

Abstract
Activation of gene expression by FOXO transcription factors can promote neuronal death in response to loss of trophic support, or oxidative stress. The predominant neuronal FOXOs, FOXO1 and FOXO3, promote the expression of pro-death genes, such as Fas Ligand, Bim and Txnip. Neuroprotective signals initiated by neurotrophins, growth factors or synaptic activity trigger the nuclear export of FOXOs via activation of the PI3K-Akt pathway. One key aspect of FOXO regulation is that once PI3K-Akt activity has returned to baseline, FOXOs return to the nucleus to resume the activation of their target genes. Thus, the FOXO-inhibiting capacity of the PI3K-Akt pathway is thought to be short-lived. However, we show here that synaptic NMDA receptor activity not only triggers FOXO export, but also suppresses the expression of FOXO1. Blockade of PI3K activity prevents both FOXO nuclear export and suppression of FOXO1 expression, raising the possibility that FOXO1 is itself a FOXO target gene. We found that FOXO3, and to a lesser extent FOXO1 transactivates the FOXO1 promoter via a consensus FOXO binding site (GTA AAC AA), and also an upstream sequence resembling a classical FOXO-binding insulin response sequence (CAA AAC AA). Activity-dependent suppression of the FOXO1 promoter is mediated through the proximal GTAAACAA sequence. Similar suppression via this site is observed by activating neuronal IGF-1 receptors by exogenous insulin. Thus, through a feed-forward inhibition mechanism, synaptic activity triggers FOXO export resulting in suppression of FOXO1 expression. These results suggest that FOXO-inactivating signals are likely to result in longer-term inhibition of FOXO target gene expression than previously thought.
Keywords: FOXO, Forkhead box, auto-regulation, Phosphoinositide-3 kinase, Akt, synaptic activity, NMDA receptor, nuclear export
Introduction
Correct redox regulation is essential in all cells, especially in post-mitotic cells such as neurons where harmful oxidative damage can accumulate. Oxidative damage and stress occurs when there is an imbalance between production of reactive oxygen species (ROS) and the cell’s capacity to neutralize them through its intrinsic antioxidant defenses. Neurons are particularly susceptible to oxidative damage due to high levels of ROS production (through respiration and metabolism) and relatively low levels of certain antioxidant enzymes, particularly catalase.1,2 Oxidative damage is implicated in the pathogenesis of several neurodegenerative diseases as well as acute cerebrovascular disorders.1,2 We recently showed that the vulnerability of neurons to oxidative death triggered by exposure to hydrogen peroxide was regulated by synaptic activity acting via N-methyl-D-aspartate (NMDA) receptor (NMDAR) signaling.3 Neurons that were experiencing (or had recently experienced) higher levels of synaptic NMDAR activity were far more likely to withstand the oxidative insult than electrically quiet neurons. Accumulation of reactive oxygen species following an oxidative insult was significantly lower in active neurons than in inactive ones. Neurons experiencing complete NMDAR blockade were highly vulnerable to peroxide-induced apoptosis in vitro, and NMDAR blockade in vivo promoted neuronal apoptosis associated with oxidative damage.
