

Abstract
Tim-3 (T cell immunoglobulin, mucin domain)-3 is a membrane protein expressed at late stages of interferon-gamma secreting CD4+ T helper type 1 (Th1) cell differentiation and constitutively on dendritic cells (DCs). Ligation of Tim-3 on Th1 cells terminates Th1 immune responses. In addition, Tim-3 plays a role in tolerance induction, though the mechanism by which this is accomplished has yet to be elucidated. While it is clear that Tim-3 plays an important role in the immune system, little is known regarding the molecular pathways that regulate Tim-3 expression. Here we have examined the role of Th1-associated transcription factors in regulating Tim-3 expression. Our experiments reveal that Tim-3 expression is regulated by the Th1-specific transcription factor T-bet. This introduces a novel paradigm into the generation of a Th1 response, whereby a transcription factor responsible for effector Th1 cell differentiation also increases the expression of a specific counter-regulatory molecule to ensure appropriate termination of pro-inflammatory Th1 immune responses.
Keywords: T cells, Autoimmunity, Transcription Factors
Introduction
Tim-3 is a type I transmembrane protein, originally identified as specifically and selectively induced upon the differentiation of naïve CD4+ T helper (Th) cells into interferon-gamma secreting Th1 effector cells [1] where it serves to promote termination of Th1 cells upon interaction with its ligand, galectin-9 [2]. Modulation of the Tim-3:galectin-9 pathway has potent effects in vivo as treatment with anti-Tim-3 antibody exacerbates the induction of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS) [1], and administration of Tim-3 fusion protein (Tim-3Ig) to immunized mice results in an uncontrolled Th1 response [3]. In addition, wild type mice treated with Tim-3Ig, as well as Tim-3-deficient mice, are refractory to tolerance induction [3, 4]. Taken together, these data demonstrate the importance of the Tim-3:galectin-9 pathway in regulating inflammatory T cell responses and states of immunological tolerance. Recently, Tim-3 expression has been demonstrated on dendritic cells (DCs) and on cells of the innate immune system [5]. However, the factors that regulate Tim-3 expression both in T cells and innate immune cells have not been identified.
Recent evidence shows a correlation between mRNA expression levels of the Th1-specific transcription factor T-bet (Tbx21; T-box expressed in T cells), IFN-γ and TIM-3 in T cell clones from both patients with Multiple Sclerosis (MS) and healthy controls [6]. In this study the linked expression of T-bet, IFN-γ and TIM-3 could simply be due to the fact that T-bet is required for IFN-γ transcription and IFN-γ–producing Th1 cells predominantly express TIM-3 on their cell surface. Direct evidence that T-bet controls TIM-3 expression was lacking from this study. Interestingly, like Tim-3, T-bet is also expressed in DCs where it controls IFN-γ production [7] and is required for optimal TLR responsiveness [8]. Given that T-bet is crucial for induction of IFN-γ and Tim-3 is expressed on Th1 cells, we investigated the role of T-bet as well as other Th1 associated transcription factors in the regulation of Tim-3 expression in murine T cells during Th1 polarization and in DCs. Our studies reveal Tim-3 expression is regulated by T-bet.
