

Abstract
Cholesterol 24-hydroxylase is a highly conserved cytochrome P450 that is responsible for the majority of cholesterol turnover in the vertebrate central nervous system. The enzyme is expressed in neurons, including hippocampal and cortical neurons that are important for learning and memory formation. Disruption of the cholesterol 24-hydroxylase gene in the mouse reduces both cholesterol turnover and synthesis in the brain but does not alter steady-state levels of cholesterol in the tissue. The decline in synthesis reduces the flow of metabolites through the cholesterol biosynthetic pathway, of which one, geranylgeraniol diphosphate, is required for learning in the whole animal and for synaptic plasticity in vitro. This review focuses on how the link between cholesterol metabolism and higher-order brain function was experimentally established.
Keywords: geranylgeraniol, learning, long-term potentiation, P450, statin, 24S-hydroxycholesterol
Introduction
The brain is a famously metabolically active tissue. Despite occupying only 2% of body mass in humans, the tissue accounts for 20% of resting oxygen consumption, receives 15% of cardiac output, and burns an estimated 4 × 1021 molecules of ATP every minute of the day (1). This massive consumption, which is fueled by glucose metabolism and used almost entirely to maintain the electrical activity of the roughly 100 billion individual but interconnected neurons of the brain, is unchanging.
An estimated half of the dry weight of the human brain is composed of lipids (2), including ∼20% of the body's total cholesterol content. A majority of this cholesterol accumulates in the brain during a brief period of development in which neuronal axons are encircled by specialized plasma membranes, termed myelin, that are elaborated by oligodendroglial cells. For many years, it was thought that once myelination was complete, there was little further metabolism of cholesterol in the brain; however, this picture of stasis has changed over the past decade, and we now know that a small subset of neurons actively synthesizes and breaks down cholesterol and that alterations in this metabolism have profound effects on higher-order brain function.
Here, we summarize current knowledge concerning cholesterol synthesis and turnover in the brain and focus on the role of cholesterol 24-hydroxylase, an enzyme that drives cholesterol metabolism in the central nervous system. Broader perspectives on cholesterol metabolism in the brain are presented in reviews by Dietschy & Turley (3, 4) and Vance et al. (5).
Cholesterol in the Brain
Early metabolic studies identified cholesterol in the brain and suggested that the sterol was both abundant and ubiquitously distributed throughout the tissue. The subsequent development of radioactive tracers and advances in analytical chemistry allowed rates of cholesterol synthesis and accumulation to be measured and provided the first evidence for a brain-specific cholesterol turnover pathway.
Cholesterol Levels in Tissues
Cholesterol is a major lipid component of mammalian cell membranes, and in general, tissues contain an amount of cholesterol that is proportional to their total membrane surface area. The average tissue concentration of cholesterol in experimental organisms such as the mouse ranges between 2 and 3 mg per gram wet weight; however, some tissues with specialized functions such as the brain, adrenal gland, and lung exceed this average (6). Of these, the brain has the highest cholesterol content at ∼15 mg/g tissue, followed by the adrenal gland at ∼9 mg/g, and the lung at ∼6 mg/g. The cholesterol content of the adrenal gland is high because this tissue stores large amounts of cholesteryl esters for use in the synthesis of steroid hormones. Similarly, the elevated cholesterol content of the lung arises from the extensive gas-exchanging surface area of the aveoli and the associated lipid-rich surfactant.
Two Pools of Cholesterol in the Brain
The cholesterol that is present in the brain exists in two pools. One pool is large, accounting for ∼70% of the total (7), metabolically stable (8), and found in the myelin membranes of white matter. The concentration of cholesterol in this pool is high (∼40 mg/g tissue) and reflects the dense packing of multiple opposed lipid bilayers in the myelin sheath. The second pool of cholesterol is smaller, representing ∼30% of total, and is found in the plasma and subcellular membranes of neurons and glial cells of gray matter. The concentration of cholesterol is lower in gray matter (∼8 mg/g), but as is discussed below, this pool is metabolically active.
Origin of Brain Cholesterol
Two sources of cholesterol are available to tissues, including an exogenous source in the form of plasma lipoproteins and an endogenous source in the form of a biosynthetic pathway of more than 20 enzymes that convert acetate into cholesterol (Figure 1a). The choice between exogenous versus endogenous cholesterol supply pathways is determined in part by the organization and extent of vascularization in individual tissues. Thus, highly vascularized tissues, such as the adrenal gland and liver, have ready access to circulating lipoprotein particles and preferentially utilize this source. The brain, although estimated to contain ∼400 miles of blood vessels (9), is an exception to this generalization and relies exclusively on the synthesis of cholesterol from acetate to meet cholesterol needs (10–13). This reliance arises as a consequence of the blood-brain barrier, which is composed of tightly opposed endothelial cells that line the extensive vasculature of the tissue and prevent access to lipoprotein particles in the plasma (14).
Figure 1.

Cholesterol metabolism in the brain. (a) An abbreviated cholesterol biosynthetic pathway is shown in which key intermediates and enzymes are highlighted. Statins reduce the flow of intermediates through the biosynthetic pathway by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase. Farnesyl diphosphate (farnesyl-PP) and geranylgeranyl diphosphate are polyisoprenoid end products of the pathway. Knockout of the cholesterol 24-hydroxylase gene causes cholesterol accumulation and activation of a negative feedback pathway that suppresses the reductase enzyme as well as others in the pathway and subsequently decreases cholesterol synthesis. Redrawn from Reference 79. (b) Cholesterol synthesis and accumulation in mouse brain as a function of age of the animal.
Experimental Methods to Study Cholesterol Metabolism in the Brain
Early insight into brain cholesterol metabolism came from measurements of steady-state levels of the sterol. These studies were done using laborious chemical methods that involved either precipitation of cholesterol from complex lipid mixtures with digitonin (15), or repeated extraction and crystallization of the molecule from organic solvents (16), followed by gravimetric or colorimetric determination of the cholesterol present. The availability of isotopically labeled precursors allowed rates of cholesterol synthesis to be estimated (16–18); however, the accuracy of these measurements was uncertain as the calculations assumed that labeled precursors entered different cell types at equal rates and that the specific activity achieved in all cells was identical. Andersen & Dietschy (19) developed methods using 3H2O as a precursor that overcame these concerns and, in so doing, ushered in the current era of research in brain cholesterol metabolism. With this method, they obtained quantitative data on rates of cholesterol synthesis in the brains of experimental organisms ranging from rodents to primates (4).
A typical cholesterol synthesis experiment in the mouse involves injection of 20–50 mCi of 3H2O into the peritoneal cavity of individual animals, followed by a one-hour incubation to allow equilibration and incorporation of the radioisotope into newly synthesized cholesterol. Tissues of interest are dissected and dissolved in alcoholic potassium hydroxide, and the lipids are extracted into petroleum ether. Sterols are precipitated from the crude mixture with digitonin, collected by centrifugation, and are separated from the precipitant by treatment with pyridine. Thereafter, the radioactivity present is determined by liquid scintillation counting, and the mass of newly synthesized cholesterol is calculated using several experimentally determined constants (19).
Cholesterol Metabolism in the Mouse Brain
Using the Dietschy method, rates of cholesterol synthesis and accumulation as a function of age in the mouse brain were measured (20). As summarized in Figure 1b, synthesis and accumulation are low in newborns but begin to increase shortly after birth. Cholesterol accumulation peaks at a rate of ∼0.26 mg/day two to three weeks after birth and then gradually decreases to ∼0.012 mg/day in adult mice. Changes in synthesis track these trends, peaking at ∼0.28 mg/day in weeks two to three of postnatal life and then decreasing to ∼0.035 mg/day as the animal ages. The fact that the rate of cholesterol synthesis in the adult brain is larger than the accumulation rate (Figure 1b) indicates that there must be a mechanism by which cholesterol is turned over.
24S-Hydroxycholesterol and Cholesterol Turnover
Clues to what this turnover mechanism might be appeared early in the literature with the identification of a dihydroxylated sterol in horse brain (21), which was shown to be cholest-5-ene-3β,24-diol or 24-hydroxycholesterol (22). This oxysterol was subsequently detected in the human (23–26), bovine (26, 27), rabbit (26), and rat brain (28) at levels of tens of micrograms per gram of tissue and was shown to consist of a single epimer, 24S-hydroxycholesterol (21). Smith and colleagues in Galveston, Texas, showed that microsomes from bovine brain (26) and rat brain (28) converted cholesterol into 24S-hydroxycholesterol in a reaction requiring NADPH, and they speculated that the enzyme involved was a cytochrome P450 (P450). The latter findings were of crucial importance as they suggested that 24S-hydroxycholesterol was an enzymatically formed metabolite of cholesterol and not a product of chemical oxidation arising from the extensive extraction procedures required to resolve the rare oxysterol from the thousands-fold more abundant cholesterol precursor.
Measurement and Metabolism
Smith's studies on 24S-hydroxycholesterol in the brain were published in the early 1970s, but interest in this metabolite languished in the ensuing two decades. The field was rejuvenated by Björkhem and colleagues working in Huddinge, Sweden, who in the 1990s developed sensitive isotope dilution gas chromatography-mass spectrometry assays to quantitate oxysterols in plasma (29). Using these assays, they documented the in vivo formation of 24S-hydroxycholesterol in rats (30–32), humans (31, 33, 34), mice (35), and guinea pigs (36). The contribution of the brain to levels of circulating 24S-hydroxycholesterol varies between species (35), but in all organisms examined, the brain is a significant source of this oxysterol in plasma. In humans, plasma levels are high in neonates and then decline by a factor of five in subjects age 10 years and above (31). In mice, serum levels of 24S-hydroxycholesterol peak at ∼75 ng/ml on postnatal day 15 and then decline to ∼20 ng/ml after postnatal day 30 (37).
