

Abstract
The Chagas’ disease parasite Trypanosoma cruzi promotes survival and differentiation of neurones by binding and activating nerve growth factor (NGF) receptor TrkA. The functional mimic of NGF in T. cruzi is a surface-bound and shed immunogenic protein [neurotrophic factor/trans-sialidase (TS)], which raised the possibility that immune response to T. cruzi in general and to neurotrophic factor/TS in particular leads to loss of immunological tolerance to host NGF and/or the NGF-binding partner TrkA. In testing this hypothesis, we found that sera of individuals with chronic Chagas’ disease bear unique IgG2 autoantibodies that bind TrkA and TrkA family members TrkB and TrkC (ATA). Binding of ATA to Trk receptors is specific because the autoantibodies did not cross-react with five other growth factor receptors, NGF and other neurotrophins, and T. cruzi. Thus, individuals with chronic Chagas’ disease produce unique antibodies that react with pan-Trk receptors, one of which (TrkA) T. cruzi exploits to inhibit host cell apoptosis and to promote cellular invasion.
Introduction
The protozoan Trypanosoma cruzi causes debilitating Chagas’ disease, which afflicts millions of people in the Americas. Trypanosoma cruzi invades a variety of cells that express receptor tyrosine kinases (RTK) TrkA, TrkB and/or TrkC, which includes astrocytes, Schwann cells, enteric glial cells, neurones, dendritic cells and smooth muscle cells [1–6]. Trk receptors A, B and C are activated primarily by the neurotrophins nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3), respectively, in development and repair of vertebrate nervous systems [7, 8]. Trks have a conserved amino acid sequence and a common structure, which consists of a multi-domain extracellular domain with a membrane-proximal immunoglobulin (Ig)-like unit that bears the neurotrophin-binding site, a single trans-membrane domain and an evolutionary conserved intracellular tyrosine kinase.
Earlier studies revealed a unique link between T. cruzi and neurotrophins: the parasite trans-sialidase (TS) moon-lights as TrkA ligand in triggering survival and differentiation of neuronal cells independently of intrinsic enzymatic activities [9–12], analogous to also sugar-binding enzyme, phosphoglucose isomerase that can additionally function as growth factor for spinal and sensory neurones [13, 14]. Because this parasite-derived neurotrophic factor (PDNF) is a competitive inhibitor of NGF binding to TrkA, and because NGF blocks PDNF/TrkA interaction [10], may be PDNF and NGF share structural determinants to compete with each other in their binding to TrkA. Given that their primary and 3D structures are seemingly unrelated to each other [15, 16] and that PDNF is immunogenic in T. cruzi-infected individuals [17, 18], we hypothesized that a subset of the PDNF antibody repertoire, shaped through trypanosome exposure to the immune system for many years, should identify subtle PDNF configurations common to NGF or NGF receptor TrkA. In testing this hypothesis we discovered a symphony of autoantibodies in the sera of chagasic sera that are specific for neurotrophin receptors TrkA, TrkB and TrkC (ATA). Because ATA react with a cell-surface neurotrophic receptor exploited by T. cruzi to promote host cell survival and invasion [10, 19], the results raise the possibility of Trk receptor autoimmunity influencing Chagas’ disease pathogenesis.
Materials and methods
Sera from T. cruzi-infected and non-infected individuals
We screened 25 non-chagasic (i.e. seronegative for T. cruzi) and 29 chagasic (i.e. seropositive for T. cruzi) sera (total = 54 sera): seven non-chagasic and 16 chagasic at an uncharacterized clinical stage (UnCh) were from Laboratório Emilio Ribas in Fortaleza, Brazil, and 31 sera, used in an earlier study [20], were from Laboratório de Pesquisa de Doença de Chagas, Goiania, Brazil, made up of 18 non-chagasic and 13 chagasic patients [one UnCh, four IND, four with megacolon (MC), and one each with megacolon and megaoesophagus (MC + ME), cardiomyopathy (Myo), megacolon and cardiomyopathy (MC + Myo), megaoesophagus, megacolon and cardiomyopathy (MC + ME + Myo)]. The age of infected patients ranged from 36 to 69 years and non-chagasic individuals from 6 to 56 years. Sexes were equally distributed between chagasic and non-chagasic donors. Ethical approval was obtained from the Human Investigation Review Committee of Tufts-New England Medical Center and Federal University of Goiàs, Brazil.