Investigations into the mechanism behind this revealed that synaptic activity exerted a number of changes to the thioredoxin-peroxiredoxin system which contributed to the activity-dependent protection.3 Synaptic activity enhanced thioredoxin activity and facilitated the reduction of hyperoxidized peroxiredoxins, an important class of antioxidant enzymes. These changes were mediated by a coordinated program of gene expression changes. One of these changes involved the transcriptional suppression of the thioredoxin inhibitor Txnip (thioredoxin-interacting protein), a protein known to enhance oxidative stress.4,5 Studies revealed that Txnip is regulated by a class of transcription factors called Forkhead box O (FOXO). Three of the four FOXO isoforms (FOXO1, FOXO3 and FOXO4) are regulated by Akt-dependent phosphorylation, triggering their export from the nucleus.6 FOXO1 and FOXO3 are the predominant neuronal FOXOs. We found that synaptic activity turns off Txnip transcription by inducing phosphoinositide 3-kinase (PI3K), which then activates Akt, triggering FOXO phosphorylation, dissociation from the Txnip promoter and export from the nucleus.3
FOXOs have important roles in many different tissues and carry out very different functions. These roles include modulating the expression of genes involved in apoptosis, cell cycle progression, differentiation, vascularization, oxidative stress responses and energy metabolism.6-8 In the nervous system, FOXO activation can promote neuronal death following excitotoxic injury, trophic factor withdrawal and oxidative stress.6,9,10 Known pro-death FOXO target genes include BH3-only genes Noxa, Bim and Puma, Fas ligand, as well as the newly discovered Txnip. Key to all aspects of FOXO function in neurons and elsewhere is that they are subject to dynamic regulation by a variety of extracellular cues, acting through intracellular signaling pathways. They are subject to inhibitory phosphorylation by several protein kinases including Akt, SGK (serum- and glucocorticoid-inducible kinase), IKK (inhibitor of κB kinase) and CDK2 (cyclin-dependent kinase 2) and are activated by both JNK (cJun N-terminal kinase) and MST1 (mammalian sterile 20-like kinase-1) in response to stress. They are also subject to other post-translational modifications including acetylation and ubiquitination which influence their activity or preference for transactivating certain genes.11,12
Post-translational modification events, particularly phosphorylation, tend to be short-lived, persisting only as long as the activity of the upstream kinase. This idea of dynamic regulation allows for little temporal summation of FOXO-inhibiting or activating signals. However, we show here that FOXO1 is itself regulated by FOXO transcription factors acting via two cis-acting FOXO consensus sites in its promoter. Synaptic activity promotes PI3K-dependent FOXO export and subsequent transcriptional suppression of FOXO1 via one of these promoter elements. Thus, FOXO-inactivating signals can suppress expression of FOXO1, in addition to the documented post-translational inhibition. Given the importance of FOXO1 in trophic factor withdrawal-induced neuronal death,13 this study indicates that synaptic NMDAR activity, or other FOXO-inactivating signals, may induce long-lasting protection by suppressing FOXO1 expression.
Results
Synaptic NMDAR activity suppresses FOXO1 expression
We recently showed that Txnip is a pro-oxidative FOXO1 target gene which is rapidly inhibited when synaptic activity triggers the nuclear export of FOXO1 and its dissociation from the Txnip promoter.3 We wanted to determine whether synaptic activity induced any longer-term changes to FOXO1 activity beyond its acute export from the nucleus. To stimulate synaptic activity in rat cortical neurons we used the established method of network disinhibition to enhance synaptic activity, by applying the GABAA receptor antagonist bicuculline, and the K+ channel antagonist 4-aminopyridine (which enhances burst frequency, hereafter BiC/4-AP14,15). Neurons were placed in trophically deprived medium and synaptic activity initiated by BiC/4-AP treatment. We found that expression of FOXO1 was strongly suppressed: after 4 h of enhanced activity levels were already lower and this was maintained at 24 h post-stimulation (Fig. 1A). The suppression of FOXO1 transcription by synaptic activity was reduced by MK-801 co-treatment (Fig. 1A), indicating that synaptic NMDAR activity is important for the suppressing function of synaptic activity. The repression of FOXO1 mRNA expression by synaptic activity was also reflected at the protein level as assayed by western blot: 24 h post-stimulation levels of FOXO1 protein are substantially lower compared to control (Fig. 1B).
Figure 1.