Rates of 24S-hydroxycholesterol production in humans can be measured by determining arteriovenous differences in the amount of the oxysterol and are found to be ∼6 mg per day (0.09 mg/day/kg body weight) (31, 34). Analysis of adult rats, maintained in an 18O2-containing atmosphere and in which the isotopically labeled oxygen atom can be incorporated into the hydroxyl groups of cholesterol and oxysterols, suggests that ∼0.02% of the pool of cholesterol in the brain is converted into 24S-hydroxycholesterol each hour (32). This rate is approximately half of that estimated for the synthesis of cholesterol, which indicates that the 24S-hydroxycholesterol pathway accounts for ∼50% of cholesterol turnover in the rat. By following cholesterol synthesis in the adult mouse brain, Dietschy and colleagues (20) determined a cholesterol synthetic rate of ∼2.2 mg/day/kg body weight and a cholesterol accretion rate of ∼0.8 mg/day/kg body weight. Subtracting these two values indicates that the 24S-hydroxycholesterol pathway may turn over as much as ∼1.4 mg cholesterol/day/kg body weight in the mouse.
Tracer experiments in humans with deuterated 24S-hydroxycholesterol indicate that the clearance rate of the oxysterol by the liver is ∼7.6 mg/day (34), which is roughly equal to the production rate measured in the brain (∼6 mg/day). The half-life of a racemic mixture of 24-hydroxycholesterol is ∼12 hours on the basis of measurements in two adult subjects. Additional studies compare steady-state levels of the oxysterol in plasma to estimated sizes of the liver and brain and show that the rate of clearance is inversely proportional to body surface area (38). Of the 24S-hydroxycholesterol taken up by the liver, approximately half is converted into primary bile acids that are secreted into bile and half is excreted as bile acid intermediates (39). A similar conversion of the oxysterol into undefined metabolites in bile was reported in the mouse (40). Small amounts of the 3-sulfate and 3-glucuronide esters of 24S-hydroxycholesterol are detected in neonatal feces (41), urine (42), and plasma (43), suggesting that sulfation and glucuronidation may represent additional pathways of 24S-hydroxycholesterol catabolism. Subsequently, the levels of these metabolites were shown to be increased in subjects with liver failure (44), which indicates that their formation may represent responses to injury rather than normal degradative pathways for the oxysterol.
Proposed Role in Cholesterol Turnover
Consideration of the above chemical and metabolic data led Björkhem and coworkers (33) to propose that formation of 24S-hydroxycholesterol by the brain represents a novel pathway of cholesterol turnover. In this pathway, the oxysterol is initially formed within the brain and then gains access to the circulation by spontaneous diffusion across cellular membranes and the blood-brain barrier (45). In plasma, 24S-hydroxycholesterol associates with lipoprotein particles, chiefly low-density and high-density lipoproteins (46), and then is cleared in a single pass by the liver (34). Once in the liver, the oxysterol is 7α-hydroxylated (47) and then further metabolized into bile acids and other intermediates before excretion into the bile. This pathway is summarized in Figure 2.
Figure 2.

Cholesterol turnover in the brain. Cholesterol is converted into 24S-hydroxycholesterol by cholesterol 24-hydroxylase and spontaneously diffuses into the circulation. The oxysterol associates with high-density and low-density lipoproteins, which are cleared by the liver. The 24S-hydroxycholesterol is metabolized into bile acids and excreted into the bile.
Cholesterol 24-Hydroxylase
24S-hydroxycholesterol is synthesized by cholesterol 24-hydroxylase, a cytochrome P450 of ∼500 amino acids that is embedded in the membranes of the smooth endoplasmic reticulum of neurons. Mutation of the cholesterol 24-hydroxylase gene (Cyp46a1) in the mouse disrupts cholesterol synthesis and turnover in the brain.
Isolation of cDNAs
Complementary DNAs encoding the mouse cholesterol 24-hydroxylase were isolated in a generalized expression screen for oxysterol biosynthetic enzymes (37). In the cloning experiment, pools containing ∼4000 individual cDNAs from a liver library were introduced into cultured human embryonic kidney 293 cells, and the ability of the transfected cells to convert radiolabeled cholesterol into oxysterol products was determined by thin-layer chromatography. The screen yielded cDNAs encoding multiple sterol hydroxylases, including sterol 12α-hydroxylase, cholesterol 25-hydroxylase (48), sterol 27-hydroxylase, and in a single pool out of 250 screened, a full-length cDNA specifying cholesterol 24-hydroxylase. This mouse cDNA was then used to isolate the human ortholog from a fetal brain cDNA library by cross-hybridization.
Catalytic Activity, Sequence Conservation, and Structure
Analyses of the oxysterols produced by cells transfected with the mouse and human cholesterol 24-hydroxylase cDNAs show that these enzymes catalyze the reaction shown in Figure 3a. In addition, smaller amounts of 25-hydroxycholesterol are synthesized at approximately one-fourth the rate of 24Shydroxycholesterol formation (37). Further studies in transfected human embryonic kidney 293 cells and with a partially purified recombinant enzyme from Escherichia coli show that cholesterol 24-hydroxylase catalyzes several different reactions (49, 50), including the formation of 24,25- and 24,27-dihydroxycholesterols, hydroxylation of other sterols and steroids on both the rings and side chains of the molecules, and the hydroxylation of drugs such as bufuranol and diclofenac. The catalytic properties of the purified full-length enzyme as well as several modified forms with N-terminal and C-terminal truncations are known (51). The Km for cholesterol is in the low micromolar range, and kcat/Km values are ∼0.002 min−1 μM−1. These in vitro biochemical studies reveal that cholesterol 24-hydroxylase is capable of utilizing a range of substrates, and they suggest that the enzyme may, in addition to cholesterol turnover, fulfill other metabolic roles in the brain.
Figure 3.
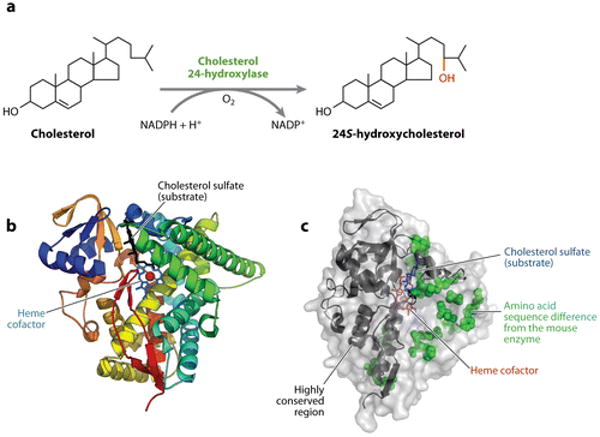
Cholesterol 24-hydroxylase. (a) The reaction catalyzed by cholesterol 24-hydroxylase. (b) The structure of the human enzyme as determined by X-ray crystallography. Twelve α-helices and four β-pleated sheets are shown in various colors together with the heme cofactor and a cholesterol sulfate substrate. Redrawn and reprinted with permission from Reference 53. (c) Surface representation of the human enzyme. Twenty amino acid sequence differences with the mouse enzyme, which are concentrated on one side of the protein, are shown as space-filling representations. Regions of the protein at the N and C termini that are more highly conserved are indicated by ribbon diagrams. Data extracted from Protein Data Bank entry 2q9f.
Conceptual translations of the protein sequences predicted from the cloned mouse and human cDNAs reveal P450s with several unique features. First, the two proteins share less than 35% sequence identity with other members of the P450 super family and thus represent a new subfamily (CYP46A1) of these ubiquitously distributed enzymes. Second, whereas most human and mouse P450 orthologs are ∼70% identical in sequence, the cholesterol 24-hydroxylases from these two species are 95% identical (37). This conservation extends across the vertebrate kingdom and underscores the potential physiological importance of the enzyme and of cholesterol turnover in the brain. Third, unlike all other P450s, the sequence ProAlaProProProProPro-Cys is found at the extreme C termini of the mouse and human proteins. This sequence, or a closely related variant, is also present in the chimpanzee, rhesus, cow, dog, horse, and rat cholesterol 24-hydroxylases but is absent from the frog, chicken, zebra fish, and platypus enzymes. Polyproline motifs of this type are often binding sites for interacting proteins (52), and the conserved presence of this unusual sequence in cholesterol 24-hydroxylase suggests that protein-protein interactions may play an important role in the function of this unusual P450.
The three-dimensional structure of amino acids 59–491 of the human cholesterol 24-hydroxylase in ligand-free and ligand-bound states was reported in 2008 by Pikuleva and colleagues (53, 54). The structures, determined to 2.4-Å (ligand-free) and 1.9-Å (ligand-bound) resolution by X-ray crystallography, reveal a typical mammalian P450 fold (55), in which the primary sequence is organized into 12α-helices and four β-pleated sheets (Figure 3b). The heme prosthetic group is positioned between two α-helices and is liganded to cysteine 437 of the protein. An active site with a small volume (∼300 Å3) is present in which a high-affinity substrate (cholesterol 3-sulfate) is bound via a combination of hydrogen bonding and hydrophobic packing with amino acids from multiple α-helices. Comparisons between the active sites of cholesterol 24-hydroxylase and the CYP2R1 vitamin D 25-hydroxylase (56, 57) predict that the sterol and secosterol substrates enter the active site of their respective hydroxylases by different channels and, once bound, have different orientations with respect to the heme cofactor (53). When amino acid sequence differences between mouse and human cholesterol 24-hydroxylases are superimposed on a surface representation of the protein's structure, most (20/22) of the differences found in the structure are grouped together whereas the N and C termini remain relatively substitution free (Figure 3c). Whether this skewed distribution affects the functions of the two orthologs is not known.