Purification of ATA and PDNF antibodies
Ten micrograms of ECD of TrkA-Fc, TrkB-Fc or TrkC-Fc, and receptors without Fc (R&D Systems, Minneapolis, MN, USA) were run by SDS-PAGE and transferred to a nitrocellulose membrane. The location of the receptor was determined by cutting thin strips from each side of the membrane and developing with alkaline phosphatase (AP)-labelled antibody to Fc (for Trk-Fc receptors) or with primary antibody to TrkA, TrkB and TrkC (for non-chimera antibodies) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and to AP-labelled secondary antibody. The membrane was cut horizontally on the edges of the region that contained the receptors, incubated with protein G-purified IgG from chagasic sera overnight at 4 °C with gentle shaking, washed twice with 50 mM Tris-buffered saline (pH 7.2) (TBS), eluted (1 min) with 50 mM glycine-HCl buffer (pH 2.5), the eluate (i.e. ATA) immediately neutralized in 0.1 M TBS (pH 8.0), concentrated by ultrafiltration (Millipore Corporation, Billerica, MA, USA), diluted in glycerol (final concentration, 30%), filtered in 0.22 μm membranes, and stored at 4 °C until use. This protocol adsorbed >90% of anti-Trk antibodies from IgG as determined by ELISA. Antibody concentration was determined by absorption at 280 nm in a spectrophotometer or ELISA (Cisbio, Bedford, MA, USA). Affinity-purified PDNF antibodies were isolated as described for ATA except bacterially produced recombinant PDNF (rPDNF) (5 μg) [9] was blotted to nitrocellulose instead of Trk. In some experiments ATA were adsorbed to T. cruzi lysates (10 μg of 1% Triton X-100 extract) blotted to nitrocellulose. The concentration of ATA was determined by: (1) ELISA (see below) and (2) immunoblotting [21]. The amount of ATA was calculated based on the human IgG standard. ATA concentration determined by the two methods did not vary by >10%.
ELISA, avidity and immunofluorescence
Microtitre wells were coated overnight (4 °C, 400 ng/ml neurotrophic factors) (R&D Systems) as described earlier [21], blocked with 5% goat serum (2 h, 37 °C), followed by chagasic sera diluted at 1:200 (unless otherwise indicated) in 5% bovine serum albumin/phosphate-buffered saline (PBS) pH 7.2, washed, and developed with AP-labelled secondary relevant antibody. ATA isotyping was performed by ELISA with commercially available kits (Sigma-Aldrich, St Louis, MO, USA) based on mouse mAb to human IgG isotypes and goat antibodies specific for IgA and IgM. To calculate the concentration of ATA by ELISA, microtitre wells were coated with various concentrations of ATA or standard human IgG (overnight, 4 °C), blocked with 5% goat serum in PBS containing 0.1% Tween-20 (37 °C, 1 h), washed in PBS/Tween-20 buffer and developed with AP-conjugated goat antihuman antibody (Promega, Madison, WI, USA). Immunofluorescence of PC12wt and PC12nnr5 cells was performed as described earlier [10] except that PC12 cells used here were differentiated with NGF (10 ng/ml, 4 days).
