

Abstract
Methylation of cytosines in the dinucleotide CpG has been shown to suppress transcription of a number of tissue-specific genes, yet the precise mechanism is not fully understood. The vertebrate globin genes were among the first examples in which an inverse correlation was shown between CpG methylation and transcription. We studied the methylation pattern of the 235-bp ρ-globin gene promoter in genomic DNA from primary chicken erythroid cells using the sodium bisulfite conversion technique and found all CpGs in the promoter to be methylated in erythroid cells from adult chickens in which the ρ-globin gene is silent but unmethylated in 5-day (primitive) embryonic red cells in which the gene is transcribed. To elucidate further the mechanism of methylation-induced silencing, an expression construct consisting of 235 bp of 5′ promoter sequence of the ρ-globin gene along with a strong 5′ erythroid enhancer driving a chloramphenicol acetyltransferase reporter gene, ρ-CAT, was transfected into primary avian erythroid cells derived from 5-day embryos. Methylation of just the 235-bp ρ-globin gene promoter fragment at every CpG resulted in a 20- to 30-fold inhibition of transcription, and this effect was not overridden by the presence of potent erythroid-specific enhancers. The ability of the 235-bp ρ-globin gene promoter to bind to a DNA Methyl Cytosine binding Protein Complex (MeCPC) was tested in electrophoretic mobility shift assays utilizing primary avian erythroid cell nuclear extract. The results were that fully methylated but not unmethylated 235-bp ρ-globin gene promoter fragment could compete efficiently for MeCPC binding. These results are a direct demonstration that site-specific methylation of a globin gene promoter at the exact CpGs that are methylated in vivo can silence transcription in homologous primary erythroid cells. Further, these data implicate binding of MeCPC to the promoter in the mechanism of silencing.
Methylation of cytosine residues in the dinucleotide CpG is the most common postsynthetic eukaryotic DNA modification. Since the reports of an inverse correlation between DNA methylation and expression of vertebrate β-type globin genes (1–3), a large body of evidence relating DNA methylation to gene expression has accumulated (4, 5). At the same time, the absence of detectable DNA methylation in some eukaryotes such as Drosophila (6) and Saccharomyces cerevisiae (7) has raised doubts about its role in normal development and tissue-specific gene expression. However a study by Li et al. (8) showing abnormal development and embryonic lethality in transgenic mice expressing decreased but not completely absent DNA methyl transferase activity following knockout of the DNA methyl transferase gene lends strong support to a critical role for DNA methylation in developmental gene regulation. Recently, a similar critical function of DNA methylation in plant development has been demonstrated by Ronemus et al. (9). However, a direct demonstration of the role of DNA methylation in suppressing transcription of a specific gene during development in normal tissues has been lacking.
In the avian β-type globin cluster (5′-ρ-βH-β-ɛ-3′), silencing of the embryonic ρ-gene occurs concomitantly with activation of the adult β-gene on day 5 of embryonic development (10). In definitive embryonic and adult chicken erythroid cells, the embryonic globin genes are nontranscribed (11, 12), and a strong inverse correlation exists between site-specific DNA methylation and expression of the chicken β-type globin genes (13). Our laboratory has shown that the normally silent embryonic ρ-globin gene in red cells of anemic adult chicken can be transcriptionally activated by treatment with the DNA methyltransferase inhibitor, 5-azacytidine (14). However, the cause and effect relationship between methylation and stage-specific globin gene transcription in normal erythroid cells has not been fully elucidated. In this report, we describe (i) the in vivo methylation pattern of all CpG dinucleotides in a 235-bp minimal promoter of the embryonically expressed avian ρ-globin gene in 5-day primitive and definitive chicken erythroid cells, (ii) the transcription inhibitory effect of physiologically precise CpG methylation on the ρ-globin gene promoter despite the presence of strong 5′ and/or 3′ erythroid-specific enhancer elements in primary embryonic erythroid cells, and (iii) the demonstration of a DNA methyl binding protein complex that offers a possible mechanism of methylation-induced transcriptional silencing of the ρ-globin gene in normal erythroid cells.
MATERIALS AND METHODS
Vector Constructs.
All transfection vectors were constructed in pUC 18. The vectors depicted in Fig. 2A were constructed by cloning a 2.2-kb ρ promoter region 5′ to a chloramphenicol acetyltransferase (CAT) reporter gene, pCAT Basic (Promega). Vectors in Fig. 2B were constructed by cloning a 3′ β/ɛ enhancer element 3′ into the ρ promoter containing CAT reporter gene constructs. Vectors in Fig. 2C were constructed by replacing the 2.2-kb ρ promoter with a 235-bp ρ-globin minimal promoter and cloning a DNA fragment containing 5′ hypersensitive sites 2 and 3 of the chicken β-globin cluster into the vector.
