

Abstract
The bacteriophage phi29 DNA packaging motor contains a protein core with a central channel comprising twelve copies of re-engineered gp10 protein geared by six copies of packaging RNA (pRNA) and a DNA packaging protein gp16 with unknown copies. Incorporation of this nanomotor into a nanodevice would be beneficial for many applications. To this end, extension and modification of the motor components are necessary for the linkage of this motor to other nanomachines. Here the re-engineering of the motor DNA packaging protein gp16 by extending its length and doubling its size using a fusion protein technique is reported. The modified motor integrated with the eGFP-gp16 maintains the ability to convert the chemical energy from adenosine triphosphate (ATP) hydrolysis to mechanical motion and package DNA. The resulting DNA-filled capsid is subsequently converted into an infectious virion. The extended part of the gp16 arm is a fluorescent protein eGFP, which serves as a marker for tracking the motor in single-molecule studies. The activity of the re-engineered motor with eGFP-gp16 is also observed directly with a bright-field microscope via its ability to transport a 2-μm-sized cargo bound to the DNA.
Keywords: DNA, nanobiotechnology, nanoparticles, packaging motors
1. Introduction
Linear dsDNA viruses translocate their genomic DNA into a preformed procapsid with remarkable speed. This entropically unfavorable movement is carried out by an adenosine triphosphate (ATP)-driven nanomotor.[1–3] Such a motor with features typical of ATPases[4] has been identified in a number of viruses including T4,[5] λ,[6] T3 and T7,[7,8] phage 21,[9] P22,[10] SPP1,[11] herpesvirus,[12,13] cytomegalovirus (CMV),[14] adenovirus,[15] and poxvirus.[16] The DNA packaging motor of the bacterial virus phi29 is particularly attractive since it is relatively simple in structure and can be assembled in vitro using purified components.[17,18] The motor is geared by six copies of an RNA molecule called pRNA (packaging RNA)[19] and the ATPase gp16.[17] The novel and ingenious design of the 30-nm-nanomotor, one of the strongest biomotors constructed to date, has inspired the engineering of an imitative DNA packaging motor for applications in nanobiotechnology.[20] The high efficiency of in vitro DNA packaging up to 90%[17,18,21,22] and the feasibility to turn the motor on and turn off with γ-S-ATP[23] has enabled the single-molecule measurement of motor velocities and forces using an optical trap.[24,25] The studies revealed that the phi29 DNA packaging motor is one of the strongest nanomotors, with a stalling forceof 57 pN.
Phi29 DNA packaging enzyme gp16 plays a central role in this motor.[4,26] It was found that gp16 binds the 5′/3′ helical double-stranded end of pRNA during DNA packaging[27] and the binding to pRNA enhances the ATPase activity of gp16.[28,29] Hydrolysis of one ATP molecule packages 2[4,25] or 2.5[30a] base pairs of genomic DNA.[30b] The study and application of gp16 has been hindered by its extreme insolubility.[17] Denaturation of the gp16 in an inclusion body followed by renaturation[17] kept the gp16 soluble and active only for 20 min. Co-expression of gp16 with groE enhanced the solubility of gp16;[31] however, the soluble gp16 aggregated after purification. The insolubility and self-aggregation of gp16 has led to contradictory data regarding ATPase activity, binding location, and the stoichiometry of gp16 on the motor.[4,28,31] Fusion of gp16 with thioredoxin has also enhanced its solubility[32,33] although polymers or organic solvents were required in order to maintain solubility over time.[32,33]
Single-molecule imaging is an important technique for elucidating the structure, function, and action mechanism of complicated biological systems. The fusing of proteins with a fluorescent peptide ensures single labeling of exact location with up to 100% labeling efficiency for single-molecule analysis. For example, the walking mechanism of myosin has been elucidated with the aid of the protein-engineering technique of fluorescent-protein extension,[34] thus enabling the measurement of the distance between the two heads.[35] A similar technique has been applied to the study of the interactions between repressor and DNA.[36] Fusion of the MotB protein in a bacterial flagella motor with fluorescent protein has also enabled stoichiometry determination and dynamic assay with single-molecule precision.[37]
Here, we report the re-engineering of the gp16 protein by extending its N-terminus with a fluorescent arm to produce soluble, stable, and active fluorescent gp16. The construction of an active gp16 with a fluorescent arm will facilitate future studies on the structure, function, distance, speed, and motion mechanism of the phi29 DNA packaging motor as well as gp16 stoichiometry determination by single-molecule imaging.
