

Summary
TGF-β plays an important role in the induction of regulatory T cells (Treg) and maintenance of immunologic tolerance, but whether other members of TGF-β superfamily act together or independently to achieve this effect is poorly understood. Although others have reported that the bone morphogenetic proteins (BMP) and TGF-β have similar effects on the development of thymocytes and T cells, in this study, we report that members of the BMP family, BMP-2 and BMP-4, are unable to induce non-regulatory T cells to become Foxp3+ Treg. Neutralization studies with Noggin have revealed that BMP-2/4 and the BMP receptor signaling pathway is not required for TGF-β to induce naive CD4+CD25- cells to express Foxp3; however, BMP-2/4 and TGF-β have a synergistic effect on the induction of Foxp3+ Treg. BMP-2/4 affects non-Smad signaling molecules including phosphorylated ERK and JNK, which could subsequently promote the differentiation of Foxp3+ Treg induced by TGF-β. Data further advocate that TGF-β is a key signaling factor for Foxp3+ Treg development. In addition, the synergistic effect of BMP-2/4 and TGF-β indicates that the simultaneous manipulation of TGF-β and BMP signaling might have considerable effects in the clinical setting for the enhancement of Treg purity and yield.
Keywords: BMP, TGF-β, Foxp3, regulatory T cells
Introduction
A subset of CD4+ regulatory T cells (Treg), expressing the IL-2 receptor α chain (CD25) and the nuclear transcription factor Foxp3, plays an important role in the maintenance of immune homeostasis and prevention of autoimmunity [1, 2]. These cells comprise thymus-derived, naturally-occurring CD4+ Treg (nTreg) and those that can be induced in the periphery (induced Treg, iTreg) [3]. Although Treg subsets may have different developmental mechanisms, they share similar phenotypes and suppressive activities [4]. These Treg populations may have a synergistic action or have different targets that maintain immune homeostasis [5].
Transforming growth factor β (TGF-β) is essential for the induction of Foxp3 expression and for suppressive activity in iTreg, as well as the maintenance of both Foxp3+ nTreg and iTreg [6-11]. It is still unclear whether other molecules except TGF-β also promote the development of Foxp3+ iTreg. In addition, the TGF-β signaling pathway is not essential for the development of nTreg since both TGF-β and TGF-β receptor II knockout mice express Foxp3+ nTreg in the thymus [10, 12, 13]. It has been suggested that other member(s) of TGF-β superfamily such as bone morphogenetic proteins (BMP) might substitute for TGF-β during the development of Treg [14].
BMP, with more than 20 family members, have been shown to regulate many fundamental biological processes including cell proliferation, differentiation, apoptosis, migration, and adhesion [15]. Furthermore, BMP are involved in the development of almost all tissues and organs, including thymocytes and hematopoietic stem cells [16]. Recent studies have revealed that transgenic expression of the BMP antagonist Noggin in thymic epithelial cells results in the development of a smaller-sized thymus [17]. In a manner similar to that of TGF-β, treatment of purified human CD34+CD38- cells with BMP-2, -4 or -7 not only affected their own proliferation and differentiation, but exerted a direct effect on human stem cell survival as well [18]. As extracellular growth factors, BMP bind to heteromeric complexes of BMP serine/threonine kinase type I (Alk2, Alk3, and Alk6) and type II receptors [19, 20]. Upon ligand-induced aggregation of the receptors, constitutively activated BMP type II receptor kinase phosphorylates and activates the type I receptor, which subsequently recognizes and phosphorylates receptor-bound BMP specific Smad proteins (Smad1, Smad5, and Smad8) on the carboxyl terminal SSXS motif. These Smads dissociate from the receptors, form complexes with Smad4, translocate into the nucleus, bind to BMP responsive element and act as transcriptional co-modulators to induce or repress BMP-target gene expression [21]. BMP-2 and BMP-4 are two common forms and typical representatives of BMP that affect the T cell development in the thymus [22]. CD4+ cells have been found to express BMP receptors I and II and treatment of BMP induced CD4+ cells to express phosphorylation of Smad1, Smad5 and Smad8 [23].
It still remains unclear whether BMP affect the development of Foxp3+ Treg. As BMP-2-/-, BMP-4-/- and their downstream molecule Smad5-/- mice all die at mid-gestation due to multiple embryonic and extraembryonic defects [24, 25], it is difficult to determine their role in the development of nTreg. To learn the role of BMP signaling in the induction of iTreg, we here have demonstrated that both BMP-2 and BMP-4 are unable to substitute for TGF-β in the differentiation of Foxp3+ iTreg. Addition of BMP-2 or BMP-4 to TCR-activated CD4+ cells failed to induce the expression of Foxp3 and the development of suppressive activity. However, the addition of BMP-2 and BMP-4 significantly increased the ability of TGF-β to promote the generation of Foxp3+ iTreg, and this synergistic effect was also dependent upon TGF-β receptor signaling, as well as ERK and/or JNK MAPK pathways. Moreover, lack of BMP receptor signaling did not impair the TGF-β-mediated induction of Foxp3+ iTreg. Our results demonstrate that TGF-β superfamily members, BMP and TGF-β, each may play a distinct role in modulating Treg development.
Results
BMP-2 and 4 are unable to induce Foxp3+ iTreg in the absence of TGF-β
We and others have reported previously that TGF-β can induce TCR-activated naïve CD4+CD25- cells to become CD4+CD25+Foxp3+ iTreg [6-9]. Because BMP-2 and BMP-4 both belong to the TGF-β superfamily and exert similar effects on the development of early thymocytes and peripheral lymphocytes as TGF-β [17, 18], we initially sought to determine if these members could mediate the induction of Foxp3+ in a manner similar to that of TGF-β. Unexpectedly, we found that the addition of BMP-2 or BMP-4 to TCR-activated naïve CD4+CD25- cells did not induce Foxp3 expression (Fig. 1A-C). In addition, the resultant CD4+CD25+ cells did not exhibit an anergic status, one of the hallmark features of Treg (Fig. 2A). Accordingly, they failed to suppress T cell proliferation (Fig. 2B&C). Conversely, addition of TGF-β1 induced the expected Foxp3 expression and these cells displayed typical anergic status and potent suppressive activities (Fig. 1A-C, Fig 2). Further experiments with time-course study showed that BMP-2 or BMP-4 alone was unable to induce Foxp3 expression over a week-long culture period (Fig. 1D), despite providing a wide variety of physiologic doses of BMP-2 or BMP-4 (Fig. 1E).
Figure 1.