Results
Defects in Tim-3 expression in T-bet- and STAT-4-deficient lymphocytes
Our previous studies demonstrated upregulation of Tim-3 expression in Th1 cells after three rounds of in vitro polarization [1]. Therefore, in order to examine the role of Th1-associated transcription factors on Tim-3 expression, we differentiated naïve CD4+CD62Lhigh T cells from wild type, T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− mice in vitro under Th1-polarizing conditions and examined both Tim-3 mRNA and protein expression after each round of differentiation. In keeping with our previous observations, we first detected Tim-3 mRNA as well as protein in wild type T cells after the third round of in vitro Th1 differentiation [1] (Fig. 1). At this time, Tim-3 mRNA and protein expression were considerably lower in T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− T cells. After the fourth round of differentiation, Tim-3 expression reached maximal levels in wild type T cells (Fig. 1A and C). At this time, Tim-3 expression was still impaired in T-bet−/− cells with STAT-4−/− T cells showing a more modest reduction in Tim-3 mRNA and little if any difference in protein expression. Importantly, T-bet−/− and T-bet−/− × STAT-4−/− T cells expressed similar levels of Tim-3 mRNA, suggesting that T-bet has a more dominant role in regulating Tim-3 expression. Overall, these data suggested that T-bet and to a lesser degree, STAT-4, were important regulators of Tim-3 expression. However, since IFN-γ is deficient in the cultures of T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− T cells (Fig. 1B), this raised the possibility that defects in Tim-3 expression could be secondary to defects in IFN-γ/IFN-γR signaling. This is unlikely for three reasons. First, T-bet−/− T cells exhibit the greatest defect in Tim-3 expression while having a considerably smaller defect in IFN-γ relative to STAT-4−/− and T-bet−/− × STAT-4−/− T cells (50% IFN-γ+ relative to 9% and 1%, respectively) (Fig. 1B). Second, Tim-3 is upregulated in IFN-γ−/− T cells that overexpress T-bet, showing that T-bet can drive expression of Tim-3 in the absence of IFN-γ (Fig. 2). Lastly, Tim-3 is detected on the surface of T cells derived from IFN-γR−/− mice undergoing Th1 differentiation at levels comparable to wild type T cells (data not shown). Thus, the IFN-γ/IFN-γR signaling pathway is dispensable for Tim-3 expression.
Figure 1. Impaired Tim-3 expression in T-bet and STAT-4 deficient Th1 cells.

(A) Naïve CD4+ (CD4+ CD62Lhigh) T cells from wild type, T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− mice were stimulated in vitro under Th1 polarizing conditions. After each round of polarization, cells were harvested and total RNA prepared for analysis of Tim-3 expression by real-time PCR. Expression of Tim-3 RNA relative to control HPRT expression is shown. Error bars indicate SEM of two replicate experiments. Data shown are representative of 2–3 independent experiments. (B) CD4+ T cells from (A) were activated with PMA/Ionomycin and then stained with mAbs to Tim-3, CD4, IFN-γ, IL-4, IL-10 and IL-17. Histograms represent Tim-3 expression (filled histogram, FMO control; solid black line, Tim-3 staining). Dot plots represent cytokine expression in CD4+ cells. Numbers indicate frequency of cytokine positive cells. Similar results were obtained in 2–3 independent experiments. (C) Median fluorescence intensity (MFI) of Tim-3 staining on CD4+ T cells from wildtype, T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− T cells after each round of in vitro Th1 differentiation. Data shown are representative of 2–3 independent experiments.
Figure 2.
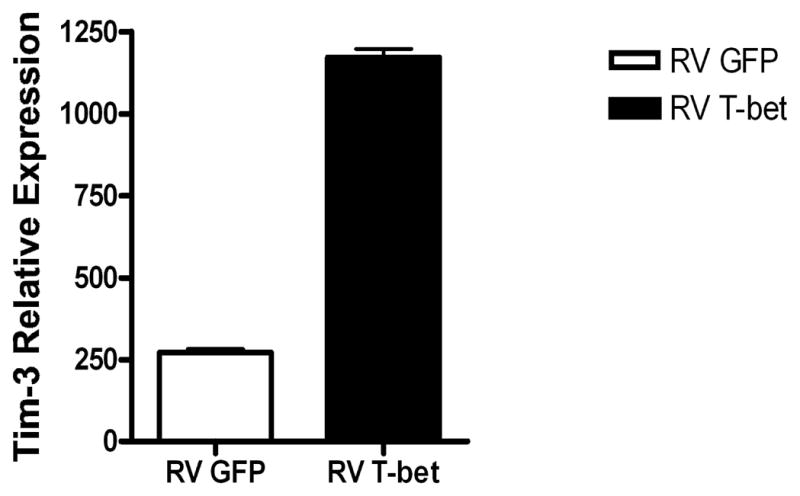
T-bet can drive the expression of Tim-3 independently of transactivation of IFN-γ. Naïve (CD4+CD62LhighCD25−) CD4+ T cells from T-bet−/− × IFN-γ−/− mice were transduced at 48 hours post activation with plate bound CD3 and CD28 with either empty (RV GFP) or T-bet (RV T-bet) expressing retrovirus and sorted for CD4 and GFP expression at day 5. The cells were rested overnight in IL-2 (100 U/ml) and RNA was extracted 24 hours later. Tim-3 RNA expression was quantified by real time PCR and normalised to β-actin. Error bars represent SEM from triplicate PCRs. Data shown are representative of 3 independent experiments.