Figure 2.

Levels of ρ-globin gene promoter activity in primary chicken erythroid cells transiently transfected with the methylated or mock-methylated ρ-globin gene constructs depicted. Complete methylation of all CpGs in a particular region of a given construct is represented by attached M. Each experiment was carried out in triplicate, and levels of CAT conversion percentages presented represent the mean values and standard deviations. Methylation of the extended 2.2-kb upstream promoter resulted in significant reduction in promoter activity in the absence (A) or presence (B) of strong erythroid 3′ β/ɛ enhancer. Methylation of the 235-bp ρ-globin gene promoter also resulted in significant reduction in promoter activity even in the presence of strong 5′ enhancer, which includes erythroid-specific hypersensitive sites 2 and 3, whereas methylation of the pUC vector alone had no effect (C).
Methylation Reactions.
Whole plasmids were methylated using Sss 1 methylase (New England BioLabs) following the manufacturer’s protocol. The extent of methylation after each reaction was determined by digestion with MspI and HpaII. Region-specific methylation was carried out by excision of the fragment of interest by restriction digestion and gel isolation of the DNA fragment to be methylated. In each case, half of the DNA was methylated with Sss 1 methylase, and in a parallel control reaction the other half was incubated with methylase in the absence of S-adenosylmethionine (mock methylated) as a control. Methylated and mock-methylated DNA were religated into the vector DNA from which they were excised.
Transfection Assays.
The protocol of Lieber et al. (15) was followed for primary avian erythroid cell transfections except that osmotic shock was in 300 mM NH4Cl (pH 7.4) for 60 min at room temperature. Cells were cultured for 40–48 h in 10 ml of Leibovitz’s culture medium. Cells were harvested, and cytoplasmic extracts were assayed for CAT activity by using a liquid scintillation assay kit (Promega) (16). After extraction, reactions were counted in a Beckman liquid scintillation counter. All transfections were carried out in at least three independent experiments and in triplicate, and results are expressed as mean values with standard errors of the mean for each construct. Controls for transfection efficiency were carried out using an Rous sarcoma virus promoter enhancer-driven β-galactosidase reporter gene construct.
Bisulfite Genomic Sequencing for Determination of DNA Methylation.
After obtaining DNA from 5-day and adult chicken erythroid cells, bisulfite genomic sequencing was performed as described by Clark et al. (17) with the following modifications: (i) the bisulfite conversion reaction was carried out by incubating DNA with a 5 M bisulfite solution and 100 mM hydroquinone, pH 5.0, at 50°C for 4 h (18), and (ii) removal of free bisulfite was achieved by using a Qiaex II gel extraction kit (Qiagen). Sequencing of PCR-amplified product was performed using the dideoxy technique, [α-35S]dATP internal labeling protocol using internal primers, primer F (5′-TGTAGGGGTGTTTTGTGTAAG-3′) and primer R (5′-CTATAAAAACACTCAAAACTTAAAAC-3′), constructed after taking into account the bisulfite conversion reaction. The sequence of the unmodified sense strand for which these primers were constructed is depicted in Fig. 1B. Improved sequencing results were, however, obtained using α-33P-labeled ddNTP terminators.
Figure 1.
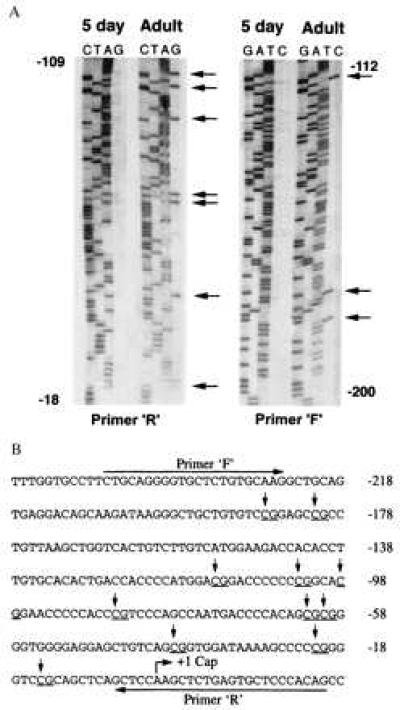
In vivo methylation of CpG dinucleotides of the rho promoter in 5-day and adult chicken erythroid cells using the bisulfite conversion technique. Arrows indicate methylated cytosines. Positions indicated are relative to the transcription start site (A). Cytosines that are not associated with CpG dinucleotides (sequence shown in B) have all been converted to thymidines in both 5-day and adult erythroid cells. (B) ρ-Globin gene promoter sequence. Arrows indicate methylated cytosines that are clearly seen in A (data not shown for CpG dinucleotide at position −15). Primers R and F indicate the sequence of ρ-globin gene promoter used for designing internal primers and for sequencing.