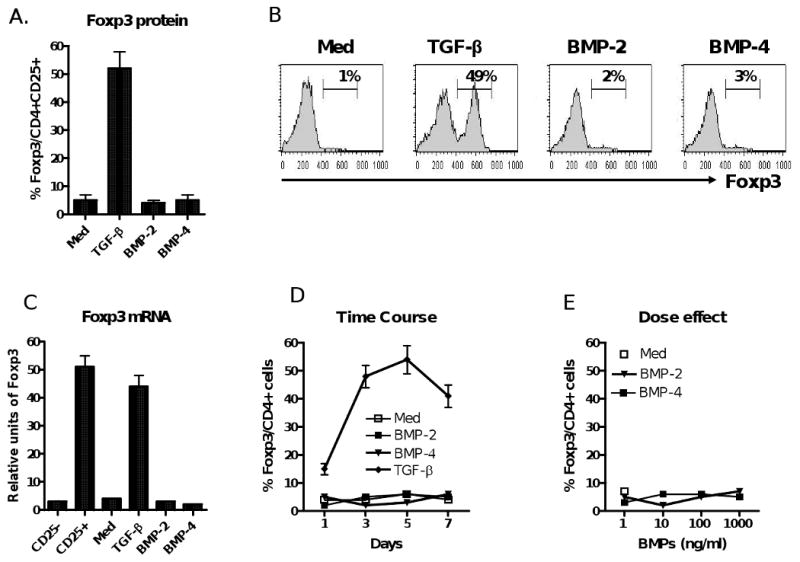
BMP-2 and BMP-4 fail to induce Foxp3 expression on CD4+ cells. (A) Splenic naive CD4+CD25- cells isolated from C57BL/6 mice were stimulated with anti-CD3/CD28 coated beads (1:5) in the presence of IL-2 (20 u/ml) and TGF-β (2 ng/ml), or BMP-2 (10 ng/ml), or BMP-4 (10 ng/ml) for 4 days. Intracellular Foxp3 protein expression was analyzed by flow cytometry. Data show mean ± SEM of five independent experiments. (B) Representative histogram of experiments described in panel (A). The percentage of CD25+ cells expressing Foxp3 is shown in the upper right of the histogram. (C) Foxp3 mRNA expression as determined by real time PCR after normalization of β-actin. Data show mean ± SEM of four independent experiments. (D) Foxp3 and CD25 expression over time using a similar experimental design as in A. (E) Dose effect of BMP-2 and BMP-4 on the induction of Foxp3 expression. Data from panel D and E show mean ± SEM of four independent experiments.
Figure 2.
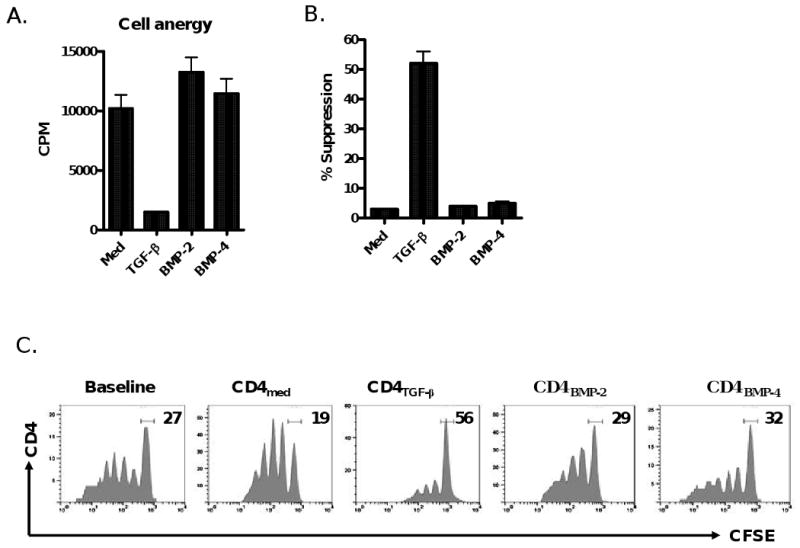
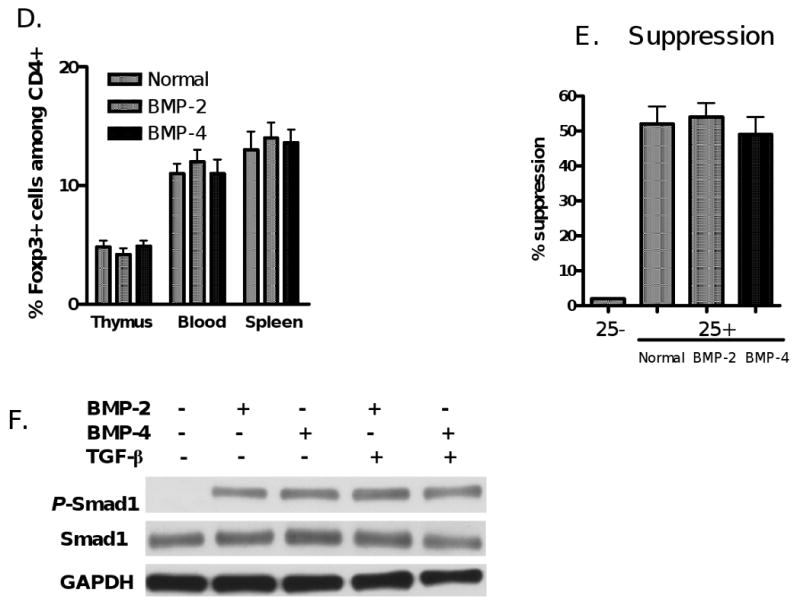
BMP-2/4-primed CD4+ cells are unable to display the phenotypes and function of suppressor cells. (A) BMP2/4- or TGF-β-primed CD4+ cells were induced as described in Fig. 1. These cells were restimulated with anti-CD3 (0.25 μg/ml) in the presence of APC (3000-rad γ irradiation) for three days and proliferation was assayed by 3H-thymidine incorporation. (B) CD4+CD25- T cells labeled with CFSE were stimulated with soluble anti-CD3 and APC, or with BMP-2-, BMP-4-, TGF-β-primed CD4+ or control (Med) cells for 4 days. The ratio of conditioned cells to responder T cells is1:4. The % suppression was calculated as previously reported [11]. (C). Cytometric data representative of four independent experiments in panel (B). (D & F). C57BL/6 mice received intraperitoneal injections of BMP-2 (0.5 mg/mouse), BMP-4 (0.5 mg/mouse) or control PBS every other day for 10 days. The frequency (D) and suppressive activities (E) of CD4+CD25+Foxp3+ cells in thymus, spleen and blood in these mice were determined at day 12 after administration using flow cytometry and a standard in vitro suppressive assay as above. Each group includes 4 mice and values indicate mean ± SEM. Note there is no significant difference between mice receiving BMP-2/4 or PBS. (F) Naïve CD4+ cells isolated from C57BL/6 mice were treated with BMP-2/4 with or without TGF-β for 2 hours and the Smad1 and phosphorylated Smad1 expression was determined by western blot. Data are representative of three independent experiments.