2. Results
2.1. Construction and Purification of the eGFP-gp16
The gene coding gp16 was engineered into the E. coli expression vector pET32 Xa/LIC, along with the 239-amino acid eGFP and a six-Histidine tag (His-tag) placed between the eGFP and gp16 for one-step purification (Figure 1A). As a result, the N-terminus of native gp16 was extended by 266 aminoacids as compared to its original form, which contains 332 amino acids (Supporting Information, Figure S1). The resultant chimeric gp16 (eGFP-gp16) therefore had 598 amino acids with a size of 68.5 kDa (Figure 1B), approximately twice as large as that of native gp16 (36.5 kDa). Using the His-tag included in the fusion protein, the expressed eGFP-gp16 was easily purified to homogeneity by one-step affinity chromatography as shown in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Figure 1C).
Figure 1.

A) Map of E. coli expression vector plasmid pET32 harboring eGFP-gp16 with His-tag. B) Schematic illustration showing the structure of the nanomotorwith eGFP-gp16. C) eGFP-gp16 purified by His-tag affinity chromatography (10% SDS-PAGE). Lane 1: Molecular-weight protein marker. Lanes 2 and 3: E. coli BL21(DE3) with the pET32-eGFP-gp16 uninduced and induced by IPTG, respectively. Lanes 4 and 5: soluble cell extracts containing over-expressed eGFP-gp16 before and after passage through an affinity chromatography column, respectively. Lane 6: purified eGFP-gp16.
2.2. Motor Function in Packaging of DNA
2.2.1. Function of EGFP-gp16 in Driving the Motor for DNA Packaging
A DNA packaging assay was performed to test the function of the purified eGFP-gp16 in the DNA packaging motor by comparing it with that of native gp16. eGFP-gp16 could package the input DNA (Figure 2A), which suggested that the extension of gp16 by fusion with eGFP did not affect the biological function of gp16 in driving the DNA packaging motor. To ensure that the procapsids packaged by the eGFP-gp16 could be transformed to infectious virions, purified gp9, 11, 12, and 13 were added to the packaged procapsid mixture. The in vitro phi29 virus assembly test showed that the packaged procapsids were converted successfully into infectious virions (Figure 2B) and both engineered gp16 and eGFP-gp16 were as efficient as the native gp16. Data indicated that the re-engineering of the gp16 to extend its N-terminus successfully increased the solubility and stability of gp16 and resulted in intact motor-driving function.
Figure 2.
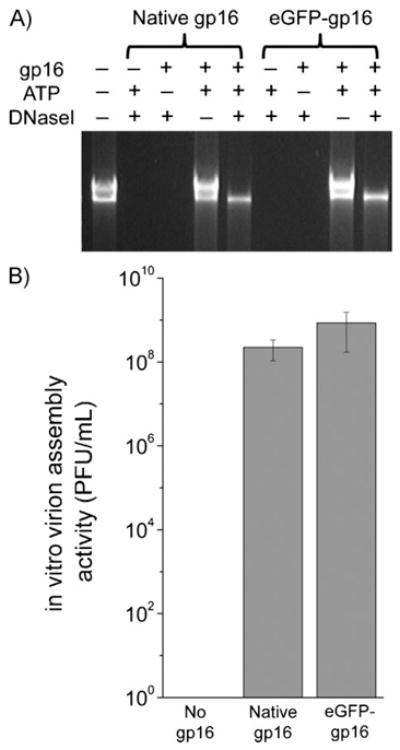
A) In vitro DNA packaging activity of eGFP-gp16 compared to that of native gp16 by DNase I protection assay. DNase I is used to digest any unpackaged DNA that can not be protected by procapsids from DNase I digestion. B) In vitro phage virus assembly activity of eGFP-gp16 compared to that of native gp16.