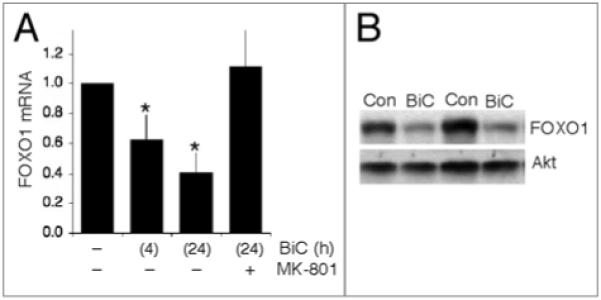
Synaptic nMdA receptor activity suppresses expression of FOXO1. (A) Qrt-PCr analysis of FOXO1 mrnA levels in neurons placed in trophically deprived medium and stimulated as indicated. Levels of FOXO1 mrnA are normalized to those of GAPdH. *p < 0.05 (AnOVA followed by Fisher’s LSd test here and throughout), n = 5. (B) Example western blot demonstrating that activity-dependent suppression of FOXO1 mrnA levels results in lower protein expression. Two sets of protein lysates are shown, taken from independent cultures, to illustrate the effect of BiC/4-AP stimulation on FOXO1 protein levels after 24 h. For comparison, Akt levels are shown to be similar.
Activity-dependent suppression of FOXO1 is PI3K-dependent
We next investigated the mechanism by which FOXO1 expression is suppressed by synaptic activity. There are relatively few transcription factors whose activity is suppressed by synaptic activity, but one of them is FOXO itself, which is subject to activity-dependent nuclear export via the PI3K-Akt pathway. Analysis of the FOXO1 promoter revealed two potential FOXO binding sites. A proximal site (GTAAACAA) at −306 nt. relative to the transcription start site which conforms to the exact FOXO-binding consensus16 and a consensus insulin-response sequence (IRS, CAAAACAA) at −483 nt. which is also known to potentially bind FOXOs.17 Both these sites are evolutionarily conserved in rodents and humans, indicative of functional importance (Fig. 2A). Before testing the significance of these promoter elements directly, we first investigated whether blockade of the PI3K-Akt pathway inhibited activity-dependent suppression of FOXO1 expression. We decided to use this initial approach because inhibition of this pathway, using the PI3K inhibitor LY294002, blocks activity-dependent export of FOXO1,18 and also FOXO3, the other major neuronal FOXO (Fig. 2B). Therefore, if LY294002 treatment did not interfere with activity-dependent suppression of FOXO1, it would argue against a role for FOXOs in FOXO1 regulation. However, pre-incubation of neurons with LY294002 completely blocked the suppression of FOXO1 expression by BiC/4-AP induced burst activity (Fig. 2C). This is very similar to the effect of LY294002 on activity-dependent suppression of Txnip expression, which we later found to be a novel FOXO target gene.
Figure 2.

Activity-induced FOXO export and suppression of FOXO1 expression is Pi3K-dependent. (A) Schematic illustration of the position and conservation of the putative FOXO-response elements within the FOXO1 promoter. Nucleotide positions refer to the rat gene. (B) Neuronal activity promotes the nuclear export of FOXO3 via Pi3K. Neurons were transfected with an expression vector encoding myc-tagged FOXO3, plus a peGFP transfection marker. The neurons were then stimulated with BiC/4-AP as indicated for 1 h in the presence or absence of the Pi3K inhibitor LY294002 (50 μM). The cells were then fixed and processed for immunofluorescence with an anti-myc antibody as described previously.30 Representative pictures are shown from the experiments performed. White arrows indicate transfected cells. (C) Qrt-PCr analysis of FOXO1 mrnA levels in neurons placed in trophically deprived medium and stimulated as indicated in the presence or absence of the Pi3K inhibitor LY294002 (50 μM). *p < 0.05 (n = 3).