We next provided further evidence that treatment of BMP-2 or BMP-4 in mice does not change CD4+Foxp3+ cell frequency and function in vivo. As shown in Fig. 2D, we observed that percentages and total numbers (data not shown) of CD4+CD25+Foxp3+ cells were not changed at day 10 following i.p. injection of BMP-2 and BMP-4 compared to mice that received a similar volume of PBS. We observed a markedly increased expression of phosphorylated Smad1 in T cells at 6-24 hours post-injection of BMP-2/4 (not shown). It has been reported that BMP signal through serine/threonine kinase receptors and activate downstream molecules of BMP receptors such as Smad1, 5 and 8 [21, 23]. We also compared the suppressive activity of CD4+CD25+ cells on T cell responses, we found that CD4+CD25+ cells sorted from mice treated with BMP or PBS displayed similar suppressive activities on T cell proliferation (Fig. 2E). Taken together, these data indicate that BMP do not affect the differentiation of CD4+Foxp3+ Treg in vitro and in vivo.
Further experiments excluded the possibility that failure of iTreg induction by BMP is due to lack of endogenous BMP signaling activity in naïve CD4+CD25- cells. As reported before, some CD4+ cell subsets express BMP receptor I and II [23]. We observed that naïve CD4+CD25- cells expressed BMP receptor I (not shown). Moreover, BMP-2- or BMP-4-primed naïve CD4+CD25- cells exhibited a distinct expression of phosphorylated Smad1 and addition of TGF-β did not alter the level of phosphorylated Smad1 expression induced by BMP-2 or BMP-4 (Fig. 2F).
Induction of Foxp3+ by TGF-β is independent upon BMP receptor signal
With evidence suggesting that BMP-2 and BMP-4 alone do not promote the differentiation of Foxp3+ iTreg, we next explored whether BMP signaling affects the induction of Foxp3+ iTreg by TGF-β. It has been reported that Noggin is a BMP antagonist that inhibits the binding of BMP to their cognate receptor [26]. In this study, we observed that addition of Noggin did not alter the TGF-β-induced Foxp3 expression (Fig. 3A) nor didn't affect their suppressive activities (Fig. 3B). In fact, we have observed that addition of Noggin significantly decreased the expression of phosphorylated Smad1 induced by BMP-4 (Fig. 3C). These results suggest that the endogenous BMP signal pathway alone plays no role in the differentiation of Foxp3+ iTreg.
Figure 3.
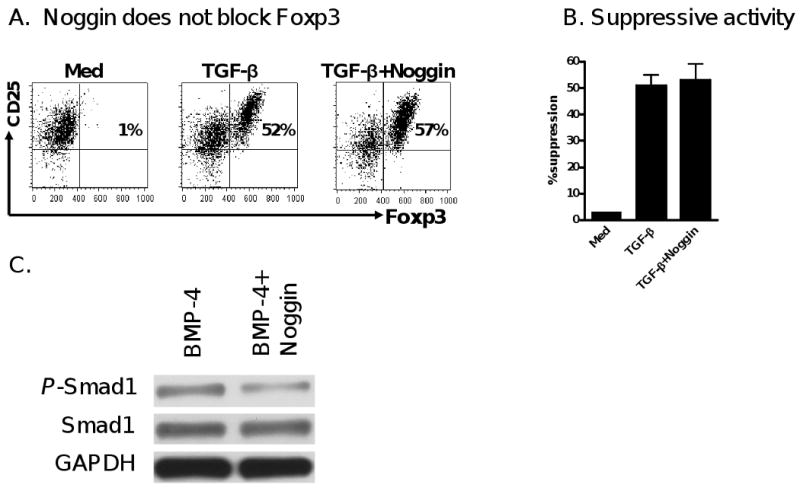
Induction of Foxp3+ by TGF-β is independent upon BMP receptor signal. (A) Foxp3 expression was determined in CD4+ cells treated with medium, TGF-β, and both TGF-β and Noggin treated CD4+ cells. Data are representative of three independent experiments. (B) The suppressive activity of these cells was analyzed by a standard suppressive assay as described in Fig 1B. (C) The effects of Noggin on the Smad1 activation of BMP-4 on CD4+ cells. Naïve CD4+ cells were stimulated with BMP-4 with or without Noggin for 2 hours and Smad1 and phosphorylated Smad1 expression were determined by western blot. Experiments were repeated twice with similar results.
Synergistic effect of exogenous BMP-2/4 and TGF-β on the induction of Foxp3+ iTreg
Unlike TGF-β, BMP-2/4 alone did not induce the differentiation of Foxp3+ iTreg. However, addition of exogenous BMP-2 or BMP-4 significantly increased the ability of TGF-β to promote the development of iTreg. As shown in Fig. 4A, naïve CD4+CD25- cells treated with BMP-2 or -4 in combination with TGF-β produced significantly higher Foxp3+ cell numbers than those treated with TGF-β alone in vitro. Moreover, CD4+ cells treated with the combination of both BMP-2/4 and TGF-β displayed more potent suppressive activity against T cell proliferation in vitro than cells treated with TGF-β alone (Fig. 4B). In an animal model of cGVHD with a typical lupus like syndrome, we found that co-transfer of BMP-4/TGF-β-treated CD4+ cells and pathogenic DBA/2 splenocytes more efficiently suppressed anti-DNA production than that of TGF-β-treated CD4+ and DBA/2 splenocytes in (DBA/2×C57BL/6) F1 mice (Fig. 4C).
Figure 4.