As the T-bet−/− × STAT-4−/− T cells in our study exhibited an increased frequency of IL-10 producing cells, this raised the possibility that IL-10 could be driving Tim-3 expression in a T-bet independent manner in these cells. To address this we neutralized IL-10 during Th1 polarization of T-bet−/− × STAT-4−/− T cells and found that Tim-3 expression is increased rather than decreased in cultures where IL-10 is neutralized (data not shown). Thus, IL-10 does not appear to drive Tim-3 expression in the absence of T-bet and STAT-4.
Since CD8+ T cells that have undergone differentiation towards Tc1 also express Tim-3 [1], we examined Tim-3 expression by wild type and T-bet−/− CD8+ T cells undergoing Tc1 differentiation. Similar to our observations for CD4+ T cells undergoing Th1 differentiation, we find that T-bet−/− CD8+ T cells express less Tim-3 mRNA (8 fold less than wild type) and protein relative to wild type CD8+ T cells (data not shown). Collectively, these data suggest that T-bet is an important regulator of Tim-3 expression in both CD4 and CD8 T cells undergoing Th1/Tc1 differentiation.
Tim-3 expression in dendritic cells is regulated by T-bet
We have recently described the constitutive expression of Tim-3 on dendritic cells in both mouse and human [5]. Interestingly, T-bet is also expressed in DCs and T-bet−/− DCs exhibit defects in responsiveness to CpG, a TLR9 agonist [7, 8]. We therefore examined whether T-bet was also involved in regulating Tim-3 expression in DCs. In agreement with our observations in T cells, T-bet−/− and T-bet−/− × STAT-4−/− DCs exhibit the greatest deficiency in Tim-3 mRNA with STAT-4−/− DCs exhibiting only a modest, if any, reduction in Tim-3 mRNA (Fig. 3). Thus, Tim-3 expression appears to be regulated similarly in both T cells and DCs. We also examined Tim-3 protein expression in DCs from all four strains by flow cytometry and found that while Tim-3 mRNA is clearly lower in T-bet−/− and T-bet−/− × STAT-4−/− DCs, these cells exhibited similar levels of Tim-3 protein to wild type mice. Since DCs constitutively express Tim-3, these data suggest that once Tim-3 protein is expressed it is stable and turns over slowly.
Figure 3. Impaired Tim-3 expression in T-bet and STAT-4 deficient dendritic cells.

CD11c+ dendritic cells were isolated by cell sorting from the collagenase digested spleens of wild type, T-bet−/−, STAT-4−/− and T-bet−/− × STAT-4−/− mice. Expression of Tim-3 RNA relative to control HPRT expression is shown. Error bars indicate SEM of two replicate experiments. Data shown are representative of 2–3 independent experiments.
Role of T-bet in Tim-3 upregulation
Our data in T-bet−/− and STAT-4−/− cells suggested that Tim-3 may be a direct target of T-bet and that the reduced expression seen in STAT-4−/− cells may be a secondary consequence of reduced T-bet levels in cells from these mice ([9] and data not shown). We therefore examined whether direct overexpression of T-bet expression would affect Tim-3 expression. CD4+ T cells from overexpressor T-bet CD2-Tg mice [10] and wild type BALB/c controls were activated under either unskewed (Th0) or Th1-inducing conditions. In the presence of the T-bet transgene, there was a significant increase in the expression of Tim-3 in the Th1-polarizing condition, and this occurred after only one round of in vitro polarization (Fig. 4B). It is noteworthy that T-bet transgenic but not wild type T cells expressed Tim-3 even when stimulated under non-polarizing conditions (Fig. 4A). While both the T-bet CD2-Tg T cells and the wild type control T cells stimulated under Th1-polarizing conditions produced significant levels of IFN-γ (Fig. 4B), only the T-bet CD2-Tg cells upregulated Tim-3, further supporting the conclusion that IFN-γ signaling does not play a role in regulating Tim-3 expression.
Figure 4. T-bet controls Tim-3 expression in vitro.