Tissue Distribution and Gene Expression
RNA and protein blotting experiments show that unlike most P450s, which are preferentially expressed in tissues active in xenobiotic and endobiotic metabolism such as the liver, kidney, and lung, cholesterol 24-hydroxylase expression is largely confined to the brain in the mouse and human (37). In the case of the mouse, small amounts of cholesterol 24-hydroxylase mRNA are detected in the testis and liver, but the mRNA does not appear to be translated into protein in the testis, and levels of the mRNA and protein are estimated to be ∼100-fold lower in the liver than in the brain. More extensive tissue surveys using real-time polymerase chain reactions (PCRs) confirm the preferential expression of the mRNA in human brain (58). There is no evidence to date that cholesterol 24-hydroxylase is expressed in the peripheral nervous system.
Ontogeny studies reveal expression of the gene in the mouse embryo as early as day 11.5 and a gradual increase in mRNA and protein in both the mouse and human postnatal brain (37, 59, 60). The regulatory sequences in the gene that underlie these tissue-selective and developmental expression patterns are under investigation (60, 61).
Cell-Type-Specific Expression Pattern
Given the proposed role of cholesterol 24-hydroxylase in the brain (Figure 2) and the high concentrations of cholesterol in white matter, the expectation is that the enzyme should be present in either oligodendrocytes that produce myelin or in support cells that are postulated to modulate intercellular cholesterol transfer (4). Despite the logic of these predictions, initial in situ mRNA hybridization and immunohistochemical experiments indicated that the cholesterol 24-hydroxylase mRNA and protein are present in neurons and not support cells (37). Expressing cells included large pyramidal neurons in various layers of the cortex and in the hippocampus as well as Purkinje cells in the cerebellum. The concordance between mRNA hybridization and immunohistochemical signals was high in these studies, which supported the surmised neuronal expression pattern.
The early immunohistochemical experiments were performed with an affinity-purified polyclonal antiserum raised in rabbits against a 17-amino acid peptide (residues 254–270 of the mouse enzyme) (37). Subsequent studies with this antiserum revealed that, under most conditions, the antibody detects two proteins in immunoblots of brain lysates, including the cholesterol 24-hydroxylase of ∼53 kDa and an unknown protein of ∼59 kDa. It is not known whether both of these antigens or others are detected by the antiserum in immunohistochemical experiments, and for this reason, it is important to include appropriate positive and negative controls to ensure proper interpretation of results obtained with this polyclonal antiserum. The issue of antibody specificity was subsequently overcome when monoclonal antibodies were produced that uniquely recognize cholesterol 24-hydroxylase from multiple species (62). The hybridoma cell lines, designated 1A7, 1F11, and 1F4, stably secrete monoclonal antibodies of different immunoglobulin G subtypes that are highly selective for cholesterol 24-hydroxylase. When these monoclonal antibodies are used in immunohistochemical and immunocytochemical experiments, two results are obtained: A single 53-kDa protein is detected on immunoblots, and strong staining is present in neurons.
An example of the neuron-specific expression of cholesterol 24-hydroxylase in mouse brain as determined by immunostaining with one of the monoclonal antibodies is shown in Figure 4a, which depicts a section from the cortex. Large pyramidal cells and their dendrites in cortical layers II, III, V, and VI are stained, whereas the nonpyramidal cells that compose layer IV do not contain signal, nor is staining detected in a cortical section from a cholesterol 24-hydroxylase knockout mouse (Figure 4b). Additional immunohistochemical experiments document the neuron-specific expression pattern of the enzyme in other subregions of the brain (62, 63), and cyclopean in situ mRNA hybridization studies by the Allen Institute for Brain Science provide a complete picture of the neuronal expression pattern of the mouse gene (http://www.brain-map.org/).
Figure 4.
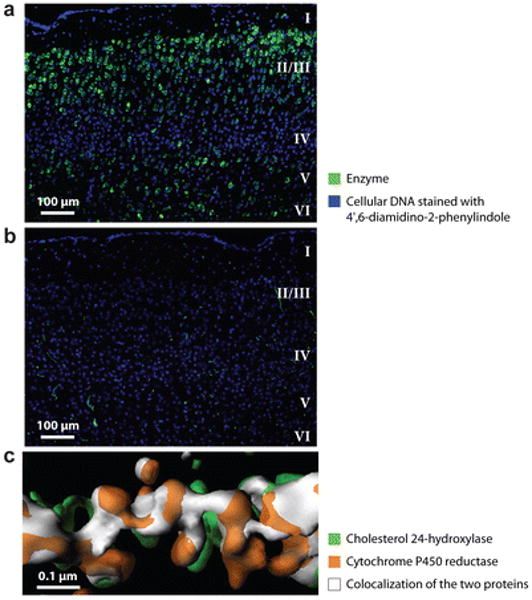
Expression of cholesterol 24-hydroxylase in the brain. (a) Wild-type mouse cortical section stained with monoclonal antibody 1A7 showing expression of the enzyme (green stain) in pyramidal neurons of layers II/III, V, and VI. (b) Section from cortex of cholesterol 24-hydroxylase knockout mouse stained with monoclonal antibody 1A7. (c) Three-dimensional reconstruction of cholesterol 24-hydroxylase staining and cytochrome P450 reductase staining showing colocalization of the two proteins in the dendritic endoplasmic reticulum of a cultured primary cortical neuron. Reprinted with permission from Reference 62.
A limitation that impeded further insight into the cell biology of cholesterol 24-hydroxylase was the dearth of readily cultured cell lines that express the enzyme. Surveys of immortalized cells have not revealed expressing cell types, with the exception of the human neuroblastoma cell line SH-SY5Y, which contains cholesterol 24-hydroxylase mRNA but does not synthesize 24S-hydroxycholesterol (60). This limitation can be overcome when expression of the enzyme is monitored in primary cortical and hippocampal neurons isolated from day 16 mouse embryos. Over a 14-day culture period, during which the neurons extend processes and form increasing numbers of synapses but do not divide, there is a gradual increase in cholesterol 24-hydroxylase mRNA and protein as judged by real-time PCR and immunoblotting. Quantification of 24S-hydroxycholesterol levels, using a sensitive liquid chromatographymass spectrometry assay (64), indicates that the oxysterol is secreted into the medium at a rate of ~80 pg/hour/μg cellular DNA and accumulates within these neurons at a rate of ~56 pg/hour/μg cellular DNA.
Immunocytochemical experiments with monoclonal antibody probes reveal that cholesterol 24-hydroxylase is located in the endoplasmic reticulum of primary neurons (62). Morphological studies show that this subcellular compartment extends throughout the cell body and into the axonal and dendritic processes of the neuron (65), and in agreement with these findings, cholesterol 24-hydroxylase is detected in the soma and dendrites. Axonal and presynaptic expression of the enzyme is not detected, presumably owing to the small amounts of endoplasmic reticulum present in these compartments. A three-dimensional reconstruction of the cholesterol 24-hydroxylase staining pattern in a dendrite together with that of a marker protein of the endoplasmic reticulum (P450 reductase) is shown in Figure 4c. Extensive colocalization is observed (white shapes) between the two proteins, and the multilobular organization of this subcellular compartment is evident.
Additional studies in primary neurons transfected with an expression vector for green fluorescent protein show that cholesterol 24-hydroxylase is also present in the endoplasmic reticulum of some dendritic spines (D.M.O. Ramirez, unpublished observations). These protuberances are found on the cell bodies, dendrites, and axonal hillocks of many different types of neurons in the vertebrate central nervous system, and a majority (>90%) of all excitatory synapses are estimated to terminate on spines (66).
Cholesterol 24-Hydroxylase Knockout Mice
Mice lacking cholesterol 24-hydroxylase were generated using standard gene targeting methods in embryonic stem cells (59). The introduced mutation eliminates expression of the cholesterol 24-hydroxylase gene by replacing exon 1 with a heterologous DNA, containing sequences from the bovine microtubule binding protein tau linked to those of E. coli β-galactosidase. Positioning the heterologous gene in exon 1 places the DNA under the control of regulatory sequences in the cholesterol 24-hydroxylase gene, thus allowing the identification of expressing cells by histochemical staining for β-galactosidase (67). In agreement with the cell-type-selective expression pattern determined for cholesterol 24-hydroxylase in adult animals, β-galactosidase staining is limited to neurons of the developing brain and is first detected in gestational day 11.5 embryos (59). These data suggest that the capacity to turn over cholesterol via the cholesterol 24-hydroxylase pathway may be acquired early during development of the central nervous system.
The knockin mutation eliminates expression of the cholesterol 24-hydroxylase mRNA and protein (59). Concentrations of 24S-hydroxycholesterol in the plasma are reduced by ∼80% in two-week-old mutant mice and by ∼60% in adult animals. In the brains of young and old mice, levels of the oxysterol are reduced to the limit of detection by gas chromatography-mass spectrometry. Although the biosynthetic origin of the residual 24S-hydroxycholesterol in the plasma of the mutant mice is not known, it is possible that this sterol arises from the sterol 27-hydroxylase, which in some (68), but not all (69), species synthesizes a variety of side-chain hydroxylated oxysterols.
Mice heterozygous and homozygous for the introduced mutation are outwardly normal (59). Parameters ranging from fertility to food consumption to body weight to circadian rhythm are unchanged in adult males and females. Although cholesterol 24-hydroxylase is expressed in the central nervous system during development, animals without the enzyme do not show gross developmental defects. The mutation was initially constructed on a mixed genetic background of the C57BL/6J and 129S6/SvEv inbred strains of mice and was later backcrossed onto an isogenic C57BL/6J background. Genetic background does not appear to have a large effect on the phenotype of the knockout animals.