Results
Identification of antibodies against the extracellular domain of Trk receptors in the sera of patient with Chagas’ disease
We tested the sera by ELISA using microtitre plates coated with recombinant extracellular domains (ECD) of Trks A, B and C, hereafter referred to as TrkA, TrkB and TrkC unless indicated otherwise. TrkA, TrkB and TrkC expressed in the mouse myeloma cell NS0 are a disulphide-linked homodimeric glycosylated protein fused to the Fc fragment of human IgG1.
We calculated a cut-off of 0.22, 0.18, and 0.31 optical density (OD) units for TrkA, TrkB, and TrkC, respectively, based on the mean + 3 SD of the values with non-chagasic sera (Fig. 1A). Statistically significant elevated ATA titres against TrkA, TrkB and TrkC were found in 26 of the 28 chagasic sera (92%) (P < 0.001), including those from IND, MC, ME and Myo patients, with mean titres of 1.37 ± 0.84, 1.32 ± 0.82 and 1.95 ± 0.96 respectively (Fig. 1A, B). The two chagasic sera seronegative for TrkA were also seronegative for Trks B and C, suggesting that antibodies against Trk receptors cross-react with each other (see below). Chagasic sera reacted with TrkB and TrkC indistinguishably from receptors devoid of Fc (data not shown). The autoantibodies were Trk-specific because the 28 chagasic sera did not react with the ECD of five other neurotrophic receptors [pan-neurotrophin receptor (p75NTR-Fc); glial cell line-derived neurotrophic factor (GDNF) family ligands (GFL) receptor α1 (GFRα1 tyrosine kinase signalling GFL co-receptor (c-Ret); transforming growth factor-β receptor II (TGF-β-II-Fc); and fibroblast growth factor receptor (FGFR-Fc)] (Fig. 1A). Furthermore, chagasic sera did not react with neurotrophins NGF, BDNF, NT-3, NT-4, and with other growth factor receptor ligands –FGF, epidermal growth factor (EGF) and TGF-β1 (data not shown).
Figure 1.

Identification of antibodies to Trk receptors in the sera of patients with Chagas’ disease. (A) Screening of antibodies to TrkA, TrkB or TrkC by ELISA in 25 chagasic sera and 29 non-chagasic sera. Chagasic sera were also screened with five other growth factor receptors (p75NTR, GFR-α1, c-Ret, TGF-βR-II and FGFR). (B) Titration by ELISA of one non-chagasic and one chagasic (megacolon) chagasic serum against TrkA, TrkB and TrkC. (C) Immunoblotting of PC12wt and PC12nnr5 cell lysates with commercial rabbit antibody to TrkA (α-TrkA) and with chagasic (megacolon) TrkA-affinity-purified antibodies (ATA). Blot shows reaction (arrows) of the antibody with the fully glycosylated (140 kDa) and precursor (110 kDa) TrkA isoforms. (D) Fluorescence microscopy of PC12wt cells (a) and PC12nnr5 cells (b) immunostained with ATA (IND phase) followed by Alexa Fluor 594-labelled goat anti-human antibody. Panels (c) and (d) show the reaction of Alexa-labelled second antibody with PC12wt cells without ATA and with chagasic affinity-purified PDNF antibodies. ×400. (E) Cross-reaction of TrkA-, TrkB- and TrkC-affinity purified ATA with Trks A, B and C, but not with c-Ret by ELISA. Antibodies were isolated from IND IgG [same donor as in (D)] on solid-phase TrkA (ATA/TrkA), TrkB (ATA/TrkB) and TrkC (ATA/TrkC), and reacted by ELISA with TrkA, TrkB, TrkC and c-Ret.