Electrophoretic Mobility Shift Assays.
HeLa cell and erythroid cell nuclear extracts were prepared according to the method of Dignam et al. (19). CG11 oligonucleotide (135 bp) containing 20 HhaI sites and 7 HpaII sites has been described (20). The rho 235 promoter fragment was obtained by restriction digestion of the plasmid rho 235-CAT. Probes were labeled using the Klenow fragment of DNA polymerase I and [α-32P]dATP. The electrophoretic mobility shift assay was performed as described (20). Micrococcus lysodeikticus DNA digested with Sau3aI was used as nonspecific competitor in all reactions.
RESULTS
Methylation Analysis of the 235-bp Minimal Promoter of the ρ-Globin Gene in Genomic DNA.
Though an inverse correlation between DNA methylation and expression of avian β-type globin genes (1, 13) has been reported, the methylation pattern of only a limited number of CpG dinucleotides has been described. Earlier techniques have depended on phosphodiester bond cleavage adjacent to cytosine residues using either chemical cleavage by hydrazine in Maxam and Gilbert sequencing reactions (21), which cleaves cytosine but not 5-methylcytosine, or methylation-sensitive restriction enzymes. As 5-methylcytosine is not cleaved in Maxam and Gilbert sequencing reactions, it is detected as a gap in a sequencing ladder, which may be difficult to interpret. Methylation-sensitive restriction enzyme-based techniques can only analyze a limited number of potentially methylated sites (22, 23). Recently, a technique has been described in which bisulfite-induced modification of genomic DNA was carried out under conditions that convert cytosine to uracil but that do not affect 5-methylcytosine. PCR amplification and sequencing of the modified DNA results in a detected sequence pattern showing conversion of all unmethylated cytosines to thymidine (since uracil is read by Taq polymerase as thymidine), whereas 5-methylcytosines remain as cytosines (17, 24). We used this technique to study the methylation pattern of CpG dinucleotides in the 235-bp minimal promoter of ρ-globin gene from erythroid cell genomic DNA of 5-day embryos and adult chickens. Previous reports have shown this promoter is capable of supporting high level, developmental specific transcription in primary avian erythroid cells (25). After bisulfite treatment, attempts to amplify a product larger than 600 bp were unsuccessful, as we were unable to detect a discrete product similar to the experience of Selker et al. (26). We were, however, able to obtain a discrete PCR product of 467 bp. Notably, similar reactions using genomic DNA not treated with bisulfite resulted in only a broad smear in ethidium-stained agarose gels. Dideoxy sequencing of the purified PCR product was carried out using initially [α-35S]dATP internal labeling protocol and subsequently with α-33P-labeled ddNTP terminators. As shown in Fig. 1A and in data not shown, sequencing of the PCR product showed the presence of cytosine residues (indicated by arrow) only in PCR products obtained from adult erythroid cell DNA. All unmethylated cytosine residues in the original sequence were converted to thymidines, demonstrating the completeness of the bisulfite conversion.
As summarized in Fig. 1B, cytosine residues in all CpG dinucleotides in the ρ-gene promoter were found to be methylated in the 235-bp minimal promoter of the ρ-globin gene in DNA from definitive adult chicken erythroid cells. In contrast, none of the cytosine residues were methylated in CpG dinucleotides from the same region in primitive 5-day embryonic erythroid cells.
Methylation of Whole Plasmid Containing the ρ-Gene 2.2-kb Upstream Promoter Driving the CAT Reporter Gene Suppresses Transcription.
We have found previously that methylation of a genomic ρ-globin gene construct containing 2.2 kb of upstream promoter sequences blocked transcription in stably transfected murine erythroleukemia cells (27). To study the cause and effect relationship between methylation and stage-specific globin gene transcription in normal erythroid cells, an expression construct consisting of 2.2 kb of ρ-globin gene promoter driving a CAT reporter gene was used to transfect primary erythroid cells from 5-day chicken embryos in a transient transfection assay system. Since all CpG dinucleotides were found to be methylated in the 235-bp minimal promoter from adult erythroid cell genomic DNA, methylation experiments were carried out using the enzyme Sss 1 methylase, which methylates all CpG dinucleotides. Methylation of the whole plasmid resulted in almost no detectable promoter activity. To assess the combination of individual components, methylation of the 2.2-kb promoter was carried out separately, and the product was religated into the unmethylated plasmid and transfected. Methylation of the 2.2-kb promoter resulted in an approximately 30-fold reduction in promoter activity compared with that from a mock-methylated control. A similar experiment, in which the CAT reporter gene alone was methylated and religated into the otherwise unmethylated plasmid construct and transfected, interestingly showed similar suppression of transcription compared with mock-methylated control (Fig. 2A).