2.2.2. eGFP-gp16 Bound to DNA
It has been previously observed that gp16 binds to both DNA and RNA (a unique feature as a motor component), without sequence specificity, but binds specifically to pRNA attached to procapsids.[27] To confirm whether the eGFP-gp16 retains its DNA binding property, we performed total internal reflection fluorescence (TIRF) imaging, as previously described.[29] The eGFP-gp16 was immobilized onto a gp16-antibody-coated surface and its presence was visualized by the fluorescence signals from eGFP (Figure 3A). Cy5-labeled DNA was then added for binding to gp16. Imaging data showed that eGFP-gp16 bound Cy5-polydT (Figure 3A), which agreed with previous observations obtained with the active gp16.[29] Non-fluorescent-engineeredgp16-immobilized surface showed similar binding of Cy5-polydT but no GFP signals (Figure 3B), while no Cy5-polydT binding was observed from the gp16-absent surface (Figure 3C). The result suggested that the eGFP-gp16 retains its nucleic acid binding activity because the re-engineering did not affect the functional domain of gp16.
Figure 3.

Binding activity of eGFP-gp16 to DNA observed by TIRF imaging microscopy. Cy5-poly dT was incubated inside a microchamber in the presence (A) or the absence (B) of immobilized eGFP-gp16on the surface. Engineered-gp16 was used as a control for DNA binding (C). The image was taken from both eGFP and Cy5 emission channels.
2.2.3. eGFP-gp16 Hydrolyzes ATP to Generate Energy for Motor Function
It is known that gp16 binds ATP and hydrolyzes the bound ATP.[17,29,38] Therefore, measuring the ATPase activity of purified gp16 is another means to determine whether the re-engineering of gp16 with extended aminoacid residues will affect the ATP binding and catalysis activity. To measure the ATPase activity of the purified eGFP-gp16, an inorganic phosphate (Pi) sensor system, MDCC-PBP (N-(2-(1-maleimidyl)ethyl)-7-(diethylamino)coumarin-3-carboxamide-phosphate binding protein), was used as it was previously shown to be successful in investigating the ATPase activity of engineered gp16.[29]
The ATP hydrolysis reaction of gp16 was initiated by adding ATP to a reaction mixture containing the purified eGFP-gp16 and MDCC-PBP. A range of ATP concentrations was used to determine the kinetic parameter of eGFP-gp16. The ATP hydrolysis assay showed a hyperbolic relationship between Vinit and the concentration of ATP (Figure 4), which implies that the eGFP-gp16 maintains its Michaelis–Menten-type enzyme functionality. The data also shows that the observed ATPase activity of eGFP-gp16 was comparable to that of the engineered gp16 used as a control. These results suggest that the extension of the N-terminus of gp16 through re-engineering did not interfere with the biological functions and that it can still bind and hydrolyze ATP. This is one of the most important checkpoints for the re-engineering of gp16 because the ATP hydrolysis is a central activity of gp16 during the DNA packaging process.
Figure 4.

Comparison of the ATPase activities between engineered gp16 and eGFP-gp16. The initial velocities Vinit for engineered gp16 (squares) andeGFP-gp16 (circles) were plotted over the tested ATP concentration by non-linear fitting (hyperbola plot) based on the Michaelis–Menten equation. Km is defined as the ATP concentration at one half of Vmax.
2.2.4. Motor with eGFP-gp16 Transports Cargo Tethered by DNA
The phi29 DNA packaging was observed with the eGFP-gp16 in a bright field using a relatively simple magneto- mechanical system, as described previously (Figure 5A).[39] The phi29 DNA packaging motor containing the eGFP-gp16 was able to package the DNA bound to a magnetic bead with a diameter of 2 μm (Figure 5B). According to the force calibration demonstrated in a previous study,[39] the magnetic bead experienced about 50 pN of magnetic force and hence applied the same force on the DNA tether when the magnet was brought near the setup. The magnetic force stretched the DNA and enabled observation of its length. The initial DNA tether length before the reactivation of the stalled motor was 6.8 μm, which is slightly longer than its reported contour length (≈6.56 μm for 19.3 kb). This could be attributed to the large applied force (>50 pN) that overstretches the DNA.[40] The plot of DNA tether length versus packaging time shows that the DNA packaging process finished in 9 minutes (Figure 5C), which is longer than the previously reported average time of 5.5 minutes.[24] This can be attributed to the periodical interruptions of the packaging by the large applied magnetic force.
Figure 5.

A) Schematic image of the bright-field observation of the phi29 DNA packaging. B) Temporal micrographs of a tethered bead showing the phi29 DNA packaging behavior. C) DNA tether length against packaging time.