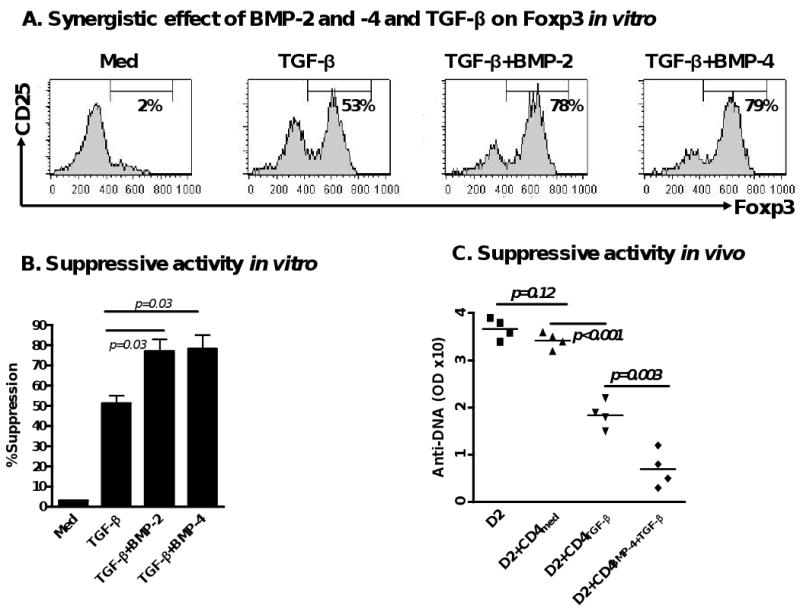
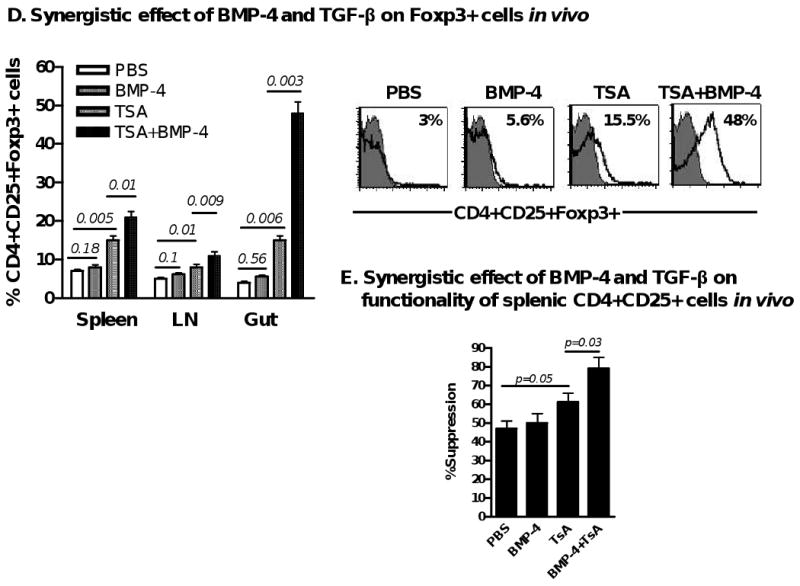
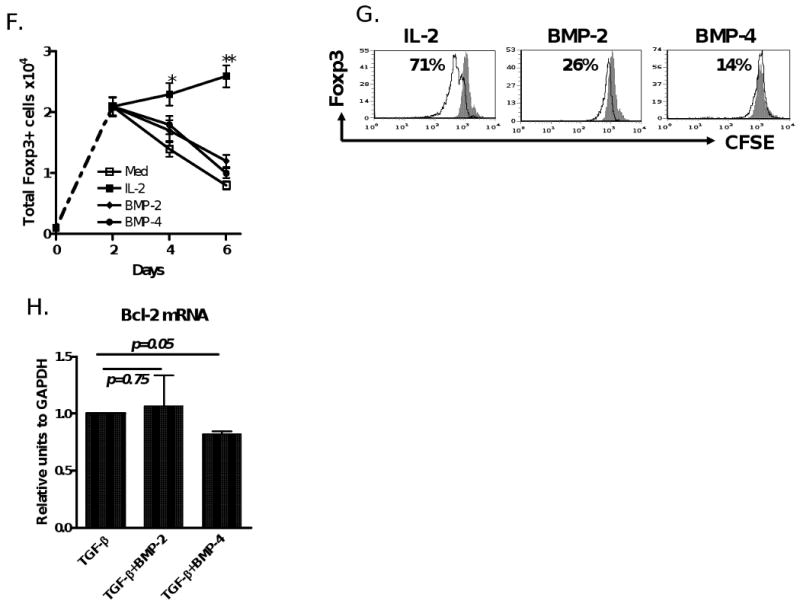
BMP-2/4 enhances TGF-β induced development of CD4+Foxp3+ Treg. (A) Effect of BMP-2/4 on the Foxp3 expression on CD4+ cells. Suppressive activities of Treg subsets against T cell proliferation in vitro (B) and anti-dsDNA production in cGVHD with a lupus syndrome model (n=4 mice each group) (C) were determined described the in the Materials and methods. Values are the mean ± SEM of three separate experiments (B). (D) The frequency of CD4+CD25+Foxp3+ cells in spleen, LN and gut of mice receiving PBS, or BMP-4, or TsA, or BMP-4+TsA at day 12 after treatment. Values indicate Mean ± SEM (left panel) of three mice in each group and representative of these experiments (right panel). Experiment was repeated with similar results. (E) Splenic CD4+CD25+ cells were sorted by FACSVantage and their suppressive activity against T cell proliferation in vitro was analyzed as panel A. (F and G). CD4+Foxp3+ cells were induced for 2 days as in Fig. 1 and labeled with CFSE, and stimulated with either IL-2 (40 units/ml), or BMP-2 (10 ng/ml), or BMP-4 (10 ng/ml) for additional 4 days. Total Foxp3+ cells were counted every two days (F) and Foxp3+ cell proliferation levels were analyzed by the dilution of CFSE dye on day 6 (G). Values indicate Mean ± SEM of three separate experiments (F) and the data presented are representative of these experiments (G). (H) Bcl-2 mRNA expression was determined by quantitative RT-PCR. Data show mean ± SEM of triplicate samples and experiment was performed with similar results.
We further investigated whether BMP and TGF-β exhibited a synergistic role in the induction of Foxp3+ Treg in vivo. As TGF-β has a very short half-life in vivo, it is not practical to evaluate synergistic effects of BMP and TGF-β following injection in vivo. However, recent studies indicated that trichostatin A (TsA), one of the histone deacetylase inhibitors, is able to promote induction and conversion of CD4+CD25- cells to Foxp3+ Treg in vivo [27, 28], we confirmed this result and found that the combination of TsA and BMP-4 administration resulted in significantly increased CD4+CD25+Foxp3+ cells in vivo than TsA treatment alone, particularly in the gut (Fig. 4D). Moreover, splenic CD4+CD25+ cells sorted from BMP-4- and TsA-treated mice displayed enhanced suppressive activity against T cell proliferation compared to TsA or BMP-4 alone treated mice (Fig. 4E). We also showed BMP-4 alone did not significantly increase Foxp3+ cell frequency in gut (Fig 4D) as well as other organs (Fig. 2D). Co-injection of BMP-2 and TsA had a similar result (data not shown). These data further support the synergistic effect of BMP on TGF-β-induced iTreg generation.
We next investigated the mechanisms by which BMP promote the Foxp3+ cell production induced by TGF-β. We previously have reported that induction of Foxp3 by TGF-β occurs only when this cytokine was added to cultures within 24 hours of TCR stimulation [11]. We now induced Foxp3+ cells for two days first and then labeled them with CFSE. These cells were extensively washed and exogenous IL-2, BMP-2, or BMP-4 were added to cultures to learn whether BMP-2 or BMP-4 can expand Foxp3+ cells in a manner like IL-2. Unlike IL-2, neither BMP-2 nor BMP-4 significantly expanded Foxp3+ cells that had been generated by TGF-β (Fig. 4F and G). Additionally, the addition of BMP-2/BMP-4 to the TGF-β did not increase the expression of Bcl-2 mRNA (Fig. 4H), one of key anti-apoptosis genes, suggesting BMP promote Foxp3+ cells through increasing conversion rather than expanding or prolonging the survival of these cells.