In addition to binding recombinant Trks, ATA also bound endogenous full-length TrkA in cell lysates and on intact cells. Affinity-purified ATA (see Materials and methods) reacted with lysates of TrkA-bearing neuronal PC12 cells by immunoblot, identifying the mature fully glycosylated 140-kDa receptor and the 110-kDa N-glycosylated precursor [22], similar to commercial rabbit anti-TrkA antibodies (Fig. 1C), and coherent with the inability of the antibodies to bind TrkA-deficient PC12nnr5 cell lysates (Fig. 1C) [23]. Immunofluorescence likewise revealed that ATA stained PC12 cells (Fig. 1D-a) but not PC12nnr5 cells (Fig. 1D-b). ATA isolated on a TrkA matrix reacted equally well with the three Trks in a binding pattern similar to that of ATA isolated from TrkB or TrkC affinity matrices (Fig. 1E); in all cases, ATA did not bind unrelated neurotrophic receptor c-Ret (Fig. 1E). The binding pattern from 12 other chagasic sera from chagasic patients in the various phases of chronic disease was analogous to the result of serum in Fig. 1E (data not shown). We conclude that sera from most chagasic individuals analysed here bear autoantibodies against Trks A, B and C but not against five other neurotrophic receptors and with a variety of growth factor receptor ligands.
ATA are IgG2κ antibodies that do not cross-react with Trk carbohydrate, T. cruzi or PDNF
Because antibody isotype may provide clues to biological function of antibodies [24], we determined the isotype of ATA by an ELISA sandwich assay in which substratum-bound Trk–antibody complexes were identified with isotype-specific mouse monoclonal and goat polyclonal antibodies. Analysis of a serum from a patient with cardiomyopathy (Myo) showed that the heavy and light chains of ATA belong to the IgG2 (Fig. 2A) and κ subtypes (data not shown), respectively, similar to the reaction of sera from patients in chronic indeterminate (asymptomatic) disease, megacolon and megaoesophagus (Fig. 2B).
Figure 2.

ATA are IgG2 antibodies. Isotyping of ATA from the sera of a patient with cardiomyopathy (A), indeterminate phase, megacolon and megaoesophagus (B), tested by ELISA in microtitre plates coated with TrkA, TrkB or TrkC, and developed with mouse monoclonal antibodies against IgG subtypes followed by alkaline phosphatase-conjugated anti-mouse antibodies, and with alkaline phosphatase-conjugated IgA- and IgM-specific antibodies.
Consistent with other human IgG2 antibodies [24, 25], ATA formed covalent dimers (i.e. resistant to 2.3% SDS and boiling) of ~300 kDa (Fig. 3A). The mean concentration of ATA in three chagasic sera was 58.8 ± 20.7 μg/ml or 0.5% IgG (Table 1), in the lower range of other autoantibodies such as soluble nuclear antigens in systemic lupus erythematosus [26]. Unlike most human IgG2 antibodies, which react with carbohydrate epitopes [24], ATA did not require such epitopes on Trks because they were equally effective in binding bacterially expressed (and thus unglycosylated) and myeloma cell-expressed (and thus glycosylated) receptors (Fig. 3B).
Figure 3.
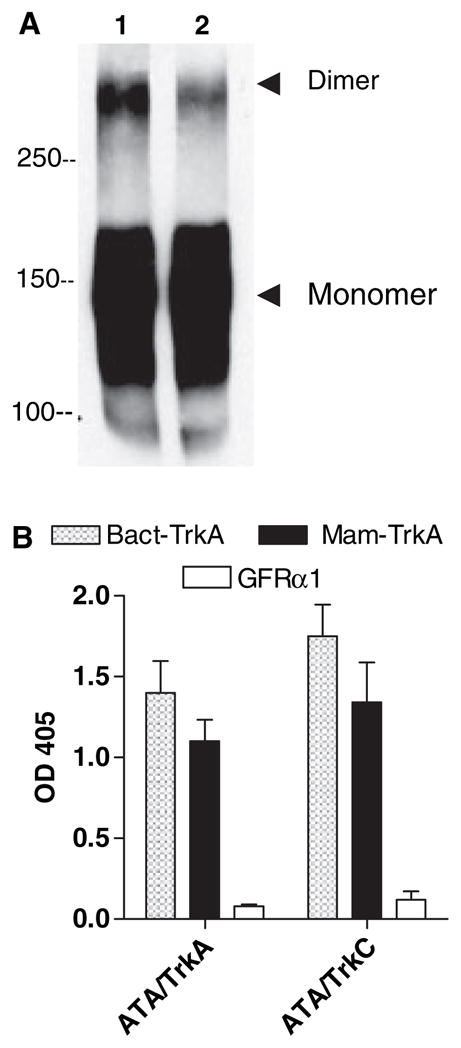
ATA form dimers and do not react with Trk-bound carbohydrate. (A) Analysis of ATA (IND) for the presence of dimers by non-reducing SDS-PAGE on 5% gels visualized on immunoblotting with goat anti-human IgG antibody followed by HRP-conjugated secondary antibody. (B) Reaction by ELISA of ATA isolated on solid-phase TrkA (ATA/TrkA) or TrkC (ATA/TrkC) with recombinant TrkA expressed in Escherichia coli (Bact-TrkA) or myeloma cells (Mam-TrkA), and with negative control GFRα1 receptor.