2.3. Single-Molecule Studies with eGFP-gp16
2.3.1. eGFP-gp16 Detection by Photobleaching Analysis
When eGFP-gp16 was visualized with a laser with a wavelength of 491 nm, bright fluorescent signals from eGFP were observed in a concentration-dependent manner (Figure 6A). To assess the fluorescence property of an individual eGFP-gp16 molecule, eGFP-gp16 was subjected to single-molecule photobleaching, as has been applied to the counting of pRNAs on the motor.[41,42] A typical fluorescence image of eGFP-gp16 immobilized by antibody on a quartz surface was obtained by TIRF microscopy (Figure 6B(a)) and each spot was analyzed individually for the change in fluorescence intensity until photo-bleaching occurred.[42] The fluorescent spots were fitted with point-spread function (PSF), as shown in Figure 6B(b). Due to the optical resolution (approximately 250 nm in this experiment), if the distance between two fluorophores is within the diffraction limit then they often appear as a single fluorescent spot in the image. The fitting of the circled spot in Figure 6B(b) gave a lateral full width at half-maximum (FWHM) of 294 nm, as expected for the microscope resolution.[36] As multiple eGFP-gp16 molecules could reside within the resolution limit when bound to the antibody surface and, hence, be detected as one spot, the number of eGFP-gp16 molecules immobilized within one spot is random. However, the number of eGFP-gp16 molecules within one fluorescent spot was revealed by the stepwise photobleaching of eGFP, as shown in Figure 6C(a) for one gp16 molecule and in Figure 6C(b) for two gp16 molecules. Each intensity drop represents the quantized photobleaching of a single eGFP molecule, which, in turn, indicates the number of single eGFP-gp16 molecules in one specific spot. The number of eGFP-gp16 molecules within one spot can therefore be accurately determined from the number of steps revealed by such plotting.
Figure 6.

A) Concentration-dependent fluorescence intensity from the extended fluorescent arm of eGFP-gp16. B, a) Enlarged single-molecule image showing individual immobilized eGFP-gp16 distributed over the observed area (70 × 70 μm). b) Point-spread-function fitting of the circled fluorescent spot. The pixel size is 160 nm. C) Photobleaching step analysis showing a) one- or b) two-step drop from the immobilized eGFP-gp16. Time 0 indicates the beginning of laser excitation. D) Distribution of fluorescence intensity of the spots that showed one (solid bars) or two (shaded bars) steps in photobleaching, respectively. Xc represents the peak value obtained from each superimposed Gaussian plot over the data points, with Xc = 5.86 (solidline) for those spots containing one gp16 molecule and Xc = 10.53 (dashed line) for those containing two gp16 molecules.
2.3.2. Total Intensity Distribution Counting Analysis of eGFP-gp16
Although the production of eGFP-gp16 was obtained with the fusion protein technique ensuring single eGFP labeling for every gp16 molecule, the fluorescence intensity of GFP has been reported to be unstable under continuous illumination.[43,44] Likewise, the fluorescence intensity of the eGFP-gp16 under continuous laser illumination dropped in seconds (Figure 6C). Although the number of copies of a given protein within a certain biocomplex can be obtained by step-wise photobleaching of GFP labels,[37,45] the complexity of GFP fluorescence makes the counting of steps challenging. To overcome this weakness in determining the eGFP-gp16 stoichiometry, we explored an alternative method of simply comparing the intensities of the fluorescent spots.[36,37,45] For each fluorescence spot, the intensity was measured by subtracting the background intensity. The intensities of the spots that showed one-step photobleaching (Figure 6C(a)) are summarized in a histogram (solid bars in Figure 6D). A Gaussian fit was performed by the OriginPro 8 software (OriginLab, USA). The peak position (Xc) at 5.86 ± 0.18 indicates the mean value of the fluorescence intensities for onestep drop, representing the unitary intensity of one gp16 molecule. The width of the Gaussian curve, in this case, was 3.36 ± 0.43, represented by W. The distribution results from variations among individual eGFP-gp16 molecules, the shot noise, the blinking of eGFP, as well as the heterogeneous illumination of the laser beam.[45]
Similarly, the intensities of the spots that showed a two-step drop (Figure 6C(b)), indicating the existence of two gp16 molecules, are summarized in a histogram (shaded bars in Figure 6D). The 2D Gaussian fitting indicated Xc at 10.53 ± 0.30, with W = 2.54 ± 0.76. Comparing the two histograms, the peak for the intensities of two gp16 molecules (Xc = 10.53) was almost twice that of one gp16 molecule (Xc = 5.86; Figure 6D). Noticeably, the two peaks were clearly separated with Xc values of 5.86 and 10.5, respectively. This agreed with our expectation that the intensity of two eGFP-gp16 molecules would be approximately twice the intensity of one eGFP-gp16 molecule. The results imply that the copy number of eGFP-gp16 within one fluorescent spot could be estimated by dividing its fluorescence intensity with the unitary intensity of a single eGFP-gp16 molecule. Such a method could be applied to a stoichiometry study of gp16 on the phi29 motor.