Naïve CD4+ (CD4+CD62LhighCD25−) T cells from T-bet CD2-Tg mice (and BALB/c wild type controls) were stimulated in vitro under Th0 (A) and Th1-polarizing conditions (B). After one round of restimulation, T cells were stimulated and then stained with mAbs to Tim-3 and CD4, and cytokine expression was detected by intracellular staining. Histograms represent Tim-3 expression (dotted line, isotype control; solid line, specific staining). Dot plots represent cytokine expression in CD4+ T cells (Th1 condition). This experiment was repeated 3 times (n=4 for both wildtype and Tg mice in each experiment). Representative data are shown.
We next addressed whether T-bet was similarly involved in regulating Tim-3 expression in vivo. As Tim-3 is not expressed on naïve T cells [2], we transferred sorted naïve CD4+ T cells from wild type or T-bet−/− mice into Rag2−/− mice. In Rag2−/− mice, transferred T cells undergo activation in response to homeostatic cues. Accordingly, we examined the frequency of cells that underwent activation by measuring incorporation of Brdu and upregulation of CD25. We found that similar frequencies of wild type and T-bet−/− cells underwent activation and trafficked to the spleen (Fig. 5A and B). However, only wild type T cells turned on expression of Tim-3 (Fig. 5C), suggesting that T-bet also regulates Tim-3 expression not only in vitro but also in vivo. The reduced expression of Tim-3 in T-bet−/− cells both in vitro and in vivo together with the enhanced Tim-3 expression in T-bet transgenic cells suggested that Tim-3 might be a direct target of T-bet.
Figure 5. T-bet controls Tim-3 expression in vivo.

1 × 106 naive (CD4+CD62LhighCD44lowCD25−) CD4+ T cells from wildtype (Balb/c) or T-bet−/− (Balb/c) were adoptively transferred i.v. into Rag2−/− (Balb/c) mice (n=5/group). Transferred mice were administered Brdu in the drinking water (0.8mg/ml). Two weeks later, splenic T cells populations were harvested. (A) Frequency of CD4+Brdu+ cells in transferred mice. (B) Frequency of CD4+CD25+ cells in transferred mice. (C) Frequency of total CD4+Tim-3+ cells in transferred mice. Error bars indicate SEM. Data shown are representative of 2 independent experiments.
T-bet binds to the Tim-3 promoter
To determine whether Tim-3 is a direct transcriptional target of T-bet, we performed chromatin immunoprecipitation (ChIP) experiments with an anti T-bet antibody and analyzed the precipitated genomic DNA by real time PCR. As a positive control, we used primer sequences to the proximal IFN-γ promoter, which showed significant enrichment from the immunoprecipitated (IP) sample compared with whole cell extract (Fig. 5A). As a negative control, T cells from T-bet−/− mice were subjected to ChIP with the same antibody (and isotype matched antibody) in parallel experiments, and no enrichment of the IFN-γ promoter was observed (Fig. 6A). To further confirm the specificity of these experiments, ChIP experiments were performed with PCR primers to the proximal portion of the IL-4 promoter. There was no enrichment of this region, as expected given that T-bet does not bind in this area [11] (Fig. 6B). ChIP experiments were then performed to analyze T-bet binding to the proximal Tim-3 promoter. There was significant enrichment in the IP sample precipitated with the T-bet antibody, while there was no enrichment seen with these primers in the T-bet−/− samples (Fig. 6C). Indeed, a sequence similar to the full T-box consensus sequence was near this enriched region on both positive and negative strands 5′ of the Tim-3 gene (5′-CAC ATGA GTG –3′, 3′-CAC TCAT GTG-5′) at ~400bp upstream of the first ATG-methionine. We also assessed whether STAT-4 bound the Tim-3 promoter. However, we could detect no binding of STAT-4 to the Tim-3 promoter (Fig. 6D), further suggesting that the reduced expression of Tim-3 in the absence of STAT-4 is perhaps indirect and could in part be due to reduced levels of T-bet in these mice [12].
Figure 6. T-bet binds to the Tim-3 promoter in vivo.

ChIP was performed on murine Th1 CD4+ T cells with a polyclonal antibody to T-bet, and the resulting genomic DNA was analyzed by real-time PCR. (A) IFN-γ promoter, (B) IL-4 promoter, (C) Tim-3 promoter and (D) Tim-3 promoter after ChIP with a STAT-4 antibody. Enrichment is expressed as the fold enrichment of IP DNA over whole cell extract. Error bars indicate SEM of triplicate reactions. Data shown are representative of 2 independent experiments.