Metabolic Consequences Arising from Loss of Cholesterol 24-Hydroxylase
Balance studies in mice lacking cholesterol 24-hydroxylase show that whole-body fatty acid and cholesterol metabolism are normal (59). Lipid synthetic rates and content in nonbrain tissues are unchanged as are bile acid pool size, intestinal cholesterol absorption, fecal sterol excretion, and plasma lipoprotein profiles. These results suggest that cholesterol 24-hydroxylase contributes little to lipid metabolism in peripheral tissues, and they indicate that a >60% reduction in plasma 24S-hydroxycholesterol levels also does not affect lipid synthesis and catabolism.
This metabolic picture is different in the knockout brain (6, 59). Although the total amount of cholesterol in the tissue remains constant at 330 mg sterol per kg body weight, de novo cholesterol synthesis is reduced by ∼40% from 1.4 mg/day/kg to 0.8 mg/day/kg (Figure 5). When synthesis is measured in subregions of the brain, such as the cortex and hippocampus, that normally express high levels of cholesterol 24-hydroxylase, rates are decreased by as much as 60%. Conversely, subregions that normally express low levels of the enzyme, such as the myelin-rich spinal cord, exhibit normal cholesterol synthetic rates. Cholesterol synthesis in the whole brain and in various subregions is reduced by 20% to 30% in mice heterozygous for the induced mutation (R.W. Halford, unpublished observations).
Figure 5.

Cholesterol synthesis and turnover in the wild-type and cholesterol 24-hydroxylase knockout mouse brains.
Several conclusions can be drawn from these studies regarding the role of cholesterol 24-hydroxylase in the brain. First, the finding that brain cholesterol levels are the same in wild-type and knockout mice agrees with studies on the regulation of the cholesterol biosynthetic pathway in which accumulation of the sterol is known to suppress further synthesis (70). Presumably, in the knockout mice, elimination of a major catabolic pathway causes cholesterol to accumulate transiently in neurons, which in turn activates the feedback pathway in these cells leading to a decrease in synthesis and the maintenance of steady-state cholesterol levels. Second, loss of cholesterol 24-hydroxylase reduces the excretion of sterols from the brain but does not abolish this removal process, thus there must be other pathways by which cholesterol is eliminated from the tissue. Third, given the mass of cholesterol 24S-hydroxycholesterol produced (∼0.6 mg/day/kg) and the facts that some but not all neurons express the enzyme (see Cell-Type-Specific Expression Pattern above) and that neurons represent ∼10% of cells in the brain (71), one can calculate that a small number of cells in the tissue are turning over cholesterol at a rate approaching that of hepatocytes in the liver (4).
Cholesterol Turnover and Learning
Apolipoprotein E (ApoE) plays a major role in cholesterol metabolism in peripheral tissues and, on the basis of expression in various cell types within the brain, is postulated to do the same in the central nervous system (72). The latter role can be demonstrated in vitro (73), but balance studies in ApoE knockout mice do not reveal alterations in brain cholesterol synthesis or accumulation in the adult animal (20), perhaps owing to the redundant functions of other lipoproteins (3). The ApoE mutant mice do exhibit deficiencies in spatial learning (74–76) and in long-term potentiation (LTP) (77), an in vitro correlate of the changes in synaptic activity that are thought to accompany learning and memory formation (78). On the basis of these observations regarding ApoE, experiments to test learning were carried out on cholesterol 24-hydroxylase knockout mice.
Behavioral Learning Deficiencies in Knockout Mice
Results from three types of behavioral learning assays indicate a role for cholesterol 24-hydroxylase in higher-order brain function (79, 80). Spatial learning, as assessed in Morris water maze tests, is markedly deficient in mice homozygous for the induced mutation in the cholesterol 24-hydroxylase gene (Figure 6a). In these experiments, normal mice quickly learn the position of a submerged platform in a tank filled with opaque water by navigating via visual cues placed around the perimeter of the tank. The knockout mice, although they swim normally and are not blind, are unable to learn the location of the platform. The performance of mice heterozygous for the mutation is intermediate between homozygous and wild-type animals. If the platform is taken away, and the number of times mice of different cholesterol 24-hydroxylase genotypes swim across the former location is counted, then crossings by wild-type animals are roughly twofold more frequent than those of knockout mice (Figure 6b). Similar but less severe deficiencies are observed in homozygous cholesterol 24-hydroxylase-deficient mice with respect to associative learning and motor learning (79).
Figure 6.
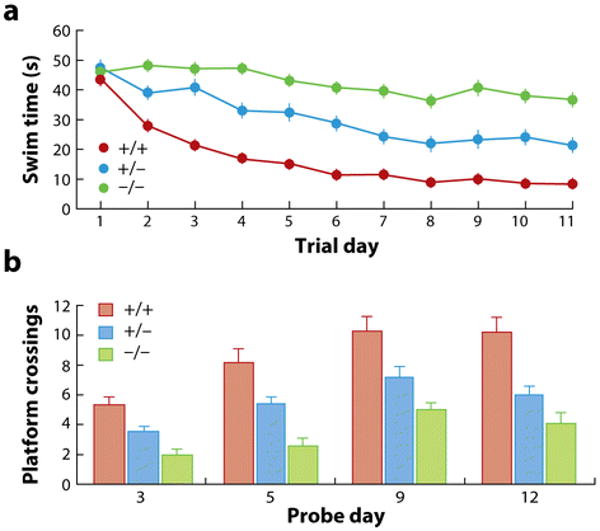
Morris water maze test. (a) Animals of the indicated cholesterol 24-hydroxylase genotypes were subjected to Morris water maze tests to assess spatial learning. Average swim time to find a submerged platform is plotted versus trial day. (b) The submerged platform was removed on the indicated day of the experiment, and the number of times animals swum across the former location of the platform were counted. Data redrawn and published with permission from Reference 80. Abbreviations: +/+, wild type for cholesterol 24-hydroxylase; +/−, heterozygous for cholesterol 24-hydroxylase mutation; −/−, homozygous for cholesterol 24-hydroxylase mutation.
Profound learning disabilities in mice, such as those observed in the cholesterol 24-hydroxylase-deficient animals, are often caused by gross anatomical defects in the subregions of the brain implicated in the behavior. For example, improper development of the hippocampus caused by loss of one or more low-density lipoprotein receptor family members gives rise to abject associative learning defects and reduced LTP (81); however, histological studies using light microscopy, fluorescent microscopy (Figure 4b), and electron microscopy do not reveal obvious anatomical shortcomings in cholesterol 24-hydroxylase knockout mice (79). This normalcy extends to embryos (59) and to primary hippocampal and cortical neurons and astrocytes cultured from day 16 knockout embryos (62). These findings suggest that the inability of the cholesterol 24-hydroxylase mutant mice to learn in common laboratory tests does not arise from anatomical or developmental defects but rather may be due to metabolic abnormalities in the animals.
Synaptic Plasticity Deficiencies
The measurement of synaptic strengthening and weakening in response to electrical stimulation of hippocampal slices is a standard in vitro approach to studying learning and memory formation (78, 82). In one version of the experiment, hippocampal slices ∼400 microns in width are prepared and placed on a stage in a chamber through which an artificial cerebrospinal fluid is circulated. An electrode is inserted into the CA1 region of the hippocampus to allow stimulation of axons that extend from pyramidal neurons in the CA3 region and form synapses with the dendrites of pyramidal neurons in the CA1 region. This synaptic circuit is referred to as the Schaffer collateral. A recording electrode is placed in the dendritic trees of the CA1 region, and a baseline response to monophasic stimulation is established over a 20-min period. Synaptic strengthening (LTP) (82) is measured after θ-burst stimulation, a series of high-frequency stimuli delivered in closely spaced intervals over a period of 1–2 min (83). Synaptic weakening, long-term depression (LTD) (84), is measured after repeated low-frequency stimulation over a period of 15 min. The biochemical mechanisms leading to LTP and LTD are varied and complex (85), but there is general agreement that these phenomena reflect, in part, the synaptic events that underlie learning in the whole animal (86).
Figure 7a shows the results from an experiment in which LTP is assessed in hippocampal slices from wild-type and cholesterol 24-hydroxylase-deficient mice (79). A θ-burst stimulation at minute 20 of the experiment leads to a strengthening of subsequent output from the CA1 pyramidal cell dendrites in wild-type slices, but to weaker potentiation in knockout slices. Other aspects of synaptic plasticity in the cholesterol 24-hydroxylase-deficient mice are apparently normal, including LTD, inhibitory responses mediated by picrotoxin-sensitive GABA receptors, short-term plasticity as judged by paired-pulse facilitation, spontaneous release of neurotransmitters as judged by measurement of miniature synaptic currents in single pyramidal CA1 neurons, and general synaptic transmission using plots of fiber volley magnitude versus excitatory postsynaptic potentials (79). The latter results suggest that the impairment of LTP in the knockout mice is selective and does not arise as a consequence of grossly aberrant neuronal function.
Figure 7.

Hippocampal long-term potentiation (LTP) in wild-type and cholesterol 24-hydroxylase knockout mice. (a) Baseline responses to monophasic stimulation were recorded for 20 min in slices from 5-week-old animals of the indicated genotype, and then the tissue was subjected to high-frequency stimulation, θ-burst (arrow). Subsequent responses to monophasic stimulation were strengthened in wild-type slices and less so in knockout slices. Insets show recorded potentials before (dashed lines) and 55 min after (solid lines) tetanization. (b) Effect of inhibition of cholesterol synthesis with a statin on hippocampal LTP. Redrawn and reprinted with permission from Reference 79. Abbreviations: fEPSP slope, slope of extracellularly recorded (field) excitatory postsynaptic potentials; +/+, wild type for cholesterol 24-hydroxylase; −/−, homozygous for cholesterol 24-hydroxylase mutation.