3. Discussion
Fusion of the fluorescent protein eGFP to the N-terminus of gp16 has approximately doubled the size of this ATPase. Remarkably, the extended fluorescent gp16 retained all its biological activity in pRNA/DNA binding, ATP hydrolysis, DNA packaging, and phi29 virion assembly. It is especially surprising that the long N terminal extension did not hinder the ATP binding and hydrolysis activity of gp16, even though the consensus ATP binding sequences known as Walker-A motif is near the N-terminus.[4] It suggests that the extended fusion partner did not affect the conformational change of gp16 catalyzing ATP hydrolysis.
With the aid of the highly sensitive single-molecule dual-view TIRF imaging system (SMDV-TIRF)[42] and utilizing the photobleaching property of the fluorophore singly attached to the pRNA, it has been possible to perform single-molecule counting of the pRNA molecules on each phi29 DNA packaging motor.[41] Using this approach, the stoichiometry of pRNA on each motor has been determined to be six.[41] The single-molecule photobleaching data reported here forecast its potential single-molecule applications.
4. Conclusions
Our studies showed that eGFP-gp16 could be detected at the single-molecule level using TIRF imaging. Figure 6 shows another possible application of the eGFP-gp16. Since the total fluorescence intensity was proportional to the number of the photo bleaching steps of the fluorescent protein, eGFP-gp16can be used as a tracer for the stoichiometry study of gp16 for the motor or the DNA packaging intermediates. However, for future stoichiometry studies using eGFP-gp16 on the packaging motor, special modification and care will be required to estimate the copy number of gp16 because of the blinking and unstable nature of GFP. It is also possible to observe the eGFP-gp16 directly and locate the gp16 immobilized on the slide. The eGFP-gp16 could serve as a reference position in a single-molecule physical dynamics study. The distance parameter or relative motion of eGFP-gp16 in relation to other motor components carrying different fluorescence labels can be deduced. It would be possible to measure distance or conformational change during dynamic interactions between two molecules by fluorescence resonance energy transfer (FRET), thus assessing the motor function and motion mechanism. Either one or two gp16 molecules are countable from the photobleaching curves.
5. Experimental Section
Re-engineering of extended-gp16
Previously, gp16 was engineered into plasmid pET32 Xa/LIC and extended with a 109 amino acid residue containing a thioredoxin gene as a fusion protein.[33] The engineered plasmid containing a gp16 gene was re-engineered by replacing the thioredoxin fusion protein with eGFP amplified from pEGFP-N2 (GenBank accession number U57608, BD Biosciences Clontech, USA) by polymerase chain reaction (PCR). The re-engineered plasmid was introduced into E.coli BL21 (DE3) for protein expression.[46]
Expression and purification of eGFP-gp16
Both engineered and re-engineered-gp16 were over-expressed as previously described,[27,33] and were harvested by centrifugation. The harvested cells were resuspended in His-binding buffer (20 mM Tris-HCl, pH 7.9, 500 mM NaCl, 15% glycerol and 5 mM imidazole), and lysed by passage through a French Press (Aminco SLM, USA). The lysate was centrifuged for 20 min at 100000 g at 4°C to remove debris, and then the post-centrifugation supernatant was filtered through a 0.45-μm filter membrane (Millipore, USA). The clarified cell lysate containing the over-expressed engineered or eGFP-gp16 was further purified using one-step affinity chromatography, as described previously.[27,29,33] The eluted eGFP-gp16 was identified by subjecting it to 10% SDS-PAGE.