We also documented that the addition of the BMP antagonist Noggin significantly abolished the enhanced Foxp3 expression induced by BMP2/4+TGF-β (Fig. 5A). The synergistic effect of BMP-2/4 and TGF-β is also fully dependent upon TGF-β receptor signaling since addition of TGF-β receptor I (ALK5) inhibitor almost completely suppressed Foxp3 expression and this effect also disappeared when naïve CD4+CD25- cells from wild type mice were substituted from TGF-β receptor II knock out mice (Fig. 5B).
Figure 5.

Noggin blocks the BMP2/4 enhancement of TGF-β induced Foxp3 expression. Naïve CD4+CD25- cells isolated from C57BL/6 wild type and TGF-β RII knock out mice were stimulated as described in Fig. 1, in some cultures, Noggin (100 ng/ml) or ALK5 (TGF-β RI) inhibitor (5 μM) or DMSO control was added to cultures and Foxp3 expression was determined as above. Values indicate the mean ± SEM three independent experiments.
It seems unlikely that BMP-2 or -4 simply enhances the role of TGF-β in the induction of iTreg through activation of TGF-β receptor related Smad molecules since BMP-2 or -4 activates Smad1, 5, 8 whereas TGF-β activates Smad2 and Samd3. However, it should be noted that the Smad independent signaling molecule, P38, one of MAP kinases could be involved in the development of Foxp3+ iTreg induced by TGF-β [29]. In fact, we have observed both Smad and non-Smad signaling pathways are required for TGF-β to induce Foxp3+ iTreg (Lu L and Zheng SG. manuscript submitted). Thus, we next tested whether BMP-2 or 4 affects the MAPK expression. As shown in Fig. 6A, TCR-activated naïve CD4+CD25- cells cultured in the presence of BMP-4 did not induce p-Smad2/3 expression and did not enhance the p-Samd2/3 expression induced by TGF-β, however they did affect the expression of phosphorylated ERK1/2 and phosphorylated JNK expression although they had little influence on phosphorylated P-38 expression. As shown in Fig. 6B, either BMP-4 or TGF-β treated CD4+ cells expressed activated JNK in 12 hours and rapidly disappeared in 24 hours following TCR stimulation. Interestingly, both BMP-4 and TGF-β together began to induce activate JNK in 4 hours and significantly increased the expression of activated JNK in 12 hours and somewhat maintained its expression until 24 hours after TCR stimulation. Unlike JNK activation, BMP-4 treatment alone induced lower level of activated ERK in 24 hours, TGF-β treatment alone slightly increased ERK activation, interestingly, combination of both resulted in a marked induction of phosphorylated ERK in 12 to 24 hours although this expression significantly decreased in 48 hours after TCR stimulation (Fig. 6C). Time-course experiments revealed that ERK1/2 or JNK activation appears to be in the early stage of TCR/TGF-β stimulated CD4+ cells, further suggesting BMP promote Foxp3+ cell conversion rather than expansion (Fig.6B and C).
Figure 6.
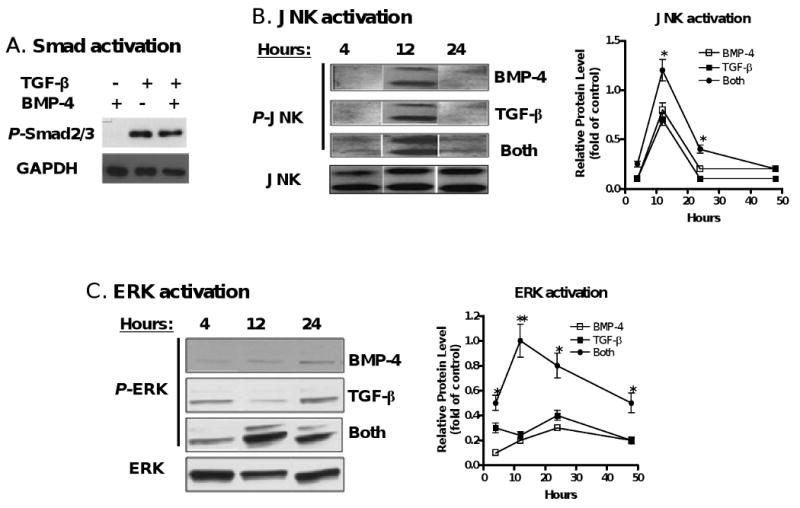
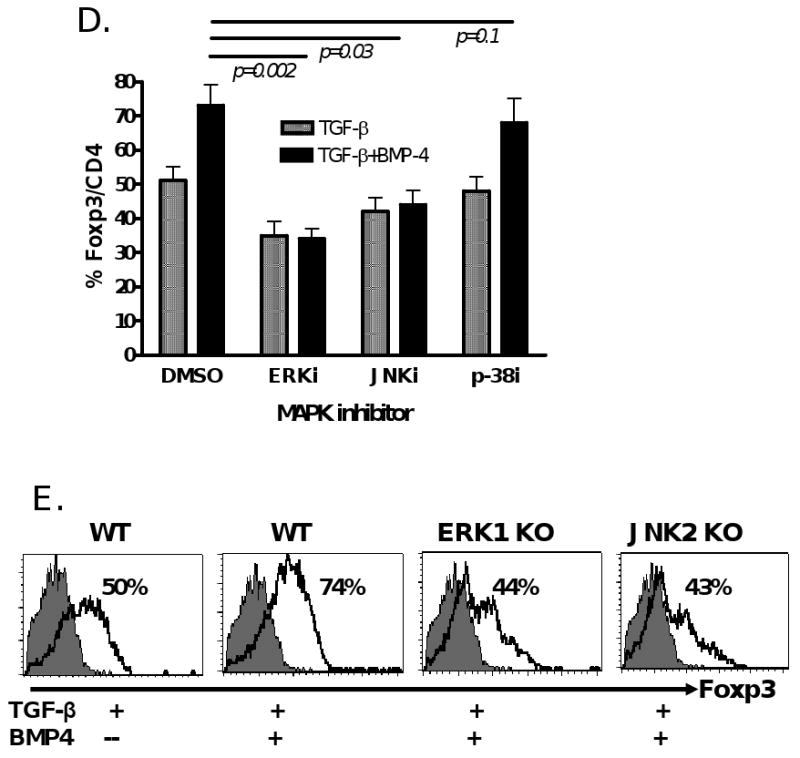

BMP2/4 on enhancement of TGF-β-induced CD4+Foxp3+ Treg development involves MAPK activation. (A) Naïve CD4+ cells were TCR (anti-CD3/CD28 coated bead 1:5) stimulated with or without BMP-4 ± TGF-β for 16 hours. Activated Smad2/3 expression was determined by western blot. These cells were stimulated with TCR in the presence of BMP-4, TGF-β or both (BMP-4+TGF-β) in various time-points as indicated and the levels of activated JNK (B) and ERK (C) were determined by western blot. (D) Foxp3 expression by TCR-stimulated CD4+ cells cultured with BMP-4 + TGF-β in the presence or absence of MAPK inhibitors. (E) Foxp3 expression by TCR-stimulated CD4+ cells cultured with BMP-4 ± TGF-β in ERK1 KO, JNK2 KO and littermate control mice for 4 days. (F) Foxp3 expression on CD4+ cells stimulated with TCR with BMP-2/4 ± TGF-β in IL-10 KO and littermate control mice for 4 days. In some experiments with wild type control mice, anti-IL-10, anti-IL-10R or control IgG were added to cultures. Data show mean ± SEM of three separate experiments or represent at least three independent experiments.
