

Abstract

Volatility and low-affinity hamper an ability to define molecular targets of the inhaled anesthetics. Photolabels have proven to be a useful approach in this regard, although none have closely mimicked contemporary drugs. We report here the synthesis and validation of azi-isoflurane, a compound constructed by adding a diazirinyl moiety to the methyl carbon of the commonly used general anesthetic isoflurane. Azi-isoflurane is slightly more hydrophobic than isoflurane, and more potent in tadpoles. This novel compound inhibits Shaw2 K+ channel currents similarly to isoflurane and binds to apoferritin with enhanced affinity. Finally, when irradiated at 300 nm, azi-isoflurane adducts to residues known to line isoflurane-binding sites in apoferritin and integrin LFA-1, the only proteins with isoflurane binding sites defined by crystallography. This reagent should allow rapid discovery of isoflurane molecular targets and binding sites within those targets.
Keywords: Anesthesia; Shaw2 K+ channel; integrin I domain; photoaffinity labeling; binding, apoferritin
The inhaled general anesthetics are generally recognized to be promiscuous pharmaceuticals whose important molecular targets underlying any of their many physiological effects are not well-defined (1). A conventional means of defining targets is through binding assays, but this has been difficult for the general anesthetics since the interactions are of low affinity and therefore extremely transient (2). This transient binding nature is further aggravated by the volatility of many of the drugs. We have previously introduced photoaffinity labeling to overcome these problems (3), and this approach has provided considerable insight into protein/anesthetic interactions. However, none of these analogs have mimicked any of the contemporary inhaled anesthetics, such as isoflurane.
The molecular targets and sites underlying the effects of isoflurane are currently of considerable interest. This stems not only from an incomplete knowledge of the targets that contribute to unconsciousness (1,4), but also from the targets underlying isoflurane’s neurotoxic5 and “preconditioning” effects (6). Thus, we describe here the synthesis and validation of a photoactive analog of isoflurane. By incorporating a diazirinyl moiety in isoflurane, we produce a molecule that demonstrates photoadduction to residues in known anesthetic binding sites, and retention of normal in vivo and in vitro actions.
Results and Discussion
Synthesis of Azi-isoflurane (1)
Preparation of azi-isoflurane (1) followed our previously described method for the preparation of similar diazirines (Scheme 1) (7). Previously described ester 2(8) was converted to its hemiacetal using excess lithium aluminum hydride at low temperature. We have previously used DIBAL-H to effect this reduction but have found that the electron-withdrawing groups in 2 prevent over-reduction by LiAlH4 at this low temperature. The crude hemiacetal was immediately condensed with tert-butylamine in refluxing benzene to form imine 3 in 75% overall yield from 2. Treatment of 3 with hydroxylamine-O-sulfonic acid (HOSA) in absolute ethanol and triethylamine produced a 50% yield of diaziridines 4 as an approximately 1:1 mixture of diastereomers as determined by capillary gc. The mixture of diastereomers 4 was converted to azi-isoflurane (1) using N-bromosuccinimide (NBS) in dichloroethane. Final purification of racemic 1 was accomplished using preparative gas chromatography. Figure 1 shows structures of isoflurane and azi-isoflurane, and NMR spectra for the synthesized compounds are contained in the Supporting Information.
Scheme 1. Preparation of Azi-isoflurane (1).

Figure 1.
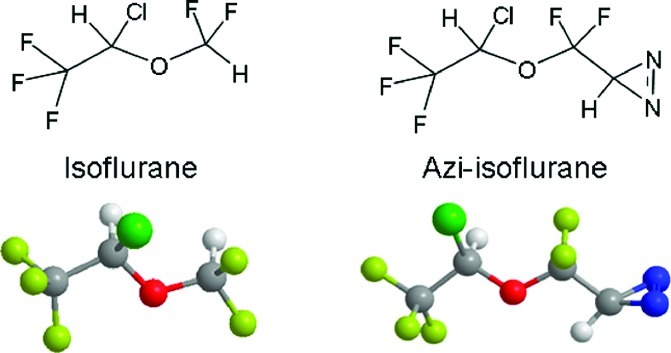
Structure comparison of the target drugs: isoflurane on the left and the photolabel analog, azi-isoflurane, on the right.