Methylation of Whole Plasmid or 2.2-kb Upstream Promoter but Not the 3′ β/ɛ Enhancer Suppresses Transcription.
To see if the presence of a strong erythroid enhancer would override methylation-induced transcriptional repression, a 480-bp fragment containing chicken β-globin cluster the β/ɛ 3′ enhancer was ligated into the ρ-CAT plasmid described in Fig. 2A. Methylation of whole plasmid or the 2.2-kb ρ-promoter alone resulted in marked reduction in promoter activity. In contrast, a similar experiment in which the 3′ β/ɛ enhancer alone was methylated showed no significant difference as compared with mock-methylated control (Fig. 2B).
Methylation of the 235-bp ρ-Gene Promoter but Not the pUC-18 Vector Suppresses Transcription Despite the Strong 5′ Enhancer (Hypersensitive Sites 2 and 3).
As described (25), the major activity of the ρ-gene promoter seems to be contained within sequences lying between −246 and the transcription start site. A construct was therefore made consisting of a ρ-gene promoter (base pairs 0 to −235) driving a CAT reporter gene in which a DNA fragment containing 5′ hypersensitive sites 2 and 3 was cloned. Methylation of the 235-bp ρ-gene promoter resulted in an approximately 20-fold decrease in promoter activity even in the presence of a strong 5′ erythroid enhancer (hypersensitive sites 2 and 3) (Fig. 2C). These 5′ hypersensitive sites have been demonstrated to have strong erythroid-specific enhancer activity in transfection assays and transgenic mice (28, 29, 43). In contrast, complete methylation of the pUC-18 vector backbone, which contains 157 CpG dinucleotides, had a negligible effect on transcription from the ρ-gene promoter (Fig. 2C).
M-rho 235 Competes for MeCPC Binding.
The reported mechanisms of methylation-induced transcriptional repression seem mainly to be either by directly preventing the binding of transcription factors to the promoter or indirectly through proteins that bind preferentially to methylated DNA (30). Among transcription factors known to be sensitive to methyl-CpG, none seems to have a canonical binding site in the ρ-gene 235-bp promoter fragment (25). The known proteins and complexes that bind preferentially to methylated DNA include methylCpG binding protein 1 (MeCP-1) (20), methylCpG binding protein 2 (MeCP-2) (31), and methylated DNA-binding protein 2 (MDBP2) (32). MDBP2 has been implicated in the transcriptional repression of the vitellogenin gene, which could be overcome by estradiol stimulation (33). Subsequently purified MDBP2 was found to be histone H1 (34). Though some investigators have proposed preferential binding of histone H1 to methylated DNA and thereby implicated it in methylation-mediated transcriptional repression (35, 36), others have found this not to be the case (37). MeCP-1 binds in vitro to DNA containing at least 12 symmetrically methylated CpGs (20), whereas MeCP-2 can bind to a single methylated CpG (31). Though MeCP-2 seems to be essential for mouse development and despite earlier findings that it did not affect transcription of methylated promoter specifically (38), it has been shown recently to block transcription in cell lines (39). MeCP-1 has been implicated in methylation-mediated transcriptional repression of several genes including the human α-globin gene in heterologous cultured cells (40, 41). F9 cells, which contain low levels of MeCP-1, cannot efficiently repress those methylated gene promoters (41).
Because the ρ-gene 235-bp promoter sequence has multiple CpGs that were found to be completely methylated in genomic DNA from adult chicken erythroid cells in which the gene is repressed, we turned our attention toward MeCP-1 as a possible mediator of transcriptional repression in the primary erythroid cell transfection assay. End-labeled methylated and mock-methylated CG11 probes (M-CG11 and CG-11, respectively) were mixed with HeLa cell nuclear extract and then subjected to electrophoretic mobility shift assays to detect MeCP-1-like complex as described (20). On autoradiography, a complex was observed with M-CG11, but none was detected with CG11 as shown in Fig. 4. Similar to the findings reported earlier (20, 40), this complex was effectively competed by an excess of cold M-CG11 as well as M-rho 235 but not by excess of cold, unmethylated CG11 or rho 235 (Fig. 3).
Figure 4.