Of interest, when ERK or JNK inhibitors (PD 98059 or SP600125) but not p-38 inhibitor (SB203580) was added to cultures, the enhancement of Foxp3 expression by BMP-4 in the presence of TGF-β was completely abolished (Fig. 6D). This suppression can not be explained by the non-specific suppression since the CD25 expression by CD4+ cell and total viable CD4+ cell numbers were comparable between groups treated with MAPK inhibitors or with DMSO control (data not shown). It is noted that Foxp3 expression by BMP-4/TGF-β was even lower than that by TGF-β alone when ERK inhibitor was added to cultures, suggesting ERK activation also involves the development of Foxp3+ Treg induced by TGF-β. To further exclude the possibility that ERK or JNK inhibitor non-specifically suppressed T cell activation and interfered with Foxp3+ cell development, we have also observed the role of MAPK pathways in Foxp3+ iTreg promotion by BMP using ERK1 and JNK2 knock out mice. As shown in Fig. 6E, deficiency of ERK1 or JNK2 completely abrogated the enhanced Foxp3 production by BMP-4 in the TGF-β-primed CD4+ cells although these deficiencies did not interfere with CD25 expression by TCR-activated CD4+ cells, suggesting the synergistic effect of BMP and TGF-β depends upon either ERK or JNK MAPK pathway.
The ERK pathway is important for the production and function of IL-10 and IL-10 induces IL-10-producing type I Treg development [30], We sought to determine whether IL-10 plays a role in the synergistic role of BMP and TGF-β in the Foxp3+ iTreg induction. As shown in Fig. 6F, addition of IL-10 to the TGF-β did not significantly increase Foxp3 production. Neutralization of either IL-10 or IL-10 receptor with respective antibodies did not alter the enhanced Foxp3+ cell induction by a combination of BMP and TGF-β. Moreover, BMP similarly promoted Foxp3+ cell production in both wild type and IL-10 knock out mice, suggesting that IL-10 is redundant towards the synergistic role of BMP and TGF-β in Foxp3+ cell development. Collectively, these results indicate that BMP-induced ERK1/2 and/or JNK might either affect activated Smad molecules induced by TGF-β or other unknown molecules that then lead to the synergistic effect between TGF-β and BMP on the differentiation of Foxp3+ iTreg.
Discussion
Although both TGF-β and BMP similarly regulate the development of thymocytes and human hematopoietic cells [17, 18], we here show that the endogenous BMP signaling pathway is not essential for the induction of Foxp3+ iTreg. The addition of exogenous BMP-2/4 alone to TCR-activated CD4+CD25- cells fails to induce Foxp3 expression and confer immune suppressive activity. TGF-β is still capable of inducing Foxp3+ expression on naïve CD4+CD25- cells by treatment with the BMP antagonist Noggin. Although the addition of BMP-2/4 to TGF-β significantly increases the ability of the latter to induce iTreg development, this synergistic effect is also TGF-β-dependent since blockade of TGF-β receptor signaling abrogates the Foxp3 induction. Both BMP and TGF-β together enhances the expression of the activated ERK and JNK MAPK to promote Foxp3+ iTreg development. These findings indicate that TGF-β rather than BMP members play an essential role in the induction of Foxp3+ Treg despite both belonging to the TGF-β superfamily.
We and others have provided considerable evidence that TGF-β induces the differentiation of iTreg that express Foxp3 and suppress T cell immune response [6-9]. Adoptive transfer of iTreg can control many autoimmune diseases and protect heart transplantation allograft survival [31, 32]. However, other stimulation without TGF-β also seems to be able to induce Foxp3+ expression [33]. As BMP belongs to the TGF-β superfamily, we investigated whether BMP might substitute for TGF-β in this capacity. Our study demonstrates that both endogenous and exogenous BMP-2 and BMP-4 are not essential for the induction of Foxp3+ expression and suppressive capacity in these cells.
CD4+ cells express BMP receptors, and treatment of the cells with BMP-2/4 activates downstream Smad1/5/8 by protein phosphorylation, suggesting that the failure of Foxp3+ iTreg induction is not due to the inability of CD4+ cells to respond to BMP-2 or BMP-4. Additionally, the synergistic action of exogenous BMP-2 or BMP-4 with TGF-β on iTreg induction also indicates CD4+ T cells do indeed response to BMP-2 or BMP-4.
The synergistic action of exogenous BMP and TGF-β on iTreg induction could be explained by promoting the Foxp3+ conversion from non-Treg, or expanding the Foxp3+ cells and prolonging the survival of Foxp3+ cells that had been induced by TGF-β. We have previously found that TGF-β mainly converts Foxp3+ cells in first 24 hours of TCR activation and TGF-β no longer exerts this conversion after 48 hours. With this system, we now observed that BMP promotes Foxp3+ cells only when BMP were added to cultures on day 0. Unlike IL-2, BMP did not expand Foxp3+ cells when they were added to cultures 48 hours after the Foxp3+ cells had been generated by TGF-β. It is known TGF-β protects T cells from apoptosis, however, addition of BMP to TGF-β treated cells did not enhance expression of the anti-apoptosis gene, Bcl-2. These results indicate that BMP mainly promote the ability of TGF-β to convert Foxp3+ cells.
We have concluded that this synergism is an effect of BMP-2 or BMP-4 on TGF-β, but not TGF-β on the BMP. It seems unlikely BMP-2 or BMP-4 activates the Smad molecules of TGF-β signal downstream (Smad2 and Smad3) since BMP-2 or BMP-4 usually activates Smad1, 5 and 8. We have confirmed that CD4+ cells treated with BMP did not induce Smad2 and Smad3 activation. We recently found that the induction of Foxp3+ iTreg by TGF-β requires multiple pathways, and that both Smad and non-Smad pathways contribute to this induction (our unpublished observation). In this regard, BMP-2 or BMP-4 might act through the activation of P38, ERK and/or JNK, thus subsequently enhancing the effect of TGF-β on the induction of Foxp3+ iTreg. It has been reported that in addition to Smad-dependent signaling and transcriptional regulation, the activated receptor complex of BMP/BMP receptor also activates non-Smad signaling pathways, such as MAPK and TAK1/MEKK1, which leads to phosphorylation of ERK and p-38 in Jurkat TAg cells or non-T cells [17, 34]. In this study, we observed that BMP-2 or BMP-4 induced the activation of ERK and JNK and addition of ERK or JNK inhibitors suppressed the effects of BMP on Foxp3 expression on CD4+ cells induced by TGF-β although it did not significantly affect the p-38 activation. Time course experiments reveal that activation of ERK and JNK appears to take place in the early stages of TCR stimulation, it is consistent with previous report that TGF-β induces CTLA-4 upregulation in first day after TCR stimulation and this early upregulation is necessary for Foxp3+ iTreg conversion [35]. We further support the importance of MAPK pathways in synergistic effects of BMP and TGF-β by using ERK1 and JNK2 knock out mice. Although ERK activation is important for the IL-10 production and function [30], Our current study indicates that IL-10 is redundant in the synergistic effect of BMP and TGF-β in the Foxp3+ cell induction. Thus, evidence established in the current study also raise the possibility that MAPK, particularly ERK and/or JNK activation could possibly involve crosstalk with activated Smad molecules induced by TGF-β, eventually leading to the synergistic effect on the induction of Foxp3+ iTreg.