Two types of experiments were performed to link the metabolic phenotypes observed in the cholesterol 24-hydroxylase knockout mice, namely absence of 24S-hydroxycholesterol and reduced de novo cholesterol synthesis, to the synaptic plasticity deficiency. In the first experiment, LTP was assessed in hippocampal slices from mice deficient in the liver X receptor (LXR) α and β genes and found to be normal (79). LXR is a nuclear receptor that activates gene transcription in response to binding oxysterols, such as 24S-hydroxycholesterol (87), and thus the LTP results suggest that loss of LXR signaling does not underlie the electrophysiological defect in the cholesterol 24-hydroxylase knockout mice. In agreement with this conclusion, no significant alterations in LXR target gene expression are detected by microarray or real-time PCR analyses of whole brain or embryonic hippocampal and cortical neuronal mRNAs from the mutant mice.
In a second experiment, LTP was assessed in wild-type hippocampal slices treated with an inhibitor of cholesterol synthesis, compactin (which is a statin that inhibits the third enzyme in the pathway, 3-hydroxy-3-methylglutaryl coenzyme A reductase) (79). The concentration of compactin used (12.5 μM) reduces de novo cholesterol synthesis in wild-type hippocampal slices by approximately the same amount as that observed in cholesterol 24-hydroxylase knockout mice (i.e., ∼ a 50% to 60% reduction in cholesterol synthesis). Under these conditions, LTP in the statin-treated wild-type slices is reduced to that observed in untreated knockout slices (Figure 7b). Incubating wild-type slices with compactin plus mevalonate, the product of the inhibited enzyme, prevents the reduction in LTP, indicating the specificity of the inhibition. Furthermore, mevalonate restores LTP to wild-type levels in hippocampal slices from cholesterol 24-hydroxylase knockout mice. The results of these experiments suggest that the LTP defect in the cholesterol 24-hydroxylase knockout mice arises from the reduction in de novo cholesterol synthesis associated with the loss of a major catabolic enzyme and not from the absence of 24S-hydroxycholesterol.
Rescue by Geranylgeraniol
That mevalonate prevents loss of LTP in compactin-treated wild-type hippocampal slices suggests that a product downstream of this intermediate is required for potentiation of synapses in the CA3-CA1 pathway. Mevalonate is converted into myriad end products ranging from nonsterol isoprenoids, such as farnesyl diphosphate and geranylgeranyl diphosphate, to sterols (Figure 1a), and thus any of these metabolites could be the active agent. Add-back experiments show that the required intermediate is a C20 isoprenoid related to geranylgeranyl diphosphate (79, 80). The data in Figure 8 show that the addition of 0.2 mM geranylgeraniol to the artificial cerebrospinal fluid at time zero restores LTP to normal levels in both cholesterol 24-hydroxylase knockout slices and in statin-treated wild-type slices. Dose-response studies show that 0.2 mM geranylgeraniol is the minimal concentration required to restore LTP in hippocampal slices from knockout mice (T. Kotti, unpublished observations).
Figure 8.
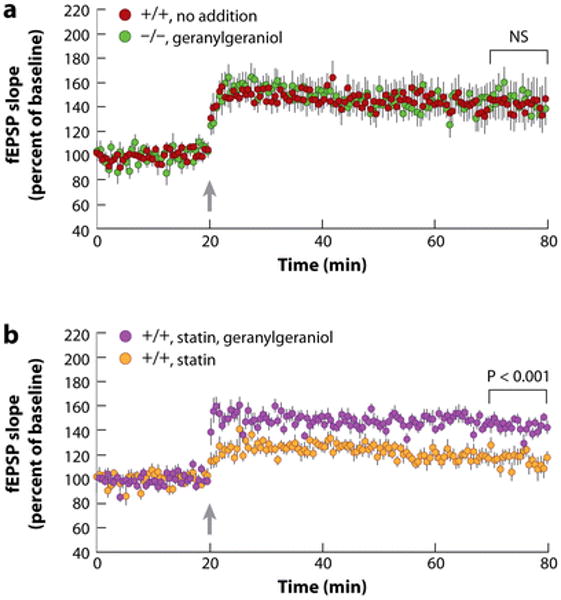
Restoration of hippocampal long-term potentiation by geranylgeraniol treatment of slices from cholesterol 24-hydroxylase knockout mice (a) and those from wild-type mice treated with a statin (b). Data in (a) redrawn and reprinted with permission from Reference 79. Abbreviations: fEPSP slope, slope of extracellularly recorded (field) excitatory postsynaptic potentials; +/+, wild type for cholesterol 24-hydroxylase; −/−, homozygous for cholesterol 24-hydroxylase mutation; arrow, time of θ-burst stimulation; NS, not significant.
As noted above, mice deficient in ApoE also exhibit defects in learning and LTP, which raises the question of whether these animals are also deficient in an aspect of cholesterol metabolism that leads to a shortage of geranylgeranyl diphosphate? The answer to this question is no; geranylgeraniol does not restore LTP in hippocampal slices from ApoE knockout mice (80). Thus, the geranylgeraniol response in cholesterol 24-hydroxylase knockout mice is genetically selective.
Phosphorylated isoprenoids are unstable and taken up poorly by mammalian cells, and for these reasons, add-back experiments utilize the alcoholic form of geranylgeranyl diphosphate, i.e., geranylgeraniol. It is currently not known whether the active form of the polyisoprenoid is the diphosphate, the alcohol, or another metabolite; however, geranylgeranyl diphosphate cannot be converted into sterols, which eliminates this class of end products. Additional experiments reveal that farnesol restores LTP, but not if a statin is included in the buffer at the same time (80). Statins block the production of the isoprenoid precursors required to elongate a C15 farnesol diphosphate to a C20 geranylgeranyl diphosphate (Figure 1b), and thus these results indicate the chemical selectivity of the LTP response. Furthermore, as the conversion of farnesol to geranylgeranyl diphosphate requires that the precursor be taken up and acted upon by intracellular enzymes, these findings indicate that either geranylgeranyl diphosphate is acting inside the cell or that the newly synthesized compound is released by some mechanism to act outside the cell.
Phenotype of Heterozygous Mice
Mice heterozygous for the induced cholesterol 24-hydroxylase mutation exhibit a hippocampal LTP phenotype that is intermediate between those of homozygous and wild-type animals and, as shown in Figure 7, exhibit an intermediate response in Morris water maze tests (80). These findings indicate that the relationship between cholesterol 24-hydroxylase activity and flux through the cholesterol biosynthetic pathway is linear rather than nonlinear as is the case in a majority of metabolic pathways (88), and they reveal that the phenotype is codominant rather than the more typical recessive phenotype associated with biosynthetic genes. The enzymes of most metabolic pathways are expressed in excess of biochemical need, and thus the 50% loss of enzyme activity associated with a mutation in heterozygous form is usually phenotypically silent. In the case of cholesterol 24-hydroxylase, the intermediate phenotype of the heterozygote suggests that levels of geranylgeraniol or metabolites derived from this compound are carefully titrated in the brain and that just enough of the isoprenoid is made to ensure normal synaptic plasticity. In agreement with this suggestion, exogenous geranylgeraniol does not increase LTP in wild-type hippocampal slices (79).
Biphasic Requirement for Geranylgeraniol in LTP
Mevalonate and geranylgeraniol restore LTP in the hippocampus when included in the artificial cerebrospinal fluid for the duration of the 80-min recording period. Additional experiments show that a 5-min pulse of geranylgeraniol delivered during the time of high-frequency stimulation conferred induction of LTP in cholesterol 24-hydroxylase knockout slices (80) but that potentiation is not maintained with this brief exposure (Figure 9a). If the isoprenoid is delivered twice, once during the time of high-frequency stimulation and again between minutes 35 and 40 of the experiment, then LTP is induced and maintained in knockout slices (Figure 9b). These findings indicate that geranylgeraniol acts within minutes and that the isoprenoid is required at two different times to effect potentiation in the hippocampus.
Figure 9.

Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation (LTP). (a) A 5-min geranylgeraniol treatment (purple bar) of slices from cholesterol 24-hydroxylase knockout mice (−/−) induces but does not maintain LTP. (b) Two 5-min geranylgeraniol treatments induce and maintain LTP. Redrawn and reprinted with permission from Reference 80. Abbreviations: fEPSP slope, slope of extracellularly recorded (field) excitatory postsynaptic potentials; +/+, wild type for cholesterol 24-hydroxylase; −/−, homozygous for cholesterol 24-hydroxylase mutation; arrow, time of θ-burst stimulation.
Mechanism of Geranylgeraniol Action in LTP
Several mechanisms may explain the ability of geranylgeraniol to affect LTP. First, both farnesyl and geranylgeranyl are attached to the C termini of hundreds of proteins by isoprenyltransferases where they direct the modified protein to different membranes within the cell (89). One or more of these prenylated proteins may be required to establish and maintain synaptic plasticity in the hippocampus, and presumably, the active protein or their attached geranylgeranyl groups are turned over rapidly to explain the acute action of geranylgeraniol in modulating LTP. Despite the attractiveness of this hypothesis, protein isoprenylation is generally a stable posttranslational modification and in agreement with this generalization, inhibitors of protein isoprenyltransferases do not have acute effects on LTP, nor is LTP abnormal in a strain of mutant mice with reduced geranylgeranyltransferase II activity (80).