Table 1.
Estimate of ATA in chagasic sera.
| Chagasic serum | α-Trk (μg/ml serum)a | α-Trk (μg/mg IgG) |
|---|---|---|
| MC + ME + Myo | 34 | 2.7 |
| IND | 42.7 | 3.8 |
| UnCh | 100 | 8.7 |
MC + ME + Myo, serum of patient with megacolon, megaoesophagus and cardiomyopathy; IND, serum of patient in the indeterminate phase; UnCh, serum of patient in an uncharacterized stage of the disease.
Mean: 58.8 ± 20.7 (μg/ml serum ± SEM) and 5.1 ± 1.8 (μg α-Trk IgG2/mg IgG ± SEM) (0.5% IgG).
Antibodies to PDNF (isolated from chagasic sera) did not bind PC12 cells and TrkA by immunofluorescence (Fig. 1D-d) and ELISA (data not shown), respectively, implying that the antibodies did not identify PDNF structures in common with TrkA. In agreement with this explanation, ATA did not react with T. cruzi by ELISA and immunofluorescence under conditions in which they reacted robustly with Trks (Fig. 4A, 4B-a and 4B-b). Maintenance of ATA titres against Trks after adsorption to T. cruzi lysate (Fig. 4A) further confirmed the inability of the autoantibodies to significantly react with T. cruzi.
Figure 4.

ATA do not cross-react with Trypanosoma cruzi and PDNF. (A) Reaction of protein G-purified non-chagasic IgG, chagasic IgG (megacolon), ATA (from same megacolon IgG) (300 ng/ml) before and after adsorption on T. cruzi lysate (T. cruzi-ads ATA) with microtitre wells coated with TrkA, TrkB, TrkC, recombinant PDNF (rPDNF/TS), endogenous PDNF/TS (ePDNF) and T. cruzi lysate. ns, statistically insignificant differences (P > 0.05) between ATA and T. cruzi-ads ATA. (B) Staining by immunofluorescence of paraformaldehyde-fixed T. cruzi with chagasic (megacolon) IgG (a) and ATA (0.85 μg/ml) (b) followed by secondary Alexa594-labelled antihuman antibody.