Physicochemical Properties
The properties of azi-isoflurane are similar to those of isoflurane (Table 1), except that it is more hydrophobic, the octanol/water partition coefficient being ∼300 compared with 125 for isoflurane. The calculated dipole is 1.5 D, while that of isoflurane, calculated the same way, is 1.67 D. The absorption spectra shows the prominent double-humped diazirine peak at 280−320 nm; thus all photolysis exposures were conducted at 300 nm. In phosphate-buffered saline and with our lamp, the disappearance rate of the diazirine has a t1/2 of 3.9 min.
Table 1.
| MW | density, g/mL | log Pa | dipole, Db | HSAF KD, μM | tadpole EC50, μM | |
|---|---|---|---|---|---|---|
| isoflurane | 184 | 1.5 | 2.1 | 1.67 | 58 ± 2 | 230 (193−275) |
| azi-isoflurane | 224 | 1.4 | 2.4 | 1.50 | 6 ± 1 | 106 (96−119) |
Octanol/water partition coefficient.
Calculated.
Binding Assays
Titration of azi-isoflurane to horse spleen apoferritin (HSAF) produced a typical exothermic enthalpogram as shown in Figure 2. Binding parameters indicate slightly enhanced affinity compared with isoflurane.
Figure 2.

Isothermal titration calorimetry. Titration of azi-isoflurane (left panel) and isoflurane (right panel) into a solution of HSAF resulted in classic exothermic enthalpograms. Fits to single-class binding models found average KA values for isoflurane of 17 260 M−1 and for azi-isoflurane of 172 200 M−1. The enthalpy change was favorable in both cases (ΔH values averaged −6 kcal for isoflurane and −9.8 kcal for azi-isoflurane), while the entropy was much more unfavorable for azi-isoflurane (ΔS = −0.3 for isoflurane and −9.6 for azi-isoflurane).
Electrophysiological Studies
The Shaw2 K+ channel has been identified as an archetypical target of n-alcohols and inhaled general anesthetics (9,10). This K+ channel was selected for this study because it is inhibited by relevant doses of inhaled anesthetics but is resistant to propofol, a typical intravenous general anesthetic. In contrast, ligand-gated ion channels do not typically exhibit this selectivity (11). Therefore, the Shaw2 K+ channel is a more stringent subject to compare the functional effects of isoflurane and azi-isoflurane on an individual target. Consistent with the modulation of Shaw2 K+ channels by halothane (not shown), whole-oocyte Shaw2 K+ currents were substantially inhibited by 1 and 2 mM of both isoflurane and azi-isoflurane (Figure 3), and azi-isoflurane seemed slightly more potent than isoflurane.
Figure 3.

Inhibition of Shaw2 K+ channels by azi-isoflurane and isoflurane. (A) Whole-oocyte Shaw2 currents evoked by step depolarizations from −100 to +60 mV delivered at 30 s intervals. Several superimposed traces are shown with the drug exposure indicated by a bar. The corresponding time course for the experiment is shown directly below the traces. Azi-isoflurane at 2 and 1 mM inhibited currents by 77% ± 0.2% (n = 2) and 27% ± 5% (n = 2), respectively, with rapid recovery on compound washout. (B) The same experiment as in panel A done with isoflurane with the corresponding time course below. Isoflurane at 2 and 1 mM inhibited currents by 67% ± 0% (n = 2) and 18% ±4% (n = 2), respectively. (C). Summarized maximal inhibition for both isoflurane and azi-isoflurane; n = 2 separate experiments for each concentration.
In Vivo Anesthetic Potency
Tadpoles were fully immobilized by both isoflurane and azi-isoflurane (Figure 4). The Hill slopes were indistinguishable, but the EC50 value for azi-isoflurane was approximately 2-fold smaller (higher potency) than for isoflurane. Recovery from both compounds, even at maximal concentrations, was rapid, and no mortality out to 24 h was noted.
Figure 4.

Tadpole potency. Shown are concentration−effect curves for both isoflurane (●) and azi-isoflurane (○). Points represent the average of two determinations each in ten tadpoles. Lines are best fit Hill plots, constraining the bottom to 0. EC50 values (95% CI) for isoflurane and azi-isoflurane, respectively, were 230 (193−275) μM and 107 (96−119), and for Hill slope, −5.7 (−11 to −1) and −7.1 (−12 to −2).