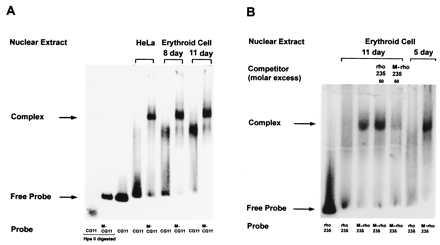
Mobility shift assay for the presence of MeCPC in chicken erythroid cell or control HeLa cell nuclear extracts. End-labeled, methylated CG11 probe (M-CG11) but not mock-methylated CG11 (CG11) forms a complex with erythroid cell nuclear extract that has similar mobility as seen with HeLa cell nuclear extract (A). Similar results were seen in 5-day chicken erythroid cell nuclear extract (data not shown). (B) Binding of MeCPC in chicken erythroid cell nuclear extract to methylated rho 235 (M-rho 235) probe. End-labeled M-rho 235 but not mock-methylated rho 235 forms a complex with erythroid cell nuclear extract. This complex was competed effectively by an excess of unlabeled methylated but not mock-methylated rho 235.
Figure 3.
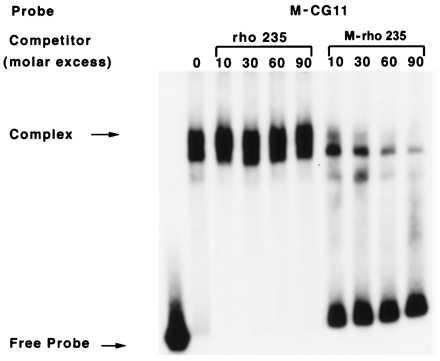
Mobility shift assay for MeCPC binding to methylated CG11 probe (M-CG11). 0.6 ng of methylated CG11 end-labeled probes were mixed with 6 μg of HeLa cell nuclear extract and varying amounts of mock-methylated or methylated rho 235 promoter DNA fragment. The reaction mixture was electrophoresed in a 1.5% agarose gel, and complexes were analyzed by autoradiography. This autoradiogram illustrates the effective competition of MeCPC binding to M-CG11 probe by methylated rho 235 but not by mock-methylated rho 235 (M-rho 235).
M-CG11 Competes for MeCP-1-Like Complex Binding by M-rho 235.
Because M-rho 235 effectively competed for MeCP-1 binding by M-CG11, the possibility that it could directly form a complex with MeCP-1 was examined. In a reverse experiment, end-labeled methylated and mock-methylated rho 235 probes were used in band shift assays with HeLa cell nuclear extract. A complex was observed with M-rho 235, but none was detected with (mock-methylated) rho 235. This complex was effectively competed by an excess of unlabeled M-rho 235 but not by an excess of unlabeled rho 235. Competition of this complex with M-CG11 and CG11 showed effective competition by M-CG11 but not by (mock-methylated) CG11 (data not shown).
MeCPC Is Present in Nuclear Extracts from Primary Erythroid Cells and a Similar Complex Binds to Methylated rho 235.
Having demonstrated the binding of MeCPC to methylated rho 235 using HeLa cell nuclear extract, its presence in the primary chicken erythroid cells used in the transient transfection functional assays was investigated. End-labeled methylated and mock-methylated CG11 probes were mixed with erythroid cell nuclear extracts derived from 8- and 11-day chicken embryos. Electrophoretic mobility shift assays showed the presence of a complex with similar mobility as seen with HeLa cell nuclear extracts, with M-CG11 but not with CG11, as shown in Fig. 4A.
Previous studies of MeCP-1 have relied on competition between methylated promoter sequences and methylated CG11 probe to demonstrate its interaction with the promoter under study. To extend these findings, we investigated directly the ability of methylated rho 235 minimal promoter to bind to MeCP-1-like methylation-dependent complex using homologous primary avian erythroid cell nuclear extracts.
As illustrated in Fig. 4B, the formation of such a complex with the rho 235-bp promoter sequence was detected in 5-day (primitive) and 11-day (definitive) avian erythroid cell nuclear extracts. This methylation-dependent complex was easily competed by an excess of unlabeled methylated rho 235 but not by an excess of unlabeled mock-methylated rho 235.