Discussion
There are about 20 families of RTKs, all of which are critically important to many features of mammalian development and adult physiology, and they share a common structure: a ligand-binding extracellular domain, a single trans-membrane domain and an evolutionarily conserved intracellular tyrosine kinase domain. Mutations in these receptors underlie an increasing number of inherited human disorders such as loss-of-function mutations in TrkA that leads to congenital insensitivity to pain with anhydrosis (CIPA), and a gain-of-function mutation in tyrosine kinase with Ig and EGF (TIE) gene, which results in venous malformation [27]. Autoimmunity to the receptors could be another potential mechanism linking altered RTK function and disease pathogenesis. Indeed, such a link has been established in some cases, as in the cause–effect relationship between autoantibodies to muscle-specific tyrosine kinase and myasthenia gravis [28]. However, autoantibodies against neurotrophic RTKs have not been reported in the literature that describes serum antibodies against a smorgasbord of central nervous system antigens in patients with a variety of neurodegenerative diseases [29]. Thus, this study identifies, for the first time, specific autoantibodies against neurotrophin receptors of the RTK family (TrkA, TrkB and TrkC) (ATA) in the sera of patients with chronic Chagas’ disease; it could be a first step to determine whether Trk antibodies modify Trk biological function and underlie in Chagas’ disease progression. Trypanosoma cruzi elicits immune responses to many autoantigens, some of which, like ribosomal P protein [30], M2 muscarinic and β1 adrenergic receptors [31], myosin [32], and undefined antigens on neurones and nerves [33, 34], are believed to mediate Chagas’ disease pathogenesis.
The rationale that led to this study was the possible structural resemblance of the PDNF of T. cruzi with NGF and/or NGF-binding site of TrkA, as both PDNF and NGF promote TrkA-dependent survival and differentiation of neurones and T. cruzi invasion, and compete with each other in binding to TrkA [10]. Yet, several types of binding assays – ELISA, immunofluorescence and immunoblotting – failed to demonstrate binding of ATA to undenatured or denatured PDNF, and live or paraformaldehyde-fixed T. cruzi.
Thus, the mechanism that triggers the production of ATA remains to be determined, but molecular mimicry, often associated with autoimmune-prone infections with microbes, including T. cruzi, may not be one [32, 35] (Figs 1D, 2A and B). Trypanosoma cruzi infection causes intense inflammatory reactions and avid parasite invasion of most tissues, including Trk-rich central and peripheral nervous systems, and non-neural tissues. This could elicit immune reaction against cryptic epitopes and result in bystander activation and epitope spreading. A corollary of this epitope-spreading mechanism, which is thought to arise independently of the initial antigenic stimulus [36], is that ATA could exist in non-T. cruzi-related inflammatory diseases of the nervous system. It will be of interest to determine whether Trk antibodies are elicited in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease and Creutzfeldt-Jakob disease [29], and in diseases characterized by microbial invasion of the nervous system such as Lyme’s disease and toxoplasmosis.
The mammalian neurotrophins NGF, BDNF, NT-3 and NT-4 activate one or more of the three Trk family members, resulting in signalling through Ras, phosphatidyl inositol-3-kinase, phospholipase C-λ1 and pathways controlled by these proteins such as mitogen-activated kinases [8, 37]. Through these signalling pathways, neurotrophin/Trk interaction control survival, development, and function of neurones in both peripheral and central nervous systems, including axon and dendrite growth, expression of ion channels, transmitter biosynthetic enzymes and neuropeptides essential for normal neural function. In addition, neurotrophin/Trk interactions are required for proper function of adult nervous system, such as synaptic function and plasticity, and maintenance of neuronal survival, morphology and differentiation. Thus, it will be of interest to determine whether ATA have agonist or antagonist actions on the Trk receptors, for this could implicate a role in Chagas’ disease. Furthermore, given that ATA bind to a T. cruzi host cell receptor (TrkA), it will also be valuable to establish whether ATA interfere in the crosstalk between T. cruzi and host cells. Experiments are in progress in our laboratory to determine whether ATA interfere with TrkA-dependent T. cruzi invasion of cells and whether the autoantibodies are generated in acute Chagas’ disease.
Acknowledgments
We thank Eugene M. Johnson Jr and Angela Vincent for reading the manuscript and for providing helpful and insightful suggestions. We also thank Milena de Melo- Jorge for bacterially expressed TrkA. This work was supported by NIH Grants NS40574 and NS42960.
Footnotes
The authors have no conflicting financial interests.