Photolabeling
For validation of photolabeling reliability, the only two proteins with crystallographically proven binding sites for isoflurane were selected (12,13). HSAF and both the wild-type (WT) and high-affinity (HA) integrin inserted (I) domain of LFA-1 (lymphocyte function associated antigen-1) peptides were incubated with buffer with or without 1 mM azi-isoflurane and exposed to 300 nm illumination for 10 min. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS−PAGE) purification, band excision, and trypsinization was followed by nano-LC/MS to identify peptides and residues that had been modified by 196 Da. Both the HSAF (Table 2) and WT I domain, but not the HA I domain (Table 3) demonstrated clear evidence of adducted peptides. In each case, these residues are the same as those found to interact with isoflurane from X-ray crystallography studies (PDB codes 1XZ3 and 3F78) (12,13), and no other adducted residues were noted. MS spectra are contained in the Supporting Information.
Table 2. Photoadducted Residues in HSAF (Sequence Coverage 53.7%) Peptide Starting at E53a.
| UV only |
UV + azi-isoflurane |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| b | y | b | y | ||||||
| E | 1 | 130.0499 | 12 | E | 1 | 130.0499 | 12 | ||
| L | 2 | 243.1339 | 1287.665 | 11 | L | 2 | 243.1339 | 1483.665 | 11 |
| A | 3 | 314.171 | 1174.581 | 10 | A | 3 | 314.171 | 1370.581 | 10 |
| E | 4 | 443.2136 | 1103.544 | 9 | E | 4 | 443.2136 | 1299.544 | 9 |
| E | 5 | 572.2562 | 974.5014 | 8 | E | 5 | 572.2562 | 1170.501 | 8 |
| K | 6 | 700.3512 | 845.4588 | 7 | K | 6 | 700.3512 | 1041.459 | 7 |
| R | 7 | 856.4523 | 717.3638 | 6 | R* | 7 | 1052.452 | 913.3638 | 6 |
| E | 8 | 985.4949 | 561.2627 | 5 | E | 8 | 1181.495 | 561.2627 | 5 |
| G | 9 | 1042.516 | 432.2201 | 4 | G | 9 | 1238.516 | 432.2201 | 4 |
| A | 10 | 1113.553 | 375.1987 | 3 | A | 10 | 1309.553 | 375.1987 | 3 |
| E | 11 | 1242.596 | 304.1615 | 2 | E | 11 | 1438.596 | 304.1615 | 2 |
| R | 12 | 175.119 | 1 | R | 12 | 175.119 | 1 | ||
Bold indicates ion detected on MS. Bold with * indicates adducted residue. Note that b and y ions represent MS “sequencing” in different directions along the peptide; b ions are those from cleavages moving from N to C terminus, while y ions are the reverse.
Table 3. Photolabeled Residues in LFA-1 WT (Sequence Coverage 98.9%) Peptide Starting at Y257a.
| UV + azi-isoflurane | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| b | y | b | y | ||||||
| Y | 1 | 164.0706 | 7 | Y* | 1 | 164.0706 | 7 | ||
| I* | 2 | 473.1547 | 796.4079 | 6 | I | 2 | 473.1547 | 796.4079 | 6 |
| I | 3 | 586.2387 | 487.3239 | 5 | I | 3 | 586.2387 | 487.3239 | 5 |
| G | 4 | 643.2602 | 374.2398 | 4 | G | 4 | 643.2602 | 374.2398 | 4 |
| I | 5 | 756.3443 | 317.2183 | 3 | I | 5 | 756.3443 | 317.2183 | 3 |
| G | 6 | 813.3657 | 204.1343 | 2 | G | 6 | 813.3657 | 204.1343 | 2 |
| K | 7 | 147.1128 | 1 | K | 7 | 147.1128 | 1 | ||
Bold indicates ion detected on MS. Bold with * indicates adducted residues.
Discussion
In this study, we have modified isoflurane by the addition of a diazrinyl group (CHN2) for total mass addition of 40 Da (about a 20% increase in MW). Although this group has some polar character, azi-isoflurane is somewhat more hydrophobic than isoflurane but with the same approximate dipole moment (Table 1).
In accordance with the Overton−Meyer relationship (14,15), azi-isoflurane is more potent than isoflurane and has a slightly larger effect on a selective ion channel. Further, it binds specifically and with a higher affinity to the general anesthetic binding site on apoferritin (16) as shown by isothermal titration calorimetry (ITC). Collectively, these studies suggest that the intact photolabel is highly analogous and somewhat more active than isoflurane itself.