FOXO1 is a FOXO target gene
We therefore decided to investigate the relevance of the putative FOXO binding sites further. We created a FOXO1-luciferase reporter construct by cloning approximately 1 kb of the FOXO1 promoter and 5′ UTR upstream of a luciferase reporter gene and created a mutant version of it with the proximal putative FOXO consensus site mutated to GTCGACAA (FOXO1(mut1)-Luc, bold underlined indicates the altered nucleotides). Neurons were transfected with FOXO1-Luc and 24 h later were treated with BiC/4-AP or left unstimulated. We found that BiC/4-AP-induced neuronal activity strongly suppressed the activity of the FOXO-Luc reporter (Fig. 3A). The effect of mutating the proximal FOXO consensus site was two-fold. Firstly, activity of the FOXO1 promoter was significantly lower, and secondly the effect of BiC/4-AP on the activity of the FOXO1 promoter was largely occluded by this mutation. We conclude from this that activity-dependent suppression of FOXO1 promoter activity is exerted by inactivating transcription driven by the proximal GTAAACAA FOXO consensus site. To determine whether the effect of synaptic activity on the FOXO1 promoter extended to other FOXO-inactivating signals we also performed the assay on neurons placed in a trophically rich medium. This medium contains insulin and activates neuronal IGF-1 receptors, promoting FOXO export.19 The effect of placing neurons in trophic medium was the same as inducing synaptic activity-strong suppression of FOXO1 promoter activity that is occluded by mutation of the FOXO consensus site.
Figure 3.

FOXO consensus sites mediate activity-dependent suppression and FOXO-mediated transactivation of the FOXO1 promoter. (A) Neurons were transfected with either FOXO1-Luc or FOXO1(mut1)-Luc, plus a pTK-renilla normalization vector. Neurons were stimulated where indicated with BiC/4-AP or placed into trophic medium containing insulin, for 24 h, after which reporter activity levels were measured (normalized to renilla levels). *p < 0.05 (n = 4). (B) Neurons were transfected with either FOXO1-Luc or FOXO1(mut1)-Luc, plus a pTK-renilla normalization vector. In addition, they were cotransfected with a vector encoding FOXO1, FOXO3, or beta-globin (control). Reporter activity levels were measured (normalized to renilla levels) at 48 h post-transfection. *p < 0.05 assessment of the effect of FOXO expression relative to appropriate control, #p < 0.05 assessment of the effect of the mutation on basal, and FOXO-induced activity of the FOXO1 promoter (n = 7). (C) Neurons were transfected with either FOXO1-Luc or mutated variants, plus a pTK-renilla normalization vector. In addition, they were cotransfected with a vector encoding either FOXO3, or beta-globin (control). Reporter activity levels were measured (normalized to renilla levels) at 48 h post-transfection. *p < 0.05 (n = 5).
We next wanted to determine whether this site is indeed a FOXO-responsive promoter element. We co-transfected neurons with either FOXO1-Luc or FOXO1(mut1)-Luc, together with vectors driving the constitutive expression of either the major neuronal FOXOs, FOXO1 and FOXO3, or a control (beta-globin). We found that the strong activity of the wild type FOXO1 promoter was further enhanced by expression of FOXO3 and, to a lesser extent FOXO1 (Fig. 3B). In contrast, expression of FOXO3 and FOXO1 increased FOXO1(mut1)-Luc activity by a smaller amount. Thus, the proximal FOXO consensus site on the FOXO1 promoter is indeed FOXO-responsive.
Interestingly, FOXO expression did still result in a small increase in activity of the FOXO1(mut1)-Luc construct, potentially suggestive of additional FOXO-responsive elements. The upstream IRS consensus sequence CAAACAA was an obvious candidate so we mutated this as well and looked at the effect of this mutation on responsiveness of the promoter to FOXO3 (since FOXO3 has the largest transactivating effect on the FOXO1 promoter). Mutation of the IRS-like element to CTAGACAA also reduced FOXO-responsiveness of the FOXO1 promoter both when introduced alone (FOXO1(mut2)-Luc) and also when combined with the first mutation (FOXO1(mut1 + 2)-Luc, Fig. 3C). Thus, both elements appear to be required for full FOXO responsiveness. Surprisingly, we found that the doubly mutated FOXO1(mut1 + 2)-Luc reporter had a higher basal activity than the singly mutated FOXO1(mut1)-Luc reporter (Fig. 3C). The reason for this is not clear, since mutation of the IRS site alone does not raise basal promoter activity (compare WT FOXO1-Luc with FOXO1(mut2)-Luc, Fig. 3C). Taken together, these data indicate that both sites are required for full activation of the FOXO1 promoter by FOXOs.
