct, 8-oxo-G. Furthermore, we observed that the increased levels of ROS stemming from curcumin exposure are responsible for the cell cycle arrest and cell death response triggered by curcumin. In sum, curcumin exposure drives ROS accumulation resulting in cellular stress in the form of genotoxic damage. Thus, curcumin functions as a genotoxic agent.
Consistent with a general response to genotoxic stress, we found that curcumin exposure activates the kinases, Chk1 and Chk2. Among these molecules, Chk1 is the principal signal transducing kinase responsible for establishment of the G2/M checkpoint in response to genotoxic stress [34-37]. In support of the importance of this kinase in triggering the checkpoint response to curcumin exposure, we observed that RNAi-induced knockdown and pharmacological inhibition of Chk1 strongly blunted G2/M arrest. Inhibition of Chk2 showed a more modest effect on the activation of this checkpoint. This outcome parallels findings observed with other types of genotoxic agents [38].
We also observed that activation of Chk1 and Chk2 in response to curcumin are MMR-dependent events. We and others have observed MMR-dependent activation of Chk1 and Chk2 and other kinases in response to SN1 methylating agents [24-26, 39]. It is widely viewed that the mutagenic O6MeG adducts resulting from SN1 methylator exposure is the lesion that triggers MMR-dependent responses [21, 23].
While it is undetermined which lesion(s) result in engagement of the MMR system in response to curcumin, this response most likely stems from base oxidation due to increased ROS since radical scavengers blunted the Chk1/Chk2 response following curcumin administration. Given that curcumin generates 8-oxo-G lesions, it is reasonable to assume that this lesion, and perhaps and other such mutagenic lesions, is recognized by MMR and processing of this damage is responsible for the generation of the DSB observed in curcumin-treated MMR-positive cells. When taken together, our observations lead us to propose a model elucidating how cells respond to curcumin (Fig 6D). In this model, we propose that at the doses employed, curcumin-induced DNA damage engages the MMR system, most likely through recognition of oxidative damage to the genome. MMR-dependent processing of these lesions subsequently leads to the development of DSBs within the genome. The cell consequently responds to these DSBs by activating the G2/M cell cycle checkpoint through the Chk1/Chk2 signal transduction pathway. Proper activation of this checkpoint response significantly blunts the cytotoxic effects of curcumin. Higher doses or longer incubation times with curcumin, however, results in the triggering of cell death in MMR+ cells through necrotic cell death (data not shown). Although the MMR system has no effect on the levels of ROS generated or consequential genome damage in response to curcumin, in the absence of this repair mechanism, cells fail to develop DSBs. Owing to reduced DSBs, MMR-deficient cells do not activate the G2/M checkpoint and, as a result, likely enter mitosis and subsequently trigger apoptosis through mitotic catastrophe [6, 12].
Our model proposes that MMR plays a significant role in eliciting cellular response to oxidative stress induced by curcumin. Of note, others have observed that MMR-deficient cells exhibit increased sensitivity to oxidative stress. Specifically, Chang et al [40] observed that MMR-deficient cells are more sensitive to oxidative damage induced by H2O2 than MMR-proficient cells. This result is similar to what we have documented in response to curcumin. Interestingly, unlike what we have observed using curcumin, H2O2 treatment resulted in activation of G2/M arrest through a MMR-independent mechanism. Oxyradicals generate a broad myriad of DNA damage [33]; consequently, there are potential differences in the nature of lesions generated by curcumin versus those generated by other sources of oxidative stress such as H2O2. Nonetheless, our results show that curcumin-induced G2/M arrest clearly proceeds in a mismatch repair-dependent manner and its abrogation by UCN-01 enhanced curcumin sensitivity. Although failure to activate cell cycle arrest probably contributes to curcumin sensitivity in MMR-deficient cells, it should be borne in mind that curcumin activates multiple signaling pathways. For example, curcumin treatment is associated with downregulation of NF-κB activation [14], decreased iNOS and DNA-PK catalytic subunit expression, upregulation of p53, p21, p27(Kip1) and Chk2 [15]. Curcumin activates c-Abl and JNK kinases that are required for apoptosis induced by this anti-cancer agent [17]. Moreover, a recent study showed that curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NFκB activity [41]. When taken together, these observations suggest that, in addition to abrogated checkpoint signaling, the increased curcumin sensitivity observed in MMR-deficient cells may be attributable to dysregulation of other signaling mechanisms. In addition to activating apoptosis, recent studies sowed that in normal human cells, and malignant glioma cells curcumin induced nonapoptotic, autophagic cell death [42]. Clearly, the involvement of autophagy, if any, in conferring curcumin sensitivity in MMR-deficient cells remains unknown.