Second, geranylgeraniol may act as a ligand for a cell-surface receptor. This idea is supported by the speed of the response, which is reminiscent of that to N-arachidonoylethanolamide and 2-arachidonoylglycerol, two endocannabinoids that are released from the postsynaptic membranes of neurons and act through a G protein–coupled receptor on the presynaptic membrane to decrease synaptic strength (90, 91). These lipids appear to be released from glycerophospholipid precursors via the actions of lipolytic enzymes and are then secreted from the postsynaptic membrane via a specific transporter (92). Whether geranylgeraniol follows a similar cleavage and release itinerary is not known.
There are other possibilities that may explain the apparent requirement for geranylgeraniol in LTP, including the isoprenoid (a) acting as a chaperone to facilitate movement of glutamatergic receptors to the postsynaptic membrane, or (b) acting as an allosteric regulator of a neurotransmitter receptor or ion channel activity, or (c) functioning by an as yet unprecedented mechanism.
Genetics of Cholesterol 24-Hydroxylase
The human gene (symbol CYP46A1) encoding cholesterol 24-hydroxylase is located on chromosome 14q32, and the mouse gene (symbol Cyp46a1) is located on chromosome 12F1. Both genes contain 15 exons (37), which is an unusually large number of exons for a mammalian P450 gene. The human gene spans ∼42 kb of DNA on chromosome 14, and the mouse ortholog encompasses ∼28 kb on chromosome 12. The organization and identities of genes flanking the cholesterol 24-hydroxylase loci in the mouse and human are syntenic. In the human, there are data to indicate that cholesterol 24-hydroxylase has an interesting genetics.
Linkage to Dementias
The role of cholesterol 24-hydroxylase in brain sterol metabolism has led to numerous genetic association studies that attempt to link polymorphisms in the human gene to neurodegenerative disorders, such as Alzheimer's disease. All of the linkage analyses to date have been done with small patient populations, and none have been replicated in large independent cohorts, and as a consequence as many studies find an association between the disease and polymorphisms in the CYP46A1 locus (93–96) as do not (97–100). In vitro evidence suggests that 24S-hydroxycholesterol can affect the processing of the amyloid precursor protein; however, there is no consensus regarding whether the oxysterol increases (101) or decreases (102) processing, and no correlation was observed between polymorphisms in CYP46A1 and the levels of amyloid β40 or β42 peptides in the brains of Alzheimer's subjects (103). Similarly, alterations in plasma and cerebrospinal fluid levels of 24S-hydroxycholesterol reported in subjects with Alzheimer's disease (38, 104, 105), multiple sclerosis (106, 107), and various dementias (108–110) are difficult to interpret as these differences may reflect changes in cholesterol 24-hydroxylase activity or a secondary aspect of the pathology arising from disruption of the blood-brain barrier (111) or neuronal cell death (6). Clarifying these and the other uncertainties associated with the genetics of human cholesterol 24-hydroxylase will require additional work.
Summary and Outlook
All tissues turn over cholesterol by one or more mechanisms. In the case of the brain, existing evidence indicates that elimination is accomplished in part by conversion of cholesterol to 24S-hydroxycholesterol followed by diffusion of the oxysterol from the tissue. This reaction is catalyzed by cholesterol 24-hydroxylase, a P450 enzyme expressed in a subset of neurons in the mammalian brain. Orthologs of the cholesterol 24-hydroxylase gene are found in many vertebrate genomes, indicating that this turnover pathway is ancient and utilized in multiple species.
Quantitative balance studies in rodents indicate that the cholesterol 24-hydroxylase pathway accounts for the turnover of ∼50% of cholesterol in the rodent brain, suggesting that additional pathways for sterol catabolism exist. Loss of cholesterol 24-hydroxylase in the mouse reduces the turnover of cholesterol and is compensated for by an equal reduction in sterol synthesis. This response maintains brain cholesterol levels within the normal range but has the unintended consequence of reducing the synthesis of a polyisoprenoid required for higher-order brain function. Current data indicate that the essential molecule is geranylgeraniol or a metabolite of this diterpenoid.
The studies reviewed here uncover an unexpected link between the turnover of brain cholesterol and learning. The challenges ahead are to delineate the mechanism by which geranylgeraniol affects synaptic plasticity in the central nervous system and to understand how this metabolic connection arose.
Summary Points.
Cholesterol 24-hydroxylase converts cholesterol, an intransigent membrane lipid, to 24S-hydroxycholesterol, an itinerant oxysterol that diffuses from the brain and is subsequently metabolized by the liver. This conversion represents a major pathway of cholesterol turnover in the vertebrate brain.
Cholesterol 24-hydroxylase is a highly conserved P450 that is expressed in some, but not all, neurons of the brain. The enzyme is found in the endoplasmic reticulum, which extends throughout neuronal cell bodies and dendrites.
Disruption of the mouse cholesterol 24-hydroxylase gene causes an ∼50% decrease in cholesterol turnover, which is compensated for by an equal decrease in the rate of de novo cholesterol synthesis. Steady-state levels of cholesterol are unchanged in the brains of the mutant mice. The decrease in cholesterol synthesis reduces the flow of metabolites through the biosynthetic pathway, producing a phenotype that is codominant.
Cholesterol 24-hydroxylase knockout mice are deficient in spatial, associative, and motor learning, and have abnormal hippocampal LTP. The defect in LTP can be reversed by geranylgeraniol, a polyisoprenoid end product of the cholesterol biosynthetic pathway.
Geranylgeraniol acts quickly (within minutes) and specifically (farnesol, a related polyisoprenol, will not substitute) to restore LTP in hippocampal slices.
Future Issues.
How does the subcellular localization of cholesterol 24-hydroxylase affect cholesterol turnover? Do binding proteins target the enzyme to defined subcellular localizations at which cholesterol turnover takes place? What transport processes bring cholesterol to the enzyme in the endoplasmic reticulum membrane? Is enzyme activity regulated by synaptic activity?
How does geranylgeraniol affect synaptic plasticity? What is the active form of the molecule? Is the polyisoprenoid stored, released, and taken up by neurons? Does geranylgeraniol act as a ligand to activate a cell-surface receptor? Is geranylgeraniol required for the intracellular movement of excitatory neurotransmitter receptors? Which intracellular signaling pathways are activated by geranylgeraniol? Do species other than the mouse require geranylgeraniol for LTP?
What is the logic behind linking cholesterol turnover with synaptic plasticity? Do all vertebrates utilize the cholesterol 24-hydroxylase pathway? What other pathways exist for cholesterol catabolism in the central nervous system? Are these pathways present in the peripheral nervous system?
What role does cholesterol turnover play in the human brain? Are there naturally occurring mutations in the human cholesterol 24-hydroxylase gene, and if so, what are their clinical consequences? Do these mutations generate a codominant phenotype?
Acknowledgments
We thank colleagues who have inspired, stimulated, and facilitated our work on cholesterol 24-hydroxylase, including Ingemar Björkhem, John Dietschy, Ron Estabrook, Joe Guileyardo, Daphne Head, Kim Huber, Erik Lund, Jeff McDonald, Steve McKnight, Brad Pfeiffer, Irena Pikuleva, Steve Turley, and Hao Zhang. Jay Horton provided critical comments on this manuscript. Work in the authors' laboratory is supported by NIH grants HL20948, GM69338, and DK81182 as well as by grants from the Robert A. Welch Foundation (I-0971) and the Perot Family Fund.
Footnotes
Disclosure Statement: The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
Contributor Information
David W. Russell, Email: david.russell@utsouthwestern.edu.
Rebekkah W. Halford, Email: rebekkah.halford@utsouthwestern.edu.
Denise M.O. Ramirez, Email: denise.ramirez@utsouthwestern.edu.
Rahul Shah, Email: rahul.shah@utsouthwestern.edu.
Tiina Kotti, Email: tiina.kotti@utsouthwestern.edu.