Discussion
We have shown here that FOXO1 is a FOXO target gene and as a result, its expression is subject to feed-forward inhibition by synaptic NMDAR activity, which promotes FOXO export from the nucleus. Thus, the suppressive effects of FOXO export on the expression of FOXO target genes may last considerably longer than previously thought due to the long-lasting effects of transcriptional suppression.
In the nervous system, FOXO activation is linked strongly to the promotion of neuronal death, although diversion of FOXO3 function from apoptosis to stress-resistance has been reported in the context of Sirt1-mediated deacetylation.11 In humans, certain FOXO3 haplotypes show increased risk of stroke.20 Excitotoxic injury in vivo results in activation of FOXO1 and FOXO3, induction of FOXO target gene Bim, and neuronal death.9 Conversely, FOXO export may contribute to the neuroprotective effects of estradiol in ischemic brain injury.21 FOXO3 activation promotes apoptosis of neuroblastoma cells via activation of BH3-only genes Noxa and Bim22 and death of cerebellar granule neurons at least in part by activation of Fas ligand expression.23 FOXO activation also contributes to oxidative stress-induced apoptosis in cerebellar granule neurons.10 Suppression of NMDAR activity promotes oxidative stress in part through the activation of the FOXO target gene and thioredoxin inhibitor Txnip.3 The protective effects of neurotrophic factors are thought to be mediated at least in part through promoting FOXO export and the subsequent suppression of FOXO-mediated gene expression, and the same may be true of synaptic NMDAR activity.24-26
Outside the nervous system, FOXOs have an extraordinarily diverse array of functions. For example, FOXOs, including FOXO1, have important roles in regulating blood-glucose levels by controlling hepatic gluconeogenesis and also pancreatic insulin production.27 FOXO1 exerts further metabolic control by controlling hypothalamic neuropeptide production that regulates food intake and energy homeostasis and regulating skeletal muscle atrophy under nutrient deprivation.6,28 Conditional deletion of FOXOs promotes tumorigenesis, and FOXOs have been found in chromosomal translocations in certain forms of leukemia.7 The roles of FOXOs in restricting angiogenesis, promoting apoptosis and cell cycle arrest6 may all contribute to the tumor-suppressing effects of FOXOs.7
The responsiveness of FOXO1 promoter to FOXOs may act as an important feed-forward mechanism designed to reinforce the effect of an initial environmental cue that acts to post-translationally modify the activity of FOXO proteins to initiate a particular physiological response. Naturally this would apply to FOXO-inactivating signals such as insulin, or neuronal activity, as well as activating signals such as oxidative stress. In tumors, where abnormal activation of the PI3K-Akt pathway is an important step in their initiation and maintenance, Akt-mediated FOXO export may then lead to suppression of FOXO1 expression, exacerbating the situation further.