DISCUSSION
The principal findings in this report are as follows. Every cytosine within a CpG dinucleotide in a 235-bp region of the embryonic ρ-globin gene promoter is methylated in normal adult (definitive) erythroid cells in which the gene is silent and completely unmethylated in 5-day (primitive) erythroid cells in which the gene is actively transcribed. Previous studies in our laboratory have shown extensive CpG methylation in a 4.6-kb fragment that includes 1.5 kb of 5′ and 3′ of ρ-gene flanking sequences in definitive erythroid cells (14). In vitro methylation of those same CpG sites in the 235-bp ρ-gene promoter fragment markedly reduces the promoter activity in a transient transfection assay in ex vivo primary avian erythroid cells, despite the presence of a strong erythroid-specific enhancer. The silencing effect of methylation is not overridden even in the nuclear environment of 5-day erythrocytes, which contain all the nuclear factors necessary for high level transcription of the ρ-gene. Finally, we show that a methyl CpG binding protein complex, which behaves like MeCP-1 (20), binds to methylated but not unmethylated ρ-promoter sequences.
Numerous reports have shown the ability of promoter DNA methylation to inhibit transcription of a wide variety of genes, and in some cases such methylation corresponds to the inactive state of the gene under study in vivo (reviewed in refs. 4 and 5). In the case of globin genes, inhibition of expression of the human γ-globin gene in nonerythroid murine L-cells was observed when all CpGs in the promoter were methylated in a stable transfection assay (42). More recently, it was shown that methylation of all CpGs in the human α-globin gene could block transcription in transfected nonerythroid HeLa cells and in cell-free in vitro transcription assays derived from the same cell type (40, 41). In neither of these studies was the in vivo methylation pattern of all CpGs of the specific globin gene in question reported, nor was the pattern of methylation or its effect on developmental stage-specific expression in primary erythroid cells tested.
In this report we show that methylation of the embryonic ρ-globin gene promoter is capable of attenuating transcription in primary erythroid cells even in the presence of strong erythroid-specific enhancers that we have shown (25, 43) to be capable of overcoming the stage-specific silencing effects of the ρ-gene promoter in primary erythroid cells. However, methylation of the 3′ β/ɛ erythroid enhancer or the pUC-18 vector backbone had no effect, demonstrating that the blocking action of CpG methylation on ρ-promoter driven transcription resides in the sequences of the transcription unit. Of interest, the level of transcription for the methylated rho 235-bp promoter was significantly higher in the presence of the strong erythroid enhancer. Quantitation of the increased transcription levels conferred by either the 3′ β/ɛ on 5′ hypersensitive site (HS) enhancers indicated that the magnitude of increase (10–20-fold) was approximately the same for methylated vs. unmethylated promoter constructs in concomitant transfection assays (data not shown). Thus, even though methylation decreased the frequency of productive transcriptional complex formation by one to two orders of magnitude, the two erythroid enhancers tested could still markedly increase that frequency in homologous primary erythroid cells. These results support the concept that tissue-specific enhancers can at least partially override repressive influences on promoters, at least as assayed in transient transfection systems.
Previous studies have shown the ability of the methylated DNA binding protein complex, MeCP-1, to block transcription of the human α-globin gene promoter even in the presence of the strong simian virus 40 enhancer (40, 41). Here, we show that a similar methylated DNA binding complex forms efficiently with the methylated ρ-gene promoter sequence and that the complex can be detected using nuclear extracts from the same primary avian erythroid cells in which methylation-mediated transcriptional inhibition was demonstrated. Thus, these results support a role for MeCP-1 or a similar complex in developmental silencing of embryonic globin genes in normal erythropoiesis. Determination of whether the complex formed between the methylated ρ-promoter and nuclear extracts from primary avian erythroid cells is the same as MeCP-1 will await full characterization of both of these large protein–DNA complexes. Direct evidence that this complex blocks transcription in this system will require additional studies, which are underway.
Alternatively, it is possible that methylation of the ρ-gene promoter directly blocks binding of one or more specific transcriptional activating DNA binding factors. This seems unlikely as none of the sequence recognition sites for the known activating factors that bind to the ρ-promoter contain CpGs in their core recognition sequences (25). Also, such a localized sequence-specific effect was not observed for the human γ-globin or α-globin genes (40, 42). Further support for some type of blocking effect on transcription complex formation, promoter clearance, or transcription elongation is provided by the experiment in which methylation of the CAT reporter cassette sequences only (which also contain multiple CpGs) inhibited transcription. Quantitation of the CAT reporter assay showed that methylation of the CAT cassette inhibited expression by 5-fold less than methylating the ρ-promoter sequences. This result, coupled with the finding that methylation of the entire expression plasmid did not inhibit CAT expression more than methylation of the 235-bp ρ-promoter alone, suggests that the promoter methylation observed in definitive erythroid cells is a major mediator of methylation-induced silencing of this gene in vivo. Although binding of MeCP-1 by the methylated CAT sequences has not yet been demonstrated, this seems a likely mechanism for the observed effect. Regardless, this result does support the notion that the repressive effect of CpG methylation need not necessarily be mediated through the promoter.