Assay for motor function in packaging of DNA
pRNA was synthesized by in vitro transcription with T7 RNA polymerase from a PCR-generated template, as described previously.[47,48] The preparation of procapsids,[17] DNA-gp3,[17] native gp16,[17] tail protein (gp9),[18,49] neck proteins (gp11 and gp12)[50] and the morphogenetic factor (gp13)[18] have been described previously. Using the purified phi29 components, in vitro DNA packaging and phage assembly was performed as described previously.[51] Phage virus assembly with neck and tail components gp9, gp11, gp12, and gp13 and plating was performed as described previously.[18]
Testing of binding of the eGFP-gp16 to DNA
The quartz surface of the perfusion chamber (15 μL internal volume) was coated with 100 μg/mL of anti-gp16 rabbit IgG (Proteintech Group, Inc) in HMS and incubated overnight at 4°C. The eGFP-gp16 was immobilized on the antibody-coated surface by antibody–antigen recognition binding. The unbound eGFP-gp16 was washed using HMS. Cy5-labeled DNA dissolved in HMS was then incubated with the immobilized eGFP-gp16 for 20 min at ambient temperature. Oxygen-depletion solution containing glucose oxidase, catalase, β-D-glucose, and 2-mercaptoethanol[52] was added to the chamber before observation to reduce the fast photobleaching of fluorescent materials. Observation of bound fluorescent DNA to gp16 was conducted by the TIRF imaging technique, as described previously.[41,42] An Olympus IX71 inverted microscope 60× oil immersion objective (PlanoApo, 60×, NA = 1.4) with additional 1.6× magnification lens was used to collect the fluorescence signals. A blue laser of 491 nm was applied for excitation, with a power of 16.9 W/cm2. A red laser beam with a 638-nm wavelength was used to excite Cy5-DNA. The beam size is about 80 μm × 180 μm at the quartz/solution interface. The fluorescence images were recorded with an exposure time of 200 ms by an EMCCD camera (iXon 897 V, Andor Technology).
Assay for chemical energy conversion by the eGFP-gp16
Purified engineered gp16 containing thioredoxin, as described previously, [33] was used to measure ATPase activity. The ATPase reaction was performed as described previously. [29] The initial velocities (Vinit) were calculated as the linear slope of released Pi (μM) from ATPase activity of gp16 over time (s). A non-linear plot was used to determine the steady-state ATP hydrolysis activity of gp16 as a function of substrate concentration according to the Michaelis–Menten equation (Vinit = Vmax [ATP]/(Km + [ATP]). In the equation, the Vinit (μM s−1) is plotted against [ATP], yielding a hyperbolic curve. Data analysis was performed using the OriginPro version 7.5 software (Origin Lab Corp, USA).
Cargo transportation by the motor with eGFP-gp16
Direct bright-field observation of magnetic-bead-tethered DNA packaging was carried out as described previously. [39] In summary, the phi29 packaging motor intermediate was first stalled by using a non-hydrolyzable ATP analogue, γ-S-ATP. Next, a suspension of NHS-activated magnetic-bead suspension (M-PVA Ak11, obtained from Chemagen, average diameter ≈1 μm) coupled with anti-gp3 antibody was used to bind the gp3 protein on the free end of the partially packaged DNA-gp3. The stalled motors with DNA-tethered magnetic beads were attached to a procapsid antibody-coated glass wall in the microfluidic chamber. A permanent magnet was placed on a translation stage beside the microfludic chamber. By bringing the magnet close to the chamber, a magnetic force was applied on the magnetic beads, stretching the tethered DNAs connected to the beads (the unbound beads were drawn away towards the magnet). To resume the stalled phi29 intermediates, the chamber was filled with the restart buffer containing 3 μL of 10 mM ATP, 6 μL of 160 nM eGFP-gp16, and 15 μL of TMS. DNA packaging was allowed to progress without a continous external force; the magnetic force was applied to the beads once every minute for 15 seconds to stretch the DNA and measure its length by recording micrographs. The change in the DNA length in discrete time intervals was recored in this fashion.