Literature Cited
- 1.Sokoloff L. Circulation and energy metabolism of the brain. In: Siegel GJ, Albers RW, Katzman R, Agranoff BW, editors. Basic Neurochemistry. Boston: Little, Brown; 1976. pp. 388–413. [Google Scholar]
- 2.Piomelli D, Astarita G, Rapaka R. A neuroscientist's guide to lipidomics. Nat Rev Neurosci. 2007;8:743–54. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- 3.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Dietschy JM, Turley SD. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–97. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16:193–212. doi: 10.1016/j.semcdb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Xie C, Lund EG, Turley SD, Russell DW, Dietschy JM. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res. 2003;44:1780–89. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Davison AN. Brain sterol metabolism. Adv Lipid Res. 1965;3:171–96. doi: 10.1016/b978-1-4831-9939-9.50011-5. [DOI] [PubMed] [Google Scholar]
- 8.Cuzner ML, Davison AN, Gregson NA. Turnover of brain mitochondrial membrane lipids. Biochem J. 1966;101:618–26. doi: 10.1042/bj1010618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Begley DJ, Brightman MW. Structural and functional aspects of the blood-brain barrier. Prog Drug Res. 2003;61:39–78. doi: 10.1007/978-3-0348-8049-7_2. [DOI] [PubMed] [Google Scholar]
- 10.Jurevics H, Morell P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem. 1995;64:895–901. doi: 10.1046/j.1471-4159.1995.64020895.x. [DOI] [PubMed] [Google Scholar]
- 11.Osono Y, Woollett LA, Herz J, Dietschy JM. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J Clin Investig. 1995;95:1124–32. doi: 10.1172/JCI117760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jurevics HA, Kidwai FZ, Morell P. Sources of cholesterol during development of the rat fetus and fetal organs. J Lipid Res. 1997;38:723–33. [PubMed] [Google Scholar]
- 13.Turley SD, Burns DK, Dietschy JM. Preferential utilization of newly synthesized cholesterol for brain growth in neonatal lambs. Am J Physiol. 1998;274:E1099–105. doi: 10.1152/ajpendo.1998.274.6.E1099. [DOI] [PubMed] [Google Scholar]
- 14.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Windaus A. Uber die entgiftung der saponine durch cholesterin. Chem Ber. 1909;42:238–46. [Google Scholar]
- 16.Rittenberg D, Schoenheimer R. Deuterium as an indicator in the study of intermediary metabolism. XI. Further studies on the biological uptake of deuterium into organic substances, with special reference to fat and cholesterol formation. J Biol Chem. 1937;121:235–53. [Google Scholar]
- 17.Waelsch H, Sperry WM, Stoyanoff VA. A study of the synthesis and deposition of lipids in brain and other tissues with deuterium as an indicator. J Biol Chem. 1940;135:291–96. [Google Scholar]
- 18.Sperry WM, Waelsch H, Stoyanoff VA. Lipid metabolism in brain and other tissues of the rat. J Biol Chem. 1940;135:281–90. [Google Scholar]
- 19.Andersen JM, Dietschy JM. Absolute rates of cholesterol synthesis in extrahepatic tissues measured with 3H-labeled water and 14C-labeled substrates. J Lipid Res. 1979;20:740–52. [PubMed] [Google Scholar]
- 20.Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res Dev Brain Res. 2003;146:87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 21.Ercoli A, Di Frisco S, de Ruggieri P. Sul cerebrostendiolo, un colestendiolo isolato dal cervello di cavallo. Gazz Chim Ital. 1953;83:78–86. [Google Scholar]
- 22.Ercoli A, DeRuggieri P. The constitution of cerebrosterol, a hydroxycholesterol isolated from horse brain. J Am Chem Soc. 1953;75:3284. [Google Scholar]
- 23.Schubert K, Rose G, Burger M. Uber das vorkommen von dihydroxysterinen im menschlichen gehrin. Hoppe-Seyler's Z Physiol Chem. 1961;326:235–41. doi: 10.1515/bchm2.1961.326.1.235. [DOI] [PubMed] [Google Scholar]
- 24.Smith LL, Ray DR, Moody JA, Wells JD, van Lier JE. 24-Hydroxycholesterol levels in human brain. J Neurochem. 1972;19:899–904. doi: 10.1111/j.1471-4159.1972.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 25.van Lier JE, Smith LL. Sterol metabolism. V. A chromatographic examination of human brain sterols. Tex Rep Biol Med. 1969;27:167–76. [PubMed] [Google Scholar]
- 26.Dhar AK, Teng JI, Smith LL. Biosynthesis of cholest-5-ene-3β,24-diol (cerebrosterol) by bovine cerebral cortical microsomes. J Neurochem. 1973;21:51–60. doi: 10.1111/j.1471-4159.1973.tb04224.x. [DOI] [PubMed] [Google Scholar]
- 27.Richter VR, Dannenberg H. Fraktionierung der “neutralen spurenlipoide” aus rinder- und kaninchengehirn. Hoppe-Seyler's Z Physiol Chem. 1969;350:1213–24. [PubMed] [Google Scholar]
- 28.Lin YY, Smith LL. Sterol metabolism XXVIII. Biosynthesis and accumulation of cholest-5-ene-3ß,24-diol (cerebrosterol) in developing rat brain. Biochim Biophys Acta. 1974;348:189–96. doi: 10.1016/0005-2760(74)90230-6. [DOI] [PubMed] [Google Scholar]
- 29.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal Biochem. 1995;225:73–80. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 30.Breuer O, Björkhem I. Use of an 18O2 inhalation technique and mass isotopomer distribution analysis to study oxygenation of cholesterol in rat: evidence for in vivo formation of 7-oxo-, 7β-hydroxy-, 24-hydroxy-, and 25-hydroxycholesterol. J Biol Chem. 1995;270:20278–84. doi: 10.1074/jbc.270.35.20278. [DOI] [PubMed] [Google Scholar]
- 31.Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA. 1996;93:9799–804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Björkhem I, Lütjohann D, Breuer O, Sakinis A, Wennmalm A. Importance of a novel oxidative mechanism for elimination of brain cholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J Biol Chem. 1997;272:30178–84. doi: 10.1074/jbc.272.48.30178. [DOI] [PubMed] [Google Scholar]
- 33.Diczfalusy U, Lund E, Lütjohann D, Björkhem I. Novel pathways for elimination of cholesterol by extrahepatic formation of side-chain oxidized oxysterols. Scand J Clin Lab Investig Suppl. 1996;226:9–17. [PubMed] [Google Scholar]
- 34.Björkhem I, Lütjohann D, Diczfalusy U, Stahle L, Ahlborg G, et al. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39:1594–600. [PubMed] [Google Scholar]
- 35.Meaney S, Lütjohann D, Diczfalusy U, Björkhem I. Formation of oxysterols from different pools of cholesterol as studied by stable isotope technique: cerebral origin of most circulating 24S-hydroxycholesterol in rats, but not in mice. Biochim Biophys Acta. 2000;1486:293–98. doi: 10.1016/s1388-1981(00)00070-6. [DOI] [PubMed] [Google Scholar]
- 36.Lütjohann D, Stroick M, Bertsch T, Kühl S, Lindenthal B, et al. High doses of simvastatin, pravastatin, and cholesterol reduce brain cholesterol synthesis in guinea pigs. Steroids. 2004;69:431–38. doi: 10.1016/j.steroids.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 37.Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a regulator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA. 1999;96:7238–43. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bretillon L, Lütjohann D, Stahle L, Widhe T, Bindl L, et al. Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J Lipid Res. 2000;41:840–45. [PubMed] [Google Scholar]
- 39.Björkhem I, Andersson U, Ellis E, Alvelius G, Ellegard L, et al. From brain to bile. Evidence that conjugation and ω-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J Biol Chem. 2001;276:37004–10. doi: 10.1074/jbc.M103828200. [DOI] [PubMed] [Google Scholar]
- 40.Saucier SE, Kandutsch AA, Clark DS, Spencer TA. Hepatic uptake and metabolism of ingested 24-hydroxycholesterol and 24 (S), 25-epoxycholesterol. Biochim Biophys Acta. 1993;1166:115–23. doi: 10.1016/0005-2760(93)90291-g. [DOI] [PubMed] [Google Scholar]
- 41.Gustafsson JA, Sjovall J. Identification of 22-, 24- and 26-hydroxycholesterol in the steroid sulphate fraction of faeces from infants. Eur J Biochem. 1969;8:467–72. doi: 10.1111/j.1432-1033.1969.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 42.Makino I, Sjovall J, Norman A, Strandvik B. Excretion of 3β-hydroxy-5-cholenoicand 3α-hydroxy-5α-cholanoic acids in urine of infants with biliary atresia. FEBS Lett. 1971;15:161–64. doi: 10.1016/0014-5793(71)80047-9. [DOI] [PubMed] [Google Scholar]
- 43.Summerfield JA, Billing BH, Schackelton CH. Identification of bile acids in the serum and urine in cholestasis. Evidence for 6α-hydroxylation of bile acids in man. Biochem J. 1976;154:507–16. doi: 10.1042/bj1540507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng LJ, Griffiths WJ, Nazer H, Yang Y, Sjovall J. High levels of (24S)-24-hydroxycholesterol 3-sulfate, 24-glucuronide in the serum and urine of children with severe cholestatic liver disease. J Lipid Res. 1997;38:926–34. [PubMed] [Google Scholar]
- 45.Meaney S, Bodin K, Diczfalusy U, Björkhem I. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J Lipid Res. 2002;43:2130–35. doi: 10.1194/jlr.m200293-jlr200. [DOI] [PubMed] [Google Scholar]
- 46.Babiker A, Diczfalusy U. Transport of side-chain oxidized oxysterols in the human circulation. Biochim Biophys Acta. 1998;1392:333–39. doi: 10.1016/s0005-2760(98)00047-2. [DOI] [PubMed] [Google Scholar]
- 47.Li-Hawkins J, Lund EG, Bronson AD, Russell DW. Expression cloning of an oxysterol 7α-hydroxylase selective for 24-hydroxycholesterol. J Biol Chem. 2000;275:16543–49. doi: 10.1074/jbc.M001810200. [DOI] [PubMed] [Google Scholar]
- 48.Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem. 1998;273:34316–48. doi: 10.1074/jbc.273.51.34316. [DOI] [PubMed] [Google Scholar]
- 49.Mast N, Norcross R, Andersson U, Shou M, Nakayama K, et al. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 2003;42:14284–92. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- 50.Bodin K, Andersson U, Rystedt E, Ellis E, Norlin M, et al. Metabolism of 4-hydroxycholesterol in humans. J Biol Chem. 2002;277:31534–40. doi: 10.1074/jbc.M201712200. [DOI] [PubMed] [Google Scholar]