Discussion
We have shown that cells lacking expression of the Th1-associated transcription factor T-bet−/−, and to a lesser extent if at all STAT-4, have defects in Tim-3 expression. We further show that T-bet overexpression results in constitutive Tim-3 expression and that T-bet binds directly to the Tim-3 promoter. Collectively, our data strongly support that Tim-3 is a direct transcriptional target of T-bet. Although we could not detect any binding of STAT-4 to the Tim-3 promoter, we did observe some reduction in Tim-3 expression in STAT-4−/− cells. Thus, whether STAT-4 plays a direct or indirect role in regulating Tim-3 expression is not clear at this time and further studies will be necessary to clarify the role of STAT-4 in Tim-3 regulation.
Given that Tim-3 expression is induced only after several rounds of in vitro differentiation, and that T-bet is upregulated early in Th1 differentiation [13], there are likely other transcription factors that play a role in inducing the expression of Tim-3, possibly by synergizing with signals emanating from the T cell receptor. Indeed, several transcription factors have predicted binding sites in the proximal Tim-3 promoter (Table S1). Nevertheless, in the absence of T-bet, Tim-3 expression is significantly impaired, demonstrating the importance of T-bet for Tim-3 transcription.
Recently, low-levels of Tim-3 expression have been reported on Th17 cells in both mouse [14, 15] and human [16]. Indeed, it has been suggested that low-level Tim-3 expression may allow escape of Th17 cells from galectin-9 mediated cell death [17], however the biological consequences of the low Tim-3 expression by Th17 cells has not been established. Since T-bet is not engaged during the Th17 differentiation program [18] and Th17 differentiation is not impaired in the absence of T-bet [19] [20], it is likely that other transcription factors are involved in regulating Tim-3 expression in Th17 cells.
Alterations in TIM-3 expression have important biological consequences in human disease. In multiple sclerosis, T cells present in the cerebrospinal fluid (CSF) of patients have lower TIM-3 expression and secrete higher levels of IFN-γ than T cells in the CSF of healthy controls. Moreover, the reduced expression of TIM-3 in the CSF T cells from MS patients is associated with the failure to induce tolerance in these cells. Interestingly, treatment with either glatiramer acetate or IFN-β, approved drugs for the treatment of MS, restores both TIM-3 expression and TIM-3-mediated immunoregulation in T cells from MS patients [21]. In contrast to the reduced TIM-3 expression observed in T cells from MS patients, TIM-3 expression is increased on a population of T cells in patients chronically infected with human immunodeficiency virus (HIV) [22] and hepatitis C virus (HCV) [23]. These T cells are dysfunctional in that they fail to produce cytokine and proliferate in response to stimulation with antigen. Blocking TIM-3 in these “exhausted” T cells restores proliferation and enhances cytokine production. Collectively, these data show that modulation of TIM-3 expression has therapeutic value in both autoimmune and infectious disease and underscore the importance of understanding the regulation of TIM-3 expression. However, it remains to be addressed whether TIM-3 is also a direct transcriptional target of T-bet in human cells.
The mechanisms by which Th1 responses are terminated in order to avoid ongoing and potentially pathogenic inflammation are still being elucidated. Here we provide evidence for a novel, counter-regulatory loop whereby the Th1-promoting transcription factors T-bet and possibly STAT-4 also regulate the expression of the Th1-inhibitory molecule Tim-3. Notably, the kinetics of these opposing pathways differ. Initial Th1 differentiation is driven by early induction of T-bet in the naïve T helper progenitor cell, with IL-12R/STAT-4 signaling stabilizing the Th1 phenotype shortly thereafter (reviewed in [24, 25]). Tim-3 expression is induced by these transcription factors; however, the expression of Tim-3 is delayed to a late stage of Th1 differentiation [1], thus ensuring that a vigorous Th1 response can occur, but that it can also be terminated. Indeed, T-bet can further promote termination of Th1 responses by the induction of Tim-3 ligand, galectin-9, which is potently induced by IFN-γ [26, 27]. Further elucidation of both the factors that regulate T-bet expression during early and late stages of Th1 differentiation and the mechanism of Tim-3 upregulation will enhance our understanding of the induction and termination of the proinflammatory Th1 response.