The results presented here also support the notion that DNA methylation inhibits globin gene expression in a stage-specific fashion, at least in part by a direct interference with transcription initiation, elongation, or promoter clearance rather than by inducing an inactive chromatin structure, as transient transfection of nonreplicating cells does not result in intact chromatin structure. It is still possible that additional effects of CpG methylation downstream of the promoter and in the context of intact chromatin may not be detected in the transient transfection assays used in this report and previous studies (40, 41). However, a direct effect on the transcriptional machinery is most consistent with the evidence from several labs, including our own, suggesting that the entire avian β-globin gene cluster is in an open chromatin domain in erythroid cells of all developmental stages (44–46).
The potent transcriptional silencing effect of physiologically accurate DNA methylation of the ρ-gene promoter even in the nuclear environment of transcriptionally active and stage-specific primary erythroid cells suggests that methylation plays a dominant role in maintaining the high level of repression of embryonic globin gene transcription in definitive or adult erythroid cells. By analogy, such a strong suppressive effect of DNA methylation suggests a role for active demethylation in relieving repression of CpG-rich promoters that are highly methylated in cells containing abundant MeCP-1 or like complexes. Recently, evidence for such sequence-specific demethylating activity has been reported (47).
Strong repression of transcription from the ρ-globin gene during the embryonic developmental switch from primitive to definitive erythroid cell lineages is not likely a prerequisite for survival. Nonetheless, it seems feasible that mechanisms analogous to those described here may control genes that are critical for embryonic differentiation and viability, as evidenced by the results obtained in DNA methylase or methyl binding protein-deficient mouse embryos (8) and embryonic stem cells (48). The results presented in this report are direct evidence of a transcriptional inhibitory role for DNA methylation in the silencing of a specific developmentally regulated gene in homologous primary embryonic cells.
Acknowledgments
We thank Joan Boyes, National Institutes of Health, for providing plasmids CG11 and M. lysodeikticus and for technical advice; we also thank Vivian Bardwell and Kathleen Conklin for critical reading of the manuscript. This work was supported by National Institutes of Health Grant R-37DK29902 and the Masonic Cancer Center Professorship Fund.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: MeCPC, methyl cytosine binding protein complex; CAT, chloramphenicol acetyltransferase.
References
- 1.McGhee J D, Ginder G D. Nature (London) 1979;280:418–420. doi: 10.1038/280419a0. [DOI] [PubMed] [Google Scholar]
- 2.Shen C K J, Maniatis T. Proc Natl Acad Sci USA. 1980;77:6634–6638. doi: 10.1073/pnas.77.11.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van der Ploeg L H T, Flavell R A. Cell. 1980;19:947–958. doi: 10.1016/0092-8674(80)90086-0. [DOI] [PubMed] [Google Scholar]
- 4.Razin A, Cedar H. Microbiol Rev. 1991;55:451–458. doi: 10.1128/mr.55.3.451-458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird A. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 6.Urieli-Shoval S, Gruenbaum Y, Sedat J, Razin A. FEBS Lett. 1982;146:148–152. doi: 10.1016/0014-5793(82)80723-0. [DOI] [PubMed] [Google Scholar]
- 7.Proffitt J H, Davie J R, Swinton D, Hattman S. Mol Cell Biol. 1984;4:985–988. doi: 10.1128/mcb.4.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li E, Bestor T H, Jaenisch R. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 9.Ronemus M J, Galbiati M, Ticknor C, Chen J, Dellaporta S L. Science. 1996;273:654–657. doi: 10.1126/science.273.5275.654. [DOI] [PubMed] [Google Scholar]
- 10.Brown J L, Ingram V M. J Biol Chem. 1974;249:3960–3972. [PubMed] [Google Scholar]
- 11.Groudine M, Peretz M, Weintraub H. Mol Cell Biol. 1981;1:281–288. doi: 10.1128/mcb.1.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landes G M, Martinson H G. J Biol Chem. 1982;257:11002–11007. [PubMed] [Google Scholar]
- 13.Ginder G D, McGhee J D. In: Globin Gene Expression and Hematopoietic Differentiation. Stamatoyannopoulos G, Nienhuis A W, editors. New York: Liss; 1981. pp. 191–201. [Google Scholar]