In addition to BMP, activin-A also belongs to TGF-β superfamily. While we prepared this manuscript, Huber S et al just reported that activin-A can convert non-Treg to Foxp3+ Treg and both actvin-A and TGF-β have a synergistic effect on the induction of Foxp3+ Treg [36]. Because both Activin-A and TGF-β similarly activate Smad2 [37], it is likely activin-A can, at least in the part, substitute TGF-β for the differentiation of iTreg since we have observed Samd2 had partial role in the regulation of Foxp3+ iTreg development (Zhou XH and Zheng SG, unpublished observation).
Our findings further demonstrate that TGF-β and its receptor signaling pathways are key factors in the development of Foxp3+ iTreg. In addition, the synergistic role between BMP and TGF-β indicates that manipulation of TGF-β signaling might have a considerable impact on therapeutic applications involving iTreg through improving iTreg purity and yield.
Materials and Methods
Mice
C57BL/6, IL-10 KO and JNK2 KO mice were purchased from Jackson laboratory (Bar harbor, ME), Floxed TGF-β receptor II (TRII) mice were provided by Dr. Harold Moses at Vanderbilt University [38]. ERK1 KO mice were provided by Dr. Gary Landreth at Case Western Reserve University. All mice were maintained in specific pathogen-free conditions according to the guidelines of the University of Southern California or Children's Hospital Los Angeles. All animals were treated according to National Institute of Health guidelines for the use of experimental animals with the approval of the University of Southern California Committee for the Use and Care of Animals.
Antibodies and reagents
Anti-CD4 (L3T4), anti-CD8 (Ly-2), anti-CD25 (PC61.5) were purchased from eBioscience, anti-Foxp3 (150D) was purchased from Biolegend. Anti-Smad1, Anti-phospho-Smad1Ser463/465, anti-phospho-Smad2/3Ser423/425, anti-phospho-P38Thr180Tyr182, anti-phospho-ENKThr202Tyr204 and anti-phospho-JNKThr183/Tyr185 were purchased from Cell Signaling Technologies. ALK-5 inhibitor, TSA and CFSE were purchased from Sigma-Aldrich. BMP-4, IL-2 and TGF-β were purchased from R&D Systems. BMP-2 and Noggin were gifts from Dr. Yuanping Han at the University of Southern California. Anti-CD3 and anti-CD28 coated beads were purchased from Invitrogen Life Technologies. AIM-V serum-free medium (Invitrogen Life Technologies) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 mM HEPES (all from Invitrogen Life Technologies) was used for the generation of CD4+ Treg or control cells. RPMI 1640 medium supplemented as just described with 10% heat-inactivated FCS (HyClone) was used for all other cultures.
Cell isolation and culture
T cells were prepared from lymph node and spleen cells by collecting nylon wool column nonadherent cells as previously described [31]. CD4+ T cells were isolated by negative selection. Briefly, T cells were labeled with PE-conjugated anti-CD8, anti-CD11b, and anti-B220 mAbs, incubated with anti-PE magnetic beads, and loaded onto MACS separation columns (Miltenyi Biotec). The CD4+ cells were further labeled with FITC-conjugated anti-CD25 mAb, and CD4+CD25−cells were obtained by positive selection using MACS beads (purity >98%).
Assessment of Treg activity in vitro and in vivo
To generate Treg, naïve CD4+CD25− cells from either wild-type mice or gene knock out mice as indicated were stimulated with anti-CD3/CD28 coated beads with or without TGF-β (2 ng/ml) in the presence of IL-2 (20 units/ml) in AIM-V serum-free medium. BMP-2, or BMP-4, or Noggin was also added to selected cultures. Various doses of CD4+ regulatory cells (CD4TGF-β) or control CD4+ cells (CD4med) were added to fresh T cells that were activated with anti-CD3 (0.25 μg/ml) and in the presence of irradiated APC. Proliferation was assayed by CFSE dilution assay or Thymidine [H3] incorporation assay as previously described [11], and the inhibition of cycling T cells was assessed. To evaluate the suppressive effect of Treg subsets in vivo, a cGVHD with a typical lupus syndrome was induced by adoptive transfer of 80 million of DBA/2 splenocytes into (DBA/2×C57BL/6) F1 mice. 5 million of CD4+ control (CD4+ cells activated with TCR without TGF-β), or CD4TGF-β, or CD4BMP-4+TGF-β cells were co-transferred with pathogenic DBA/2 splenocytes into F1 mice and anti-dsDNA was measured with an ELISA every two weeks post-transfer as before [31].
BMP and TsA administration
C57BL/6 mice was intraperitoneally administered with either TsA (1 mg/kg/) each day for 7 days, and/or BMP-2 (0.5 mg/mouse), BMP-4 (0.5 mg/mouse) or control PBS each other days for 10 days. The frequency of CD4+CD25+Foxp3+ cells in thymus, spleen, blood and gut at 12 days after treatment were determined by flow cytometry. Splenic CD4+CD25+ cells in various groups of mice were sorted by FACSVantage (BD Biosciences) and their suppressive activities against T cells from wild type mice was assayed as above.
Western blot analysis
A total of 5 × 106 various treated CD4+ cells were lysed in RIPA buffer. Specific proteins were detected by immunobloting following the method published previously [39]. Briefly, equal amounts (10 μg) of total cell lysate proteins were separated in NuPAGE 4-12% gradient SDS-PAGE gels using a MOP buffering system (Invitrogen). After protein was transferred into PVDF membrane, proteins of interest were detected by specific antibodies.