Materials and Methods
Neuronal cultures and stimulations
Cortical rat neurons were cultured as described31 from E21 rats except that growth medium was supplemented with B27 (Invitrogen). Stimulations were done in both cases after a culturing period of 8–10 days during which cortical neurons develop a network of processes, express functional NMDA-type and AMPA/kainate-type glutamate receptors, and form synaptic contacts. Stimulations were performed after transferring neurons into defined medium lacking trophic support “TMo”:15 10% MEM (Invitrogen), 90% Salt-Glucose-Glycine (SGG) medium (ref. 32; SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM Glycine, 30 mM Glucose, 0.5 mM sodium pyruvate, 0.1% Phenol Red; osmolarity 325 mosm/l, hereafter TMo).“Trophic medium” contained added insulin–transferrin–selenite supplement (ITS; 7.5 μg insulin/ml; 7.5 μg, transferrin/ml and 7.5 ng sodium selenite/ml; Sigma, St. Louis, MO). Bursts of action potential firing were induced by treatment of neurons with 50 μM bicuculline, and burst frequency was enhanced by addition of 250 μM 4-amino pyridine.14
Plasmids
For construction of FOXO1-Luc, approximately 1,100 bp of the rat FOXO1 upstream region was amplified from genomic DNA using Picomaxx DNA Polymerase with the following amplimers: forward, 5′-ATC TCG AGT CTC TAA ACA CTC TCC TCT GAC C-3′; reverse, 5′-ATG ATA TCA ACT TAA CTT CGC TGG GTC AC-3′. The resulting PCR product was digested with XhoI and EcoRV (sites within primers italicized) and cloned into the corresponding sites upstream of the firefly luciferase construct in PGL4.10. Site-directed mutagenesis was performed with the QuikChange II XL site-directed mutagenesis kit (Stratagene), following the manufacturer’s instructions. The proximal FOXO site on FOXO1 was mutated to GTCGACAA (bold indicates nucleotides changed, underlined sequence indicates creation of SalI diagnostic site). The distal IRS-like site on FOXO1 was mutated to TCTAGACAAA (bold indicates nucleotides changed, underlined sequence indicates creation of XbaI diagnostic site). All mutants were verified by sequencing. pFOXO1myc and pFOXO-3myc33 were a gift from Domenico Accili. pTK-RL was from Promega.
Western blotting and antibodies
Total cell lysates were boiled at 100°C for 5 min in 1.5x sample buffer (1.5 M Tris pH 6.8; Glycerol 15%; SDS 3%; β-mercaptoethanol 7.5%; bromophenol blue 0.0375%). Gel electrophoresis and western blotting were performed using Xcell Surelock system (Invitrogen) using precast gradient gels (4–20%) according to the manufacturer’s instructions. The gels were blotted onto PVDF membranes, which were then blocked for 1 hour at room temperature with 5% (w/v) non-fat dried milk in TBS with 0.1% Tween 20. The membranes were then incubated at 4°C overnight with the primary antibodies diluted in blocking solution: Anti-FOXO1 (1:1,000, Cell signalling), Akt (1:500, Cell Signalling). For visualisation of western blots, HRP-based secondary antibodies were used followed by chemiluminescent detection on Kodak X-Omat film. Western blots were analysed by digitally scanning the blots, followed by densitometric analysis (ImageJ). All analysis involved normalizing to a loading control (Akt).
RNA isolation, RT-PCR and qPCR
RNA was isolated using the Qiagen RNeasy isolation reagents (including the optional Dnase treatment) following passage of the cells through a QiaShredder column. For qPCR, cDNA was synthesized from 1–3 μg RNA using the Stratascript QPCR cDNA Synthesis kit (Stratagene, Amsterdam, Netherlands) according to the manufacturer’s instructions. Briefly, the required amount of RNA (up to 3 μg) was diluted in RNase-free water (up to 7 μl final volume) and mixed on ice with 2x cDNA Synthesis master mix (10 μl), random primers: oligo-dT primers 3:1 (total 2 μl–200 ng) and either 1 μl RT/RNase block enzyme mixture (for RT reactions) or 1 μl water (for No RT control reactions). Reaction mixtures were mixed and spun down and incubated for 2 min at 25°C, 40 min at 42°C and 5 min at 95°C. cDNA was stored at −20°C.