References
- 1.Brener Z. Biology of Trypanosoma cruzi. Annu Rev Microbiol. 1973;27:347–82. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 2.Donovan MJ, Miranda RC, Kraemer R, et al. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am J Pathol. 1995;147:309–24. [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg I, Prioli RP, Ortega-Barria E, Pereira ME. Stage-specific phospholipase C-mediated release of Trypanosoma cruzi neuraminidase. Mol Biochem Parasitol. 1991;46:303–5. doi: 10.1016/0166-6851(91)90054-a. [DOI] [PubMed] [Google Scholar]
- 4.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–81. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 5.Tafuri WL. Pathogenesis of lesions of the autonomic nervous system of the mouse in experimental acute Chagas’ disease. Light and electron microscope studies. Am J Trop Med Hyg. 1970;19:405–17. doi: 10.4269/ajtmh.1970.19.405. [DOI] [PubMed] [Google Scholar]
- 6.Vega JA, Garcia-Suarez O, Hannestad J, Perez-Perez M, Germana A. Neurotrophins and the immune system. J Anat. 2003;203:1–19. doi: 10.1046/j.1469-7580.2003.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–37. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- 8.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–42. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 9.Chuenkova MV, Pereira MA. A trypanosomal protein synergizes with the cytokines ciliary neurotrophic factor and leukemia inhibitory factor to prevent apoptosis of neuronal cells. Mol Biol Cell. 2000;11:1487–98. doi: 10.1091/mbc.11.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuenkova MV, PereiraPerrin M. Chagas’ disease parasite promotes neuron survival and differentiation through TrkA nerve growth factor receptor. J Neurochem. 2004;91:385–94. doi: 10.1111/j.1471-4159.2004.02724.x. [DOI] [PubMed] [Google Scholar]
- 11.Chuenkova MV, PereiraPerrin M. A synthetic peptide modeled on PDNF, Chagas’ disease parasite neurotrophic factor, promotes survival and differentiation of neuronal cells through TrkA receptor. Biochemistry. 2005;44:15685–94. doi: 10.1021/bi0512039. [DOI] [PubMed] [Google Scholar]
- 12.Chuenkova MV, PereiraPerrin M. Enhancement of tyrosine hydroxylase expression and activity by Trypanosoma cruzi parasite-derived neurotrophic factor. Brain Res. 2006;1099:167–75. doi: 10.1016/j.brainres.2006.04.128. [DOI] [PubMed] [Google Scholar]
- 13.Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24:8–11. doi: 10.1016/s0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- 14.Petsko GA, Ringe D. Protein Structure and Function. London: New Science Press Ltd; 2004. pp. 156–7. [Google Scholar]
- 15.Buschiazzo A, Amaya MF, Cremona ML, Frasch AC, Alzari PM. The crystal structure and mode of action of trans-sialidase, a key enzyme in Trypanosoma cruzi pathogenesis. Mol Cell. 2002;10:757–68. doi: 10.1016/s1097-2765(02)00680-9. [DOI] [PubMed] [Google Scholar]
- 16.McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature. 1991;354:411–4. doi: 10.1038/354411a0. [DOI] [PubMed] [Google Scholar]
- 17.Leguizamon MS, Campetella O, Russomando G, et al. Antibodies inhibiting Trypanosoma cruzi trans-sialidase activity in sera from human infections. J Infect Dis. 1994;170:1570–4. doi: 10.1093/infdis/170.6.1570. [DOI] [PubMed] [Google Scholar]