Real-time RT-PCR
Total cellular RNAs were isolated from snap-frozen lung tissue using RNeasy kit (Qiagen, Valencia, CA). The quality was checked by Experion™ automated electrophoresis system using Experion RNA HighSens analysis kit (Bio-Rad laboratories, Hercules, CA). Synthesis of cDNA and quantitative RT-PCR analysis were performed using iScript cDNA Synthesis Kit and SYBR Green I dye on iCycler-iQ system (Bio-Rad), as reported previously (39). The PCR primers for Foxp3, Bcl-2 and β-actin are: Foxp3, 5′-ACT GGG GTC TTC TCC CTC AA-3′, 5′-CGT GGG AAG GTG CAG AGT AG-3′; Bcl-2, 5′-CCT GGC TGT CTC TGA AGA CC-3′ Bcl2-R: 5′-CTC ACT TGT GGC CCA GGT AT-3′; β-actin: 5′-TGA CAG GAT GCA GAA GGA GA-3′, 5′-GTA CTT GCG CTC AGG AGG AG-3′, respectively. β-actin was used to normalize equal loading of template cDNA.
Statistical analysis
Statistical comparison between various groups was performed by the t test using GraphPad PRISM software (GraphPad). p< 0.05 was considered significant.
Acknowledgments
This work was supported by Grant NIH R01 (HL068597), ACR Research and Education Foundation's Within Our Reach, Arthritis Foundation, Outstanding Youth Scientist Investigator Award from National Natural Science Foundation of China (30728007), Webb Foundation and Key Project of Medical Leading Talents of Jiangsu Province in China (2007-2-07).
Abbreviations used in this paper
- BMP
Bone morphogenic protein
- TGF-β
Transforming growth factor beta
- CFSE
Carboxyfluorescein succinimidyl ester
Footnotes
Conflict of interest: D.A. Horwitz is a consultant for Becton Dickinson Bioscience, San Jose, CA. The remaining authors declare no financial or commercial conflict of interest.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J Biol Chem. 2001;276:37672–37679. doi: 10.1074/jbc.M104521200. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 4.Horwitz DA, Zheng SG, Gray JD, Wang JH, Ohtsuka K, Yamagiwa S. Regulatory T cells generated ex vivo as an approach for the therapy of autoimmune disease. Semin Immunol. 2004;16:135–143. doi: 10.1016/j.smim.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–435. doi: 10.1016/j.it.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25- precursors. J Immunol. 2002;169:4183–4189. doi: 10.4049/jimmunol.169.8.4183. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 9.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 10.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 12.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med. 2002;196:237–246. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–4571. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 14.von Boehmer H, Nolting J. What turns on Foxp3? Nat Immunol. 2008;9:121–122. doi: 10.1038/ni0208-121. [DOI] [PubMed] [Google Scholar]
- 15.Sporn MB, Roberts AB. The transforming growth factor bs. In: Sporn MB, Roberts AB, editors. Peptide growth factors and their receptor: Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 1991. pp. 419–472. [Google Scholar]
- 16.Hogan BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 17.Bleul CC, Boehm T. BMP signaling is required for normal thymus development. J Immunol. 2005;175:5213–5221. doi: 10.4049/jimmunol.175.8.5213. [DOI] [PubMed] [Google Scholar]
- 18.Bhatia M, Bonnet D, Wu D, Murdoch B, Wrana J, Gallacher L, Dick JE. Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. J Exp Med. 1999;189:1139–1148. doi: 10.1084/jem.189.7.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 20.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 21.Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 22.Licona-Limón P, Soldevila G. The role of TGF-beta superfamily during T cell development: new insights. Immunol Lett. 2007;109:1–12. doi: 10.1016/j.imlet.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Sivertsen EA, Huse K, Hystad ME, Kersten C, Smeland EB, Myklebust JH. Inhibitory effects and target genes of bone morphogenetic protein 6 in Jurkat Tag cells. Eur J Immunol. 2007;37:2937–2948. doi: 10.1002/eji.200636759. [DOI] [PubMed] [Google Scholar]
- 24.Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 25.Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126:1631–1642. doi: 10.1242/dev.126.8.1631. [DOI] [PubMed] [Google Scholar]
- 26.Rifas L. The role of noggin in human mesenchymal stem cell differentiation. J Cell Biochem. 2007;100:824–834. doi: 10.1002/jcb.21132. [DOI] [PubMed] [Google Scholar]
- 27.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 28.Reilly CM, Thomas M, Gogal R, Jr, Olgun S, Santo A, Sodhi R, Samy ET, Peng SL, Gilkeson GS, Mishra N. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J Autoimmun. 2008;2:123–130. doi: 10.1016/j.jaut.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Huber S, Schrader J, Fritz G, Presser K, Schmitt S, Waisman A, Lüth S, Blessing M, Herkel J, Schramm C. P38 MAP kinase signaling is required for the conversion of CD4+CD25- T cells into iTreg. PLoS ONE. 2008;3:e3302. doi: 10.1371/journal.pone.0003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O'Garra A. Interleukin-10 Production by Th1 Cells Requires Interleukin-12-Induced STAT4 Transcription Factor and ERK MAP Kinase Activation by High Antigen Dose. Immunity. 2009;29 doi: 10.1016/j.immuni.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng SG, Wang JH, Koss MN, Quismorio F, Jr, Gray JD, Horwitz DA. CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-beta suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J Immunol. 2004;172:1531–1539. doi: 10.4049/jimmunol.172.3.1531. [DOI] [PubMed] [Google Scholar]
- 32.Zheng SG, Meng L, Wang JH, Watanabe M, Barr ML, Cramer DV, Gray JD, Horwitz DA. Transfer of regulatory T cells generated ex vivo modifies graft rejection through induction of tolerogenic CD4+CD25+ cells in the recipient. Int Immunol. 2006;18:279–289. doi: 10.1093/intimm/dxh368. [DOI] [PubMed] [Google Scholar]
- 33.Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH. De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+CD25- cells. Proc Natl Acad Sci USA. 2005;102:4103–4108. doi: 10.1073/pnas.0407691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 35.Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. 2006;176:3321–3329. doi: 10.4049/jimmunol.176.6.3321. [DOI] [PubMed] [Google Scholar]
- 36.Huber S, Stahl FR, Schrader J, Lüth S, Presser K, Carambia A, Flavell RA, Werner S, Blessing M, Herkel J, Schramm C. Activin a promotes the TGF-beta-induced conversion of CD4+CD25- T cells into Foxp3+ induced regulatory T cells. J Immunol. 2009;182:4633–4640. doi: 10.4049/jimmunol.0803143. [DOI] [PubMed] [Google Scholar]
- 37.Utsugisawa T, Moody JL, Aspling M, Nilsson E, Carlsson L, Karlsson S. A road map toward defining the role of Smad signaling in hematopoietic stem cells. Stem Cells. 2006;24:1128–1136. doi: 10.1634/stemcells.2005-0263. [DOI] [PubMed] [Google Scholar]
- 38.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 39.Shi W, Chen H, Sun J, Buckley S, Zhao J, Anderson KD, Williams RG, Warburton D. TACE is required for fetal murine cardiac development and modeling. Dev Biol. 2003;261:371–380. doi: 10.1016/s0012-1606(03)00315-4. [DOI] [PubMed] [Google Scholar]