Dilutions of this cDNA were subsequently used for real-time PCR (cDNA equivalent to 6 ng of initial RNA per 15 μl qPCR reaction for all genes except GAPDH; cDNA equivalent to 3 ng initial RNA per 15 μl reaction for GAPDH). qPCR was performed in an Mx3000P QPCR System (Stratagene) using Brilliant SYBR Green QPCR Master Mix (Stratagene) according to the manufacturer’s instructions. Briefly, the required amount of template was mixed on ice with 1x Brilliant SYBR Green Master Mix, the required concentration of forward and reverse primers, 30 nM ROX passive reference dye and water to the required reaction volume. Technical replicates as well as no template and no RT negative controls were included and at least three biological replicates were studied in each case. The sequence and concentration of the primers used are as follows: FOXO1-F: 5′ CCG ACC TCA TCA CCA AGG-3′ 200 nM, −R: 5′ TCT CCA GGA CCC TCT TGC 200 nM; GAPDH-F: 5′-AGA AGG CTG GGG CTC ACC-3′ 200 nM, −R: 5′-AGT TGG TGG TGC AGG ATG C-3′ 100 nM. The cycling programme was 10 min at 95°C; 40 cycles of 30 sec at 95°C, 40 sec at 60°C with detection of fluorescence and 30 sec at 72°C; 1 cycle (for dissociation curve) of 1 min at 95°C and 30 sec at 55°C with a ramp up to 30 sec at 95°C (ramp rate: 0.2°C/sec) with continuous detection of fluorescence on the 55–95°C ramp. The data were analysed using the MxPro QPCR analysis software (Stratagene). Expression of the gene of interest was normalized to GAPDH.
Transfection, luciferase assays and immunofluorescence
Neurons were transfected at DIV8 using Lipofectamine 2000 (Invitrogen) according to the manufacturers instruction. Firefly luciferase-based reporter gene constructs (FOXO1-Luc and mutant variants) were transfected along with a renilla expression vector (pTK-RL), and also, where relevant, other expression vectors. Neurons were stimulated (where appropriate) 24 h after transfection (for 24 h). Luciferase assays were performed using the Dual Glo assay kit (Promega) with Firefly luciferase-based reporter gene activity normalized to the Renilla control (pTK-RL plasmid) in all cases. Immunofluorescence was performed as described.34 9E10 Anti-myc antibody (Santa Cruz) was used (1:200) and visualized using biotinylated secondary antibody/cy3-conjugated streptavidin. Nuclei were counter-stained with DAPI.
Statistical analysis, equipment and settings
Statistical testing involved a 2-tailed paired student t-test. For studies employing multiple testing (e.g., the use of two pairs of siRNA, or comparisons between multiple deletion/mutant luciferase reporters constructs) we used a one-way ANOVA followed by Fisher’s LSD post-hoc test. For western blots we used chemiluminescent detection on Kodak X-Omat film. Appropriate exposures were taken such that bands were not saturated. For figure preparation of example western blots, linear adjustment of brightness/contrast was applied (Photoshop) equally across the entire image, taking care to maintain some back-ground intensity. Pictures of transfected neurons were taken on a Leica AF6000 LX imaging system, with a DFC350 FX digital camera. The DFC350 FX digital camera is a monochrome camera, and so coloured images (e.g., of green fluorescent protein) essentially involve taking a black and white image (using the appropriate filter set) and applying a colour to the image after capture. All luminescent assays were performed on a FLUOstar OPTIMA (BMG Labtech, Aylesbury, UK). Light collection time and gain were set such that counts were substantially lower than the maximum level collectible.
Concluding Comments
While this manuscript was in preparation a study on FOXO regulation in fibroblasts also came to the conclusion that FOXO1 is a FOXO target gene.29 This study reported that FOXO3 was a better activator of the FOXO1 promoter than FOXO1 itself, similar to our observations (Fig. 3B). They identified the same proximal FOXO consensus site (GTA AAC AA) as mediating a large portion of the observed FOXO responsiveness, and which was responsible for growth factor-induced transcriptional suppression. Interestingly, they found that mutation of this proximal site reduced, but did not abolish, activation of the promoter by overexpression of FOXO3. This raises the possibility that the IRS-like sequence identified in this study may be mediating the remaining induction. It is therefore likely that FOXO-inactivating signals are capable of suppressing FOXO1 expression in a variety, if not all, cell types.