- 18.Pereira-Chioccola VL, Schenkman S, Kloetzel JK. Sera from chronic Chagasic patients and rodents infected with Trypanosoma cruzi inhibit trans-sialidase by recognizing its amino-terminal and catalytic domain. Infect Immun. 1994;62:2973–8. doi: 10.1128/iai.62.7.2973-2978.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Melo-Jorge M, PereiraPerrin M. The Chagas’ disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell Host Microbe. 2007;1:251–61. doi: 10.1016/j.chom.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Luquetti AOaR A. Diagnóstico laboratorial da infecção pelo Trypanosoma cruzi (Diagnosis of Trypoanosoma cruzi infection by serological assays) Chapter 17. Rio de Janeiro, Brazil: Guanabara Koogan; 2000. pp. 344–78. [Google Scholar]
- 21.Saavedra E, Herrera M, Gao W, Uemura H, Pereira MA. The Trypanosoma cruzi trans-sialidase, through its COOH-terminal tandem repeat, upregulates interleukin 6 secretion in normal human intestinal microvascular endothelial cells and peripheral blood mononuclear cells. J Exp Med. 1999;190:1825–36. doi: 10.1084/jem.190.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jullien J, Guili V, Reichardt LF, Rudkin BB. Molecular kinetics of nerve growth factor receptor trafficking and activation. J Biol Chem. 2002;277:38700–8. doi: 10.1074/jbc.M202348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green SH, Rydel RE, Connolly JL, Greene LA. PC12 cell mutants that possess low- but not high-affinity nerve growth factor receptors neither respond to nor internalize nerve growth factor. J Cell Biol. 1986;102:830–43. doi: 10.1083/jcb.102.3.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabat EA. Structural Concepts in Immunology and Immunochemistry. New York: Holt, Rihehart and Winston; 1976. p. 547. [Google Scholar]
- 25.Yoo EM, Wims LA, Chan LA, Morrison SL. Human IgG2 can form covalent dimers. J Immunol. 2003;170:3134–8. doi: 10.4049/jimmunol.170.6.3134. [DOI] [PubMed] [Google Scholar]
- 26.Maddison PJ, Reichlin M. Quantitation of precipitating antibodies to certain soluble nuclear antigens in SLE. Arthritis Rheum. 1977;20:819–24. doi: 10.1002/art.1780200310. [DOI] [PubMed] [Google Scholar]
- 27.Robertson SC, Tynan J, Donoghue DJ. RTK mutations and human syndromes: when good receptors turn bad. Trends Genet. 2000;16:368. doi: 10.1016/s0168-9525(00)02077-1. [DOI] [PubMed] [Google Scholar]
- 28.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365–8. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- 29.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease – a double-edged sword. Neuron. 2002;35:419–32. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 30.Mesri EA, Levitus G, Hontebeyrie-Joskowicz M, Dighiero G, Van Regenmortel MH, Levin MJ. Major Trypanosoma cruzi antigenic determinant in Chagas’ heart disease shares homology with the systemic lupus erythematosus ribosomal P protein epitope. J Clin Microbiol. 1990;28:1219–24. doi: 10.1128/jcm.28.6.1219-1224.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borda ES, Sterin-Borda L. Antiadrenergic and muscarinic receptor antibodies in Chagas’ cardiomyopathy. Int J Cardiol. 1996;54:149–56. doi: 10.1016/0167-5273(96)02592-2. [DOI] [PubMed] [Google Scholar]
- 32.Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity. 2006;39:41–54. doi: 10.1080/08916930500485002. [DOI] [PubMed] [Google Scholar]
- 33.Khoury EL, Ritacco V, Cossio PM, et al. Circulating antibodies to peripheral nerve in American trypanosomiasis (Chagas’ disease) Clin Exp Immunol. 1979;36:8–15. [PMC free article] [PubMed] [Google Scholar]
- 34.Ribeiro dos Santos R, Marquez JO, Von Gal Furtado CC, Ramos de Oliveira JC, Martins AR, Koberle F. Antibodies against neurons in chronic Chagas’ disease. Tropenmed Parasitol. 1979;30:19–23. [PubMed] [Google Scholar]
- 35.Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108:1097–104. doi: 10.1172/JCI14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 37.Reichardt LF. Neurotrophin-regulated signaling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]