Fluorescent detection of the eGFP-gp16
The quartz surface of the perfusion chamber (15 μL internal volume) was coated with 100 μg/mL of anti-gp16 rabbit IgG (Proteintech Group, Inc) in HMS incubated overnight at 4°C. Three different concentrations, 10 pM, 100 pM, and 1 nM of eGFP-gp16 in HMS buffer were infused in the antibody-coated flow chamber. After washing with HMS, TIRF imaging was carried out as described above to observe the bound fluorescent eGFP-gp16 on the surface. Photobleaching traces were recorded and intensities of single-step drop and two-step drop were measured using Andor iQ version 1.8 software (Andor Technology, USA), and summarized in the histograms. A 2D Gaussian curve fitting was then applied to the histograms by OriginPro 8 (OriginLab, USA).
Supplementary Material
Acknowledgments
We thank Dr. Anne Vonderheide, Dr. Wenjuan Wang, Chad Schwartz, and Mollie R. Johnson for their assistance in the preparation of this manuscript. This work was supported by National Institutes of Health grant (R01-GM59944) and PN2 EY018230 from NIH Nanomedicine Development Center for Phi29 DNA Packaging Motor for Nanomedicine through NIH Roadmap for Medical Research. Funding for open access: National Institutes of Health grant R01-GM59944. P.G. is the co-founder of Kylin Therapeutics Inc.
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
References
- 1.Black LW. Ann Rev Microbiol. 1989;43:267–292. doi: 10.1146/annurev.mi.43.100189.001411. [DOI] [PubMed] [Google Scholar]
- 2.Guo P, Lee TJ. Mol Microbiol. 2007;64:886–903. doi: 10.1111/j.1365-2958.2007.05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao VB, Feiss M. Annu Rev Genet. 2008;42:647–681. doi: 10.1146/annurev.genet.42.110807.091545. [DOI] [PubMed] [Google Scholar]
- 4.Guo P, Peterson C, Anderson D. J Mol Biol. 1987;197:229–236. doi: 10.1016/0022-2836(87)90121-5. [DOI] [PubMed] [Google Scholar]
- 5.Rao VB, Black LW. J Mol Biol. 1988;200:475–488. doi: 10.1016/0022-2836(88)90537-2. [DOI] [PubMed] [Google Scholar]
- 6.Tomka MA, Catalano CE. Biochemistry. 1993;32:11992–11997. doi: 10.1021/bi00096a008. [DOI] [PubMed] [Google Scholar]
- 7.Hamada K, Fujisawa H, Minagawa T. Virology. 1986;151:119–123. doi: 10.1016/0042-6822(86)90109-1. [DOI] [PubMed] [Google Scholar]
- 8.Serwer P, Masker WE, Allen JL. J Virol. 1983;45:665–671. doi: 10.1128/jvi.45.2.665-671.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feiss M, Frackman S, Sippy J. J Mol Biol. 1985;183:239–249. doi: 10.1016/0022-2836(85)90216-5. [DOI] [PubMed] [Google Scholar]
- 10.Strobel E, Behnish W, Schmieger H. Virology. 1984;133:158–165. doi: 10.1016/0042-6822(84)90434-3. [DOI] [PubMed] [Google Scholar]
- 11.Oliveira L, Alonso JC, Tavares P. J Mol Biol. 2005;353:529–539. doi: 10.1016/j.jmb.2005.08.063. [DOI] [PubMed] [Google Scholar]
- 12.Yu D, Weller SK. Virology. 1998;243:32–44. doi: 10.1006/viro.1998.9041. [DOI] [PubMed] [Google Scholar]
- 13.Salmon B, Baines JD. J Virol. 1998;72:3045–3050. doi: 10.1128/jvi.72.4.3045-3050.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. Nucleic Acids Res. 2002;30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Low JA, Christensen JB, Imperiale MJ. J Virol. 2001;75:10446–10454. doi: 10.1128/JVI.75.21.10446-10454.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cassetti MC, Merchlinsky M, Wolffe EJ, Weiserg AS, Moss B. J Virol. 1998;72:5769–5780. doi: 10.1128/jvi.72.7.5769-5780.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo P, Grimes S, Anderson D. Proc Natl Acad Sci USA. 1986;83:3505–3509. doi: 10.1073/pnas.83.10.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CS, Guo P. J Virol. 1995;69:5018–5023. doi: 10.1128/jvi.69.8.5018-5023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo P, Erickson S, Anderson D. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 20.Guo P. J Nanosci Nanotechnol. 2005;5:1964–1982. doi: 10.1166/jnn.2005.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F, Lemieux S, Wu X, St-Arnaud S, McMurray CT, Major F, Anderson D. Mol Cell. 1998;2:141–147. doi: 10.1016/s1097-2765(00)80123-9. [DOI] [PubMed] [Google Scholar]