- 14.Ginder G D, Whitters M J, Pohlman J K. Proc Natl Acad Sci USA. 1984;81:3954–3958. doi: 10.1073/pnas.81.13.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieber M R, Hesse J E, Nickol J M, Felsenfeld G. J Cell Biol. 1987;105:1055–1065. doi: 10.1083/jcb.105.3.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafson K S, Ginder G D. J Biol Chem. 1996;271:20035–20046. doi: 10.1074/jbc.271.33.20035. [DOI] [PubMed] [Google Scholar]
- 17.Clark S J, Harrison J, Paul C L, Frommer M. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raizis A M, Schmitt F, Jost J-P. Anal Chem. 1995;226:161–166. doi: 10.1006/abio.1995.1204. [DOI] [PubMed] [Google Scholar]
- 19.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meehan R R, Lewis J D, McKay S, Kleiner E L, Bird A P. Cell. 1989;58:499–507. doi: 10.1016/0092-8674(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 21.Maxam A, Gilbert W. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 22.Singer J, Roberts-Ems J, Riggs A D. Science. 1979;203:1019–1021. doi: 10.1126/science.424726. [DOI] [PubMed] [Google Scholar]
- 23.Singer-Sam J, Grant M, LeBon J M, Okuyama K, Chapman V, Monk M, Riggs A D. Mol Cell Biol. 1990;10:4987–4989. doi: 10.1128/mcb.10.9.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frommer M, McDonald L E, Millar D S, Collis C M, Watt F, Grigg G W, Molloy P L, Paul C L. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minie M E, Kimura T, Felsenfeld G. Development. 1992;115:1149–1164. doi: 10.1242/dev.115.4.1149. [DOI] [PubMed] [Google Scholar]
- 26.Selker E U, Fritz D Y, Singer M J. Science. 1993;262:1724–1728. doi: 10.1126/science.8259516. [DOI] [PubMed] [Google Scholar]
- 27.Wandersee N. Ph.D. thesis. Iowa City: Univ. of Iowa; 1996. [Google Scholar]
- 28.Reitman M, Lee E, Westphal H. Nucleic Acids Res. 1995;23:1790–1794. doi: 10.1093/nar/23.10.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason M M, Lee E, Westphal H, Reitman M. Mol Cell Biol. 1995;15:407–414. doi: 10.1128/mcb.15.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tate P H, Bird A P. Curr Opin Gen Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 31.Lewis J D, Meehan R R, Henzel W J, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 32.Pawlak A, Bryans M, Jost J-P. Nucleic Acids Res. 1991;19:1029–1034. doi: 10.1093/nar/19.5.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jost J P, Saluz H P, Pawlak A. Nucleic Acids Res. 1991;19:5771–5775. doi: 10.1093/nar/19.20.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jost J P, Hofsteenge J. Proc Natl Acad Sci USA. 1992;89:9499–9503. doi: 10.1073/pnas.89.20.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine A, Yeivin A, Ben-Asher E, Aloni Y, Razin A. J Biol Chem. 1993;268:21754–21759. [PubMed] [Google Scholar]
- 36.Johnson C, Goddard J, Adams R. Biochem J. 1995;305:791–798. doi: 10.1042/bj3050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bird, A., Tate, P., Nan, X., Campoy, J., Meehan, R., Cross, S., Tweedie, S., Charlton, J. & Macload, D. (1995) J. Cell Sci. 19, Suppl., 37–39. [DOI] [PubMed]
- 38.Meehan R R, Lewis J D, Bird A P. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nan X, Campoy F J, Bird A. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 40.Boyes J, Bird A. EMBO J. 1992;11:327–333. doi: 10.1002/j.1460-2075.1992.tb05055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyes J, Bird A. Cell. 1991;64:1123–1134. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- 42.Busslinger M, Hurst J, Flavell R A. Cell. 1983;34:197–206. doi: 10.1016/0092-8674(83)90150-2. [DOI] [PubMed] [Google Scholar]
- 43.Wandersee N J, Ferris R C, Ginder G D. Mol Cell Biol. 1996;16:236–246. doi: 10.1128/mcb.16.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stalder J, Larsen A, Engel J D, Dolan M, Groudine M, Weintraub H. Cell. 1980;20:451. doi: 10.1016/0092-8674(80)90631-5. [DOI] [PubMed] [Google Scholar]
- 45.Wood W I, Felsenfeld G. J Biol Chem. 1982;257:7730–7736. [PubMed] [Google Scholar]
- 46.Brotherton T W, Reneker J, Ginder G D. Nucleic Acids Res. 1990;18:2011–2016. doi: 10.1093/nar/18.8.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiss A, Keshet I, Razin A, Cedar H. Cell. 1996;86:709–718. doi: 10.1016/s0092-8674(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 48.Lei H, Oh S P, Okano M, Jüttermann R, Goss K A, Jaenisch R, Li E. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]