Materials and Methods
Mice
STAT-4−/− mice on the C57BL/6 background were kindly provided by Dr. Michael Grusby [12]. For some experiments T-bet−/− mice [28] (Balb/c background) were backcrossed onto the C57BL/6 background for six generations. T-bet−/− mice were crossed with STAT-4−/− mice to generate T-bet−/− × STAT-4−/− mice. All deficient mice were bred with mice expressing a T cell receptor specific for MOG 35–55 (2D2) on the C57BL/6 background [29]. Mice overexpressing T-bet under the control of the human CD2 promoter were previously described [10]. All animals were housed according to the guidelines established by the Harvard Committee on Animals.
In vitro T cell differentiation
Naïve CD4+ T (CD62Lhigh) were isolated by cell sorting (BD FACSAria; BD Biosciences) and cultured (1×106/well) with syngeneic irradiated splenic APC (5×106/well) in the presence of MOG 35–55 peptide (10–50 μg/ml) and rIL-12 (10 ng/ml, BD Biosciences) and anti-IL-4 (11B11; 10 g/ml). After two days, Th1 cultures were supplemented with medium containing IL-2. After each round of polarization, T cells were harvested and restimulated with 5 ng/ml PMA (Sigma) and 0.5 μg/ml ionomycin (Sigma) in the presence of monensin (Golgi Stop, BD Biosciences) for 4h at 37°C. Cells were analyzed by flow cytometry for surface expression of CD4 and Tim-3 (clone 5D12 or 2C12 for experiments with T-bet CD2 Tg) and intracellular expression of IFN-γ, IL-4, IL-10, and IL-17. Flow cytometry data was collected on a BD FACSCalibur (BD Biosciences) and analyzed with FlowJo Software (Tree Star).
Quantitative Taqman RT-PCR
Total RNA was made by TRIzol (Sigma) reagent according to the manufacturer’s protocol, reverse transcribed to cDNA and real-time RT-PCR was performed using the following primers/probe: Tim-3-Probe 6FAM-AGACACTGGTGACCCTCCATAATAACAATGGAA–TAMRA, Tim-3 F-5′CGGAGAGAAATGGTTCAGAGACA3′ and Tim-3 R-5′TTCATCAGCCCATGTGGAAAT-3′. HPRT-Probe 6FAM-TGTTGGATACAGGCCAGACTTTGTTGGAT-TAMRA, HPRT F-5′ CTGGTGAAAAGGACCTCTCG3′ and HPRT R-5′TGAAGTACTCATTATAGTCAAGGGCA3′. The Tim-3 CT value was normalized using the formula ΔCT = CTTim-3−CTHPRT.
Retroviral Gene Transduction
Naïve CD4+ T cells from lymph nodes of 4–6 week old T-bet−/− × IFN-γ−/− mice (BALB/c background) were transduced at 48 hours post activation with plate bound CD3 and CD28 with either empty (RV GFP) or T-bet (RV T-bet) expressing retrovirus and sorted for CD4 and GFP expression at day 5. The cells were rested overnight in IL-2 (100 U/ml) and RNA was extracted 24 hours later.
Chromatin Immunoprecipitation
ChIP was performed as previously described [11], and the resulting genomic DNA was analyzed by real time PCR with primers designed to span equidistant regions of the proximal promoter of the specific gene. The primers used for analysis of the specific promoters were; IFN-γ: Forward-GCCCAAGGAGTCGAAAGGA, Reverse- GGGATTACGTATTTTCACAAGTTTTTT, TIM-3: Forward- GCAGGGTGTATCTAGTGTGTCATTACA, Reverse- GAGTGCTAACCACACCCACAGA. Genomic DNA was normalized to genomic β-actin and is expressed as relative DNA quantity of IP DNA to that of whole cell extract as fold enrichment for the specific promoter. As a negative control, ChIP was performed in parallel on CD4+ T cells from T-bet−/− mice with the same antibody (anti T-bet polyclonal) and on wild type and T-bet−/− mice with an isotype matched irrelevant antibody.