- 22.Guo P, Zhang C, Chen C, Trottier M, Garver K. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- 23.Guo P, Peterson C, Anderson D. J Mol Biol. 1987;197:219–228. doi: 10.1016/0022-2836(87)90120-3. [DOI] [PubMed] [Google Scholar]
- 24.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. Nature. 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- 25.Chemla YR, Aathavan K, Michaelis J, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Cell. 2005;122:683–692. doi: 10.1016/j.cell.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 26.Hagen EW, Reilly BE, Tosi ME, Anderson DL. J Virol. 1976;19:501–517. doi: 10.1128/jvi.19.2.501-517.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee TJ, Guo P. J Mol Biol. 2006;356:589–599. doi: 10.1016/j.jmb.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 28.Grimes S, Anderson D. J Mol Biol. 1990;215:559–566. doi: 10.1016/s0022-2836(05)80168-8. [DOI] [PubMed] [Google Scholar]
- 29.Lee TJ, Zhang H, Liang D, Guo P. Virology. 2008;380:69–74. doi: 10.1016/j.virol.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Moffitt JR, Chemla YR, Aathavan K, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Nature. 2009;457:446–450. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen C, Guo P. J Virol. 1997;71:3864–3871. doi: 10.1128/jvi.71.5.3864-3871.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibarra B, Valpuesta JM, Carrascosa JL. Nucleic Acids Res. 2001;29:4264–4273. doi: 10.1093/nar/29.21.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang LP, Guo P. Virology. 2003;312:449–457. doi: 10.1016/s0042-6822(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 33.Huang LP, Guo P. J Virol Methods. 2003;109:235–244. doi: 10.1016/s0166-0934(03)00077-6. [DOI] [PubMed] [Google Scholar]
- 34.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 35.Balci H, Ha T, Sweeney HL, Selvin PR. Biophys J. 2005;89:413–417. doi: 10.1529/biophysj.105.060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang YM, Tegenfeldt JO, Reisner W, Riehn R, Guan XJ, Guo L, Golding I, Cox EC, Sturm J, Austin RH. Proc Natl Acad Sci USA. 2005;102:9796–9801. doi: 10.1073/pnas.0502917102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, Armitage JP. Nature. 2006;443:355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 38.Shu D, Guo P. Virology. 2003;309:108–113. doi: 10.1016/s0042-6822(03)00011-4. [DOI] [PubMed] [Google Scholar]
- 39.Chang CL, Zhang H, Shu D, Guo P, Savran CA. Appl Phys Lett. 2008;93:153902–153903. doi: 10.1063/1.3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 41.Shu D, Zhang H, Jin J, Guo P. EMBO J. 2007;26:527–537. doi: 10.1038/sj.emboj.7601506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang H, Shu D, Huang F, Guo P. RNA. 2007;13:1793–1802. doi: 10.1261/rna.587607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce DW, HomBooher N, Vale RD. Nature. 1997;388:338. doi: 10.1038/41009. [DOI] [PubMed] [Google Scholar]
- 44.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 45.Haggie PM, Verkman AS. J Biol Chem. 2008;283:23510–23513. doi: 10.1074/jbc.C800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Meth Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 47.Zhang CL, Lee CS, Guo P. Virology. 1994;201:77–85. doi: 10.1006/viro.1994.1267. [DOI] [PubMed] [Google Scholar]
- 48.Zhang CL, Trottier M, Guo P. Virology. 1995;207:442–451. doi: 10.1006/viro.1995.1103. [DOI] [PubMed] [Google Scholar]
- 49.Garcia JA, Carrascosa JL, Salas M. Virology. 1983;125:18–30. doi: 10.1016/0042-6822(83)90060-0. [DOI] [PubMed] [Google Scholar]
- 50.Carrascosa JL, Camacho A, Vinuela E, Salas M. FEBS Lett. 1974;44:317–321. [PubMed] [Google Scholar]
- 51.Lee CS, Guo P. Virology. 1994;202:1039–1042. doi: 10.1006/viro.1994.1434. [DOI] [PubMed] [Google Scholar]
- 52.Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S. Nature. 2002;419:638–641. doi: 10.1038/nature01083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.