The results presented here have clear potential therapeutic value. Microsatellite instability (MSI) positive phenotype is commonly observed in several tumor types including colorectal, endometrial, gastric and ovarian cancer [43]. This feature is associated with inactivation of the MMR system [44]. Currently, curcumin is being evaluated clinically for the treatment of colorectal tumors [1, 45]. Our findings clearly suggest that this anti-cancer agent may be especially useful in the treatment of MSI+ tumors. Additionally, our findings suggest that combining curcumin with Chk1 inhibitors such as UCN-01 may potentially sensitize MSI negative (MMR-proficient) tumor cells consequently enhancing cell death. Although curcumin bioavailability is sub-optimal, liposomal curcumin, curcumin nanoparticles, and curcumin phospholipid complexes are being tested as alternate approaches for enhanced bioavailability [46].
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants to R.B (GM 60945) J.C.Y (CA90787) and K.D.B (CA102289).
Grant Support: NIH grants GM 60945 (RB, ZJ), CA90787 (J.C.Y, SJ) and CA102289 (KB).
Abbreviations
- Curc
Curcumin
- shRNA
short hairpin RNA
- NAC
N-acetyl cysteine
- ROS
reactive oxygen species
- PARP
poly (ADP-ribose) polymerase
References
- 1.Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41:1955–68. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Chauhan DP. Chemotherapeutic potential of curcumin for colorectal cancer. Curr Pharm Des. 2002;8:1695–706. doi: 10.2174/1381612023394016. [DOI] [PubMed] [Google Scholar]
- 3.Jiang MC, Yang-Yen HF, Yen JJ, et al. Curcumin induces apoptosis in immortalized NIH 3T3 and malignant cancer cell lines. Nutr Cancer. 1996;26:111–20. doi: 10.1080/01635589609514468. [DOI] [PubMed] [Google Scholar]
- 4.Song G, Mao YB, Cai QF, et al. Curcumin induces human HT-29 colon adenocarcinoma cell apoptosis by activating p53 and regulating apoptosis-related protein expression. Braz J Med Biol Res. 2005;38:1791–8. doi: 10.1590/s0100-879x2005001200007. [DOI] [PubMed] [Google Scholar]
- 5.Karunagaran D, Rashmi R, Kumar TR. Induction of apoptosis by curcumin and its implications for cancer therapy. Curr Cancer Drug Targets. 2005;5:117–29. doi: 10.2174/1568009053202081. [DOI] [PubMed] [Google Scholar]
- 6.Holy JM. Curcumin disrupts mitotic spindle structure and induces micronucleation in MCF-7 breast cancer cells. Mutat Res. 2002;518:71–84. doi: 10.1016/s1383-5718(02)00076-1. [DOI] [PubMed] [Google Scholar]
- 7.Nitta M, Kobayashi O, Honda S, et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 2004;23:6548–58. doi: 10.1038/sj.onc.1207873. [DOI] [PubMed] [Google Scholar]
- 8.Dziadyk JM, Sui M, Zhu X, et al. Paclitaxel-induced apoptosis may occur without a prior G2/M-phase arrest. Anticancer Res. 2004;24:27–36. [PubMed] [Google Scholar]
- 9.Tseng CJ, Wang YJ, Liang YC, et al. Microtubule damaging agents induce apoptosis in HL 60 cells and G2/M cell cycle arrest in HT 29 cells. Toxicology. 2002;175:123–42. doi: 10.1016/s0300-483x(02)00073-2. [DOI] [PubMed] [Google Scholar]
- 10.Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- 11.Wolanin K, Magalska A, Mosieniak G, et al. Curcumin affects components of the chromosomal passenger complex and induces mitotic catastrophe in apoptosis-resistant Bcr-Abl-expressing cells. Mol Cancer Res. 2006;4:457–69. doi: 10.1158/1541-7786.MCR-05-0172. [DOI] [PubMed] [Google Scholar]
- 12.Magalska A, Sliwinska M, Szczepanowska J, et al. Resistance to apoptosis of HCW-2 cells can be overcome by curcumin- or vincristine-induced mitotic catastrophe. Int J Cancer. 2006;119:1811–18. doi: 10.1002/ijc.22055. [DOI] [PubMed] [Google Scholar]
- 13.Park C, Kim GY, Kim GD, et al. Induction of G2/M arrest and inhibition of cyclooxygenase-2 activity by curcumin in human bladder cancer T24 cells. Oncol Rep. 2006;15:1225–31. [PubMed] [Google Scholar]
- 14.Plummer SM, Holloway KA, Manson MM, et al. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signaling complex. Oncogene. 1999;18:6013–20. doi: 10.1038/sj.onc.1202980. [DOI] [PubMed] [Google Scholar]
- 15.Zheng M, Ekmekcioglu S, Walch ET, et al. Inhibition of nuclear factor-kappa B and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res. 2004;114:165–71. doi: 10.1097/01.cmr.0000129374.76399.19. [DOI] [PubMed] [Google Scholar]
- 16.Bhaumik S, Anjum R, Rangaraj N, et al. Curcumin mediated apoptosis in AK-5 tumor cells involves the production of reactive oxygen intermediates. FEBS Lett. 1999;456:311–14. doi: 10.1016/s0014-5793(99)00969-2. [DOI] [PubMed] [Google Scholar]