Acknowledgements
We thank Domenico Accili for the FOXO expression vectors, and Sofia Papadia for comments on the manuscript. This work was funded by the Wellcome Trust, a Royal Society University Research Fellowship (Giles Hardingham) and the BBSRC.
Footnotes
Addendum to: Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 2008; 11:476–87; PMID: 18344994; DOI: 10.1038/nn2071.
References
- 1.Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–58. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 3.Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–87. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–74. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida T, Nakamura H, Masutani H, Yodoi J. The involvement of thioredoxin and thioredoxin binding protein-2 on cellular proliferation and aging process. Ann N Y Acad Sci. 2005;1055:1–12. doi: 10.1196/annals.1323.002. [DOI] [PubMed] [Google Scholar]
- 6.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–36. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18:421–9. doi: 10.1016/j.tcb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Maiese K, Chong ZZ, Shang YC. OutFOXOing disease and disability: the therapeutic potential of targeting FoxO proteins. Trends in molecular medicine. 2008;14:219–27. doi: 10.1016/j.molmed.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, et al. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004;113:1059–68. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 11.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 12.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–87. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 13.Yuan Z, Lehtinen MK, Merlo P, Villen J, Gygi S, Bonni A. Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem. 2009;284:11285–92. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–7. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 15.Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–87. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629–34. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–92. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 18.Martel MA, Soriano FX, Baxter P, Rickman C, Duncan R, Wyllie DJ, Hardingham GE. Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels (Austin), Tex. 2009;3:12–5. doi: 10.4161/chan.3.1.7864. [DOI] [PubMed] [Google Scholar]
- 19.Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–18. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuningas M, Magi R, Westendorp RG, Slagboom PE, Remm M, van Heemst D. Haplotypes in the human Foxo1a and Foxo3a genes; impact on disease and mortality at old age. Eur J Hum Genet. 2007;15:294–301. doi: 10.1038/sj.ejhg.5201766. [DOI] [PubMed] [Google Scholar]
- 21.Won CK, Ji HH, Koh PO. Estradiol prevents the focal cerebral ischemic injury-induced decrease of forkhead transcription factors phosphorylation. Neurosci Lett. 2006;398:39–43. doi: 10.1016/j.neulet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 22.Obexer P, Geiger K, Ambros PF, Meister B, Ausserlechner MJ. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007;14:534–47. doi: 10.1038/sj.cdd.4402017. [DOI] [PubMed] [Google Scholar]
- 23.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 24.Gan L, Zheng W, Chabot JG, Unterman TG, Quirion R. Nuclear/cytoplasmic shuttling of the transcription factor FoxO1 is regulated by neurotrophic factors. J Neurochem. 2005;93:1209–19. doi: 10.1111/j.1471-4159.2005.03108.x. [DOI] [PubMed] [Google Scholar]
- 25.Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–8. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiese K, Chong ZZ, Shang YC. “Sly as a FOXO”: new paths with Forkhead signaling in the brain. Current neurovascular research. 2007;4:295–302. doi: 10.2174/156720207782446306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Current diabetes reports. 2009;9:208–14. doi: 10.1007/s11892-009-0034-5. [DOI] [PubMed] [Google Scholar]
- 28.Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–6. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- 29.Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284:10334–42. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mckenzie GJ, Stephenson P, Ward G, Papadia S, Bading H, Chawla S, et al. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J Neurochem. 2005;93:171–85. doi: 10.1111/j.1471-4159.2005.03010.x. [DOI] [PubMed] [Google Scholar]
- 31.Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–4. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 32.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–6. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 33.Nakae J, Barr V, Accili D. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000;19:989–96. doi: 10.1093/emboj/19.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKenzie G, Ward G, Stallwood Y, Briend E, Papadia S, Lennard A, et al. Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals. BMC cell biology. 2006;7:10. doi: 10.1186/1471-2121-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]