- 51.Mast N, Andersson U, Nakayama K, Björkhem I, Pikuleva IA. Expression of human cytochrome P450 46A1 in Escherichia coli: effects of N- and C-terminal modifications. Arch Biochem Biophys. 2004;428:99–108. doi: 10.1016/j.abb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci Signal. 2003 doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- 53.Mast N, White MA, Björkhem I, Johnson EF, Stout CD, Pikuleva IA. Crystal structure of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci USA. 2008;105:9546–51. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White MA, Mast N, Björkhem I, Johnson EF, Stout CD, Pikuleva IA. Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr D Biol Crystallogr. 2008;64:487–95. doi: 10.1107/S0907444908004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otyepka M, Skopalik J, Anzenbacherova E, Anzenbacher P. What common features and variations of mammalian P450s are known to date? Biochim Biophys Acta. 2006;1770:376–89. doi: 10.1016/j.bbagen.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 56.Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1, a microsomal vitamin D 25-hydroxylase. J Biol Chem. 2003;278:38084–93. doi: 10.1074/jbc.M307028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. 2004;101:7711–15. doi: 10.1073/pnas.0402490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcriptase PCR. Yakugaku Zasshi. 2003;125:369–75. doi: 10.1248/yakushi.123.369. [DOI] [PubMed] [Google Scholar]
- 59.Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, et al. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–88. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- 60.Ohyama Y, Meaney S, Heverin M, Ekström L, Brafman A, et al. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem. 2005;281:3810–20. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- 61.Milagre I, Nunes MJ, Gama MJ, Silva RF, Pascussi JM, et al. Transcriptional regulation of the human CYP46A1 brain-specific expression by Sp transcription factors. J Neurochem. 2008;106:835–49. doi: 10.1111/j.1471-4159.2008.05442.x. [DOI] [PubMed] [Google Scholar]
- 62.Ramirez DMO, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol. 2008;507:1676–93. doi: 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bretillon L, Diczfalusy U, Björkhem I, Maire MA, Martine L, et al. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr Eye Res. 2007;32:361–66. doi: 10.1080/02713680701231857. [DOI] [PubMed] [Google Scholar]
- 64.McDonald JG, Thompson BT, McCrum EC, Russell DW. Extraction and analysis of sterols in biological matrices by high-performance liquid chromatography electrospray ionization mass spectrometry. Methods Enzymol. 2007;432:143–68. doi: 10.1016/S0076-6879(07)32006-5. [DOI] [PubMed] [Google Scholar]
- 65.Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–53. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- 67.Callahan CA, Thomas JB. Tau-β-galactosidase, an axon-targeted fusion protein. Proc Natl Acad Sci USA. 1994;91:5972–76. doi: 10.1073/pnas.91.13.5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lund E, Björkhem I, Furster C, Wikvall K. 24-, 25-, and 27-Hydroxylation of cholesterol by a purified preparation of 27-hydroxylase from pig liver. Biochim Biophys Acta. 1993;1166:177–82. doi: 10.1016/0005-2760(93)90094-p. [DOI] [PubMed] [Google Scholar]
- 69.Pikuleva IA, Babiker A, Waterman MR, Björkhem I. Activities of recombinant human cytochrome P450c27 (CYP27) which produces intermediates of alternate bile acid biosynthetic pathways. J Biol Chem. 1998;273:18153–60. doi: 10.1074/jbc.273.29.18153. [DOI] [PubMed] [Google Scholar]
- 70.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 71.Kandel ER. Nerve cells and behavior. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. Norwalk: Appleton & Lange; 1991. pp. 18–32. [Google Scholar]
- 72.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 73.Pfrieger FW. Role of cholesterol in synapse formation and function. Biochim Biophys Acta. 2003;1610:271–80. doi: 10.1016/s0005-2736(03)00024-5. [DOI] [PubMed] [Google Scholar]
- 74.Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci Lett. 1995;199:1–4. doi: 10.1016/0304-3940(95)12006-p. [DOI] [PubMed] [Google Scholar]
- 75.Oitzl MS, Mulder M, Lucassen PJ, Havekes LM, Grootendorst J, et al. Severe learning deficits in apolipoprotein E-knockout mice in a water maze task. Brain Res. 1997;752:189–96. doi: 10.1016/s0006-8993(96)01448-5. [DOI] [PubMed] [Google Scholar]
- 76.Masliah E, Samuel W, Veinbergs I, Mallory M, Mante M, Saitoh T. Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res. 1997;751:307–14. doi: 10.1016/s0006-8993(96)01420-5. [DOI] [PubMed] [Google Scholar]
- 77.Krugers HJ, Mulder M, Korf J, Havekes L, de Kloet ER, Joëls M. Altered synaptic plasticity in hippocampal CA1 area of apolipoprotein E deficient mice. NeuroReport. 1997;8:2505–10. doi: 10.1097/00001756-199707280-00018. [DOI] [PubMed] [Google Scholar]
- 78.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 79.Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci USA. 2006;103:3869–74. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kotti T, Head DD, McKenna CE, Russell DW. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci USA. 2008;105:11394–99. doi: 10.1073/pnas.0805556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, et al. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–52. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- 82.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–50. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- 84.Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature. 1977;266:737–39. doi: 10.1038/266737a0. [DOI] [PubMed] [Google Scholar]
- 85.Sweatt JD. Mechanisms of Memory. Amsterdam: Elsevier Academic; 2003. p. 400. [Google Scholar]
- 86.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–74. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 87.Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–91. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 88.Kacser H, Burns JA. The molecular basis of dominance. Genetics. 1981;97:639–66. doi: 10.1093/genetics/97.3-4.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leung KF, Baron R, Seabra MC. Geranylgeranylation of Rab GTPases. J Lipid Res. 2006;47:467–75. doi: 10.1194/jlr.R500017-JLR200. [DOI] [PubMed] [Google Scholar]
- 90.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–82. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- 91.Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- 92.Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci USA. 2007;104:20564–69. doi: 10.1073/pnas.0706873104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kolsch H, Lütjohann D, Ludwig M, Schulte A, Ptok U, et al. Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer's disease. Mol Psychiatry. 2002;7:899–902. doi: 10.1038/sj.mp.4001109. [DOI] [PubMed] [Google Scholar]
- 94.Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, et al. Increased brain β-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol. 2003;60:29–35. doi: 10.1001/archneur.60.1.29. [DOI] [PubMed] [Google Scholar]
- 95.Li Y, Chu LW, Chen YQ, Cheung BM, Leung RY, et al. Intron 2 (T/C) CYP46 polymorphism is associated with Alzheimer's disease in Chinese patients. Dement Geriatr Cogn Disord. 2006;22:399–404. doi: 10.1159/000095723. [DOI] [PubMed] [Google Scholar]
- 96.Ma SL, Tang NL, Lam LC, Chiu HF. Polymorphisms of the cholesterol 24-hydroxylase (CYP46A1) gene and the risk of Alzheimer's disease in a Chinese population. Int Psychogeriatr. 2006;18:37–45. doi: 10.1017/S1041610205003108. [DOI] [PubMed] [Google Scholar]
- 97.Desai P, DeKosky ST, Kamboh MI. Genetic variation in the cholesterol 24-hydroxylase (CYP46) gene and the risk of Alzheimer's disease. Neurosci Lett. 2002;328:9–12. doi: 10.1016/s0304-3940(02)00443-3. [DOI] [PubMed] [Google Scholar]
- 98.Wang F, Jia J. Polymorphisms of cholesterol metabolism genes CYP46 and ABCA1 and the risk of sporadic Alzheimer's disease in Chinese. Brain Res. 2007;1147:34–38. doi: 10.1016/j.brainres.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 99.Juhász A, Rimanóczy A, Boda K, Vincze G, Szlávik G, et al. CYP46 T/C polymorphism is not associated with Alzheimer's dementia in a population from Hungary. Neurochem Res. 2005;30:943–48. doi: 10.1007/s11064-005-5979-4. [DOI] [PubMed] [Google Scholar]
- 100.Shibata N, Kawarai T, Lee JH, Lee HS, Shibata E, et al. Association studies of cholesterol metabolism genes (CH25H, ABCA1 and CH24H) in Alzheimer's disease. Neurosci Lett. 2006;391:142–46. doi: 10.1016/j.neulet.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 101.Famer D, Meaney S, Mousavi M, Nordberg A, Björkhem I, et al. Regulation of α- and β-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the α-secretase pathway. Biochem Biophys Res Commun. 2007;359:46–50. doi: 10.1016/j.bbrc.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 102.Brown J, III, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, et al. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J Biol Chem. 2004;279:34674–81. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- 103.Ingelsson M, Jesneck J, Irizarry MC, Hyman BT, Rebeck GW. Lack of association of the cholesterol 24-hydroxylase (CYP46) intron 2 polymorphism with Alzheimer's disease. Neurosci Lett. 2004;367:228–31. doi: 10.1016/j.neulet.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 104.Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41:195–98. [PubMed] [Google Scholar]
- 105.Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, et al. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer's disease compared to healthy controls. Neurosci Lett. 2002;324:83–85. doi: 10.1016/s0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- 106.Leoni V, Masterman T, Diczfalusy U, De Luca G, Hillert J, Björkhem I. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett. 2002;331:163–66. doi: 10.1016/s0304-3940(02)00887-x. [DOI] [PubMed] [Google Scholar]
- 107.Karrenbauer VD, Leoni V, Lim ET, Giovannoni G, Ingle GT, et al. Plasma cerebrosterol and magnetic resonance imaging measures in multiple sclerosis. Clin Neurol Neurosurg. 2006;108:456–60. doi: 10.1016/j.clineuro.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 108.Papassotiropoulos A, Lütjohann D, Bagli M, Locatelli S, Jessen F, et al. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36:27–32. doi: 10.1016/s0022-3956(01)00050-4. [DOI] [PubMed] [Google Scholar]
- 109.Kolsch H, Heun R, Kerksiek A, Bergmann KV, Maier W, et al. Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci Lett. 2004;368:303–8. doi: 10.1016/j.neulet.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 110.Leoni V, Shafaati M, Salomon A, Kivipelto M, Björkhem I, et al. Are the CSF levels of 24S-hydroxycholesterol a sensitive biomarker for mild cognitive impairment? Neurosci Lett. 2006;397:83–87. doi: 10.1016/j.neulet.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 111.Leoni V, Masterman T, Patel P, Meaney S, Diczfalusy U, et al. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J Lipid Res. 2003;44:793–99. doi: 10.1194/jlr.M200434-JLR200. [DOI] [PubMed] [Google Scholar]