- 17.Kamath R, Jiang Z, Sun G, et al. c-Abl kinase regulates curcumin-induced cell death through activation of c-Jun N-terminal kinase. Mol Pharmacol. 2007;71:61–72. doi: 10.1124/mol.106.026575. [DOI] [PubMed] [Google Scholar]
- 18.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 19.Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- 20.Li GM. The role of mismatch repair in DNA damage-induced apoptosis. Oncol Res. 1999;11:393–400. [PubMed] [Google Scholar]
- 21.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signaling. DNA Repair (Amst) 2004;3:1091–101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Aebi S, Kurdi-Haidar B, Gordon R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–90. [PubMed] [Google Scholar]
- 23.Ni TT, Marsischky GT, Kolodner RD. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol Cell. 1999;4:439–44. doi: 10.1016/s1097-2765(00)80346-9. [DOI] [PubMed] [Google Scholar]
- 24.Adamson AW, Beardsley DI, Kim WJ, et al. Methylator-induced, Mismatch Repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol Biol Cell. 2005;16:1513–26. doi: 10.1091/mbc.E04-02-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stojic L, Mojas N, Cejka P, et al. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–44. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc Natl Acad Sci USA. 2003;100:15387–92. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshioka KI, Yoshioka Y, Hseih P. ATR kinase activation mediated by MutSα and MutLα in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–10. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koi M, Umar A, Chauhan DP, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N'nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;(54):4308–12. [PubMed] [Google Scholar]
- 29.Umar A, Koi M, Risinger JI, Glaab WE, et al. Correction of hypermutability, N-methyl-N'-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997;57:3949–55. [PubMed] [Google Scholar]
- 30.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95:8698–702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stojic L, Cejka P, Jiricny J. High Doses of S(N)1 Type Methylating Agents Activate DNA Damage Signaling Cascades that are Largely Independent of Mismatch Repair. Cell Cycle. 2005;3:473–74. doi: 10.4161/cc.4.3.1528. [DOI] [PubMed] [Google Scholar]
- 32.Graves PR, Yu L, Schwarz JK, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–05. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- 33.Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997;272:25409–12. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 34.Macip S, Kosoy A, Lee SW, et al. Oxidative stress induces a prolonged but reversible arrest in p53-null cancer cells, involving a Chk1-dependent G2 checkpoint. Oncogene. 2006;25:6037–47. doi: 10.1038/sj.onc.1209629. [DOI] [PubMed] [Google Scholar]
- 35.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell-cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–97. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 38.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–29. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 39.Kim WJ, Rajasekaran B, Brown KD. MLH1 and ATM-dependent MAP kinase signaling is activated through C-Abl in response to the alkylator N-methyl-N'-nitro-N-nitrosoguanidine. J Biol Chem. 2007;282:32021–31. doi: 10.1074/jbc.M701451200. [DOI] [PubMed] [Google Scholar]
- 40.Chang DK, Goel A, Ricciardiello L, et al. Effect of H(2)O(2) on cell cycle and survival in DNA mismatch repair-deficient and -proficient cell lines. Cancer Lett. 2003;195:243–51. doi: 10.1016/s0304-3835(03)00145-9. [DOI] [PubMed] [Google Scholar]
- 41.Sandur SK, Deorukhkar A, Pandey MK, et al. Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-kappaB activity. Int J Radiat Oncol Biol Phys. 2009;75:534–42. doi: 10.1016/j.ijrobp.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aoki H, Takada Y, Kondo S, et al. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 43.Lynch HT, Casey MJ, Lynch J, et al. Genetics and Ovarian carcinoma. Semin Oncol. 1998;57:3253–57. [PubMed] [Google Scholar]
- 44.Muller A, Fishel R. Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC) Cancer Invest. 2002;20:102–09. doi: 10.1081/cnv-120000371. [DOI] [PubMed] [Google Scholar]
- 45.Garcea G, Berry DP, Jones DJ, et al. Consumption of the putative chemo-preventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev. 2005;14:120–5. [PubMed] [Google Scholar]
- 46.Anand P, Kunnumakkara AB, Newman RA, et al. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–18. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.