Supplementary Material
Acknowledgments
This work is supported by research grants from the National Multiple Sclerosis Society MS (RG2571D9 and FG1478A1) and the National Institutes of Health (NSO45937, NS35685, NS30843, A144880, AI139671, NS38037 (VKK); NSO54096 (ACA); GM20927 (CAS); and NS38037 and CA112663 (LHG). GML is a Medical Research Council (UK) Clinician Scientist. VKK is a recipient of the Javits Neuroscience investigator award from the National Institutes of Health. The authors would like to thank Rucha Chandwaskar and Jenna Sullivan for technical assistance.
Abbreviations
- CSF
cerebrospinal fluid
- IP
immunoprecipitate
- Tim-3
T cell immunoglobulin mucin domain-3
- Tim-3Ig
Tim-3 fusion protein
Footnotes
Disclosure of Conflicts of Interest
VKK works with Telos Inc. which has funded part of this work. CAS and VKK hold a patent related to Tim-3 and Tim-3L. LHG has equity in and is on the corporate board of Bristol-Myers Squibb Company and is a paid consultant for HealthCare Ventures LLC Scientific Advisory Board and Mannkind Corporation. She has equity in MannKind Corporation and has filed patents that have been licensed by them.
References
- 1.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 2.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 3.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ, Kuchroo VK. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, Gutierrez-Ramos JC, Coyle AJ, Strom TB. TIM-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 5.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, Bruce JN, Kane LP, Kuchroo VK, Hafler DA. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318:1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 6.Koguchi K, Anderson DE, Yang L, O’Connor KC, Kuchroo VK, Hafler DA. Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med. 2006;203:1413–1418. doi: 10.1084/jem.20060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-γ and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci USA. 2003;100:7749–7754. doi: 10.1073/pnas.1332767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lugo-Villarino G, Ito S, Klinman DM, Glimcher LH. The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc Natl Acad Sci USA. 2005;102:13248–13253. doi: 10.1073/pnas.0506638102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 2001;14:205–215. doi: 10.1016/s1074-7613(01)00103-0. [DOI] [PubMed] [Google Scholar]
- 10.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–494. doi: 10.1016/s1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 13.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein WM, Churakova T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 16.Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, Zheng XX, Strom TB, Hafler DA, Kuchroo VK. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39:2492–2501. doi: 10.1002/eji.200939274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson AC, Anderson DE. Tim-3 in autoimmunity. Curr Opin Immunol. 2006;18:665–669. doi: 10.1016/j.coi.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, Littman DR. Transcriptional regulatory networks in Th17 differentiation. curr Opin Immunol. 2009;21:146–152. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin-17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 21.Yang L, Anderson DE, Kuchroo J, Hafler DA. Lack of TIM-3 immunoregulation in Multiple Sclerosis. J Immunol. 2008;180:4409–4414. doi: 10.4049/jimmunol.180.7.4409. [DOI] [PubMed] [Google Scholar]
- 22.Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, Gyenes G, Vali B, Hyrcza MD, Yue FY, Kovacs C, Sassi A, Loutfy M, Halpenny R, Persad D, Spotts G, Hecht FM, Chun TW, McCune JM, Kaul R, Rini JM, Nixon DF, Ostrowski MA. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, Castelblanco N, Kuchroo V, Gretch DR, Rosen HR. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–9130. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocytes grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- 25.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 26.Imaizumi T, Kumagai M, Sasaki N, Kurotaki H, Mori F, Seki M, Nishi N, Fujimoto K, Tanji K, Shibata T, Tamo W, Matsumiya T, Yoshida H, Cui XF, Takanashi S, Hanada K, Okumura K, Yagihashi S, Wakabayashi K, Nakamura T, Hirashima M, Satoh K. Interferon-gamma stimulates the expression of galectin-9 in cultured human endothelial cells. J Leukoc Biol. 2002;72:486–491. [PubMed] [Google Scholar]
- 27.Asakura H, Kashio Y, Nakamura K, Seki M, Dai S, Shirato Y, Abedin MJ, Yoshida N, Nishi N, Imaizumi T, Saita N, Toyama Y, Takashima H, Nakamura T, Ohkawa M, Hirashima M. Selective eosinophil adhesion to fibroblast via IFN-gamma-induced galectin-9. J Immunol. 2002;15:5912–5918. doi: 10.4049/jimmunol.169.10.5912. [DOI] [PubMed] [Google Scholar]
- 28.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 29.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.