Of greatest interest is whether azi-isoflurane can efficiently report sequence-level binding in its molecular targets. To validate this, we used the only two isoflurane binding proteins confirmed with crystal structures that have been deposited in the PDB. In both HSAF and the integrin LFA-1 I-domain, the only adducted resides found with LC/MS are those implicated by the crystal structure. In the case of HSAF, this is arginine-59, which is positioned at the entrance to the interfacial cavity where anesthetics bind. The side chain of Arg-59 is facing solvent, thus we suspect that adduction occurs to the carbonyl oxygen, facing the cavity. In the integrin I domain, the adduction occurs at tyrosine-257, the side chain of which is positioned directly against isoflurane in 3F78(13). As crystallographically defined (17) (PDB 1MQA), the HA mutant of the integrin I-domain is engineered to distort the allosteric cavity where isoflurane was found to bind in the WT domain and thereby remove the inhibitory effect of isoflurane (18). We were able to show that this is indeed due to a loss of binding at this site, in that we could not find the adduct in the same peptide that was easily detected in the WT domain. This is the first clear example of a conformational change induced by mutagenesis that actually precludes anesthetic binding, removing its effect.
An optimal photolabel should not show selectivity toward amino acids to give confidence that equilibrium sites are being reliably reported. For example, prior labels that relied on carbon-centered radicals appeared to show selectivity toward aromatics like tyrosine (19) and tryptophan (20). In this study, we find evidence for azi-isoflurane adduction to arginine, tyrosine, and isoleucine, and in prior studies, a similar compound labeled serine and leucine (7). The side chains of these residues exhibit little chemical similarity, demonstrating a comforting lack of adduction selectivity. An alternative explanation is that activated azi-isoflurane may target backbone atoms, such as the carbonyl oxygen, explaining the lack of selectivity. Regardless of targeted moiety, photochemical promiscuity is a desirable feature in a photolabel.
Detection of adducted proteins and amino acids in this study employed mass spectrometry. However, since the adduction itself renders the peptide more hydrophobic, it is possible that some adducted peptides will either not transit the HPLC column or not volatilize in the mass analyzer. It might therefore be desirable to radiolabel azi-isoflurane to allow a different detection methodology. Azi-isoflurane has the advantage of having an exchangeable hydrogen on the chlorine-bearing carbon, which should allow tritiation. Consistent with this possibility, preliminary studies have shown efficient deuterium incorporation at this position under basic conditions.
In conclusion, we have synthesized a photoactive analog of isoflurane that is more potent and of higher affinity than isoflurane, binds to the same protein sites as isoflurane, and demonstrates rapid and nonselective photoincorporation into proteins. This reagent should provide for rapid progress on identification of molecular targets underlying isoflurane’s many effects.
Methods
Materials
Horse spleen apoferritin (HSAF) was obtained from Sigma (St. Louis, MO). All other chemicals were of reagent grade or better and were obtained from Sigma or Aldrich (St. Louis, MO) . Isoflurane was obtained from Butler (Dublin, OH). Caution: All diazirines are potentially explosive and should be treated with care. The summary NMR and high-resolution mass spectra given below are consistent with the assigned structures. Detailed spectra are provided in the Supporting Information. Final purified products were racemic and >98% pure by gas chromatographic (GC) analysis using a 30 m dimethylsilicone capillary column and flame ionization detection. WT and HA integrin LFA-1 α inserted (I) domains were bacterially expressed as inclusion bodies, refolded, and purified to homogeneity as previously described (17).
Preparation of tert-Butyl-(2,2-difluoro-2-(1-chloro-2,2,2-trifluoroethoxy)ethylidene) Amine (3)
A mixture of 1.5 g (39 mmol) of LiAlH4 powder and 50 mL of dry ether were stirred at RT for 1 h under N2. This mixture was added to an addition funnel attached directly to a 250 mL round-bottom (rb) flask previously filled with 15.2 g (56.3 mmol) of ester 2, 30 mL of dry ether, and a magnetic stir bar. The contents of the flask were cooled to −78 °C with stirring under N2. The LiAlH4 mixture was added dropwise over the course of 5 min. After being stirred for an additional 30 min at −78 °C, the mixture was poured into a cold solution of 5 mL of concentrated H2SO4 and 500 mL of water. The mixture was stirred for several minutes until all the solid had dissolved and was then extracted with ether (3 × 200 mL). The combined ether extracts were concentrated using a rotary evaporator at RT. The resulting oil was dissolved in 50 mL of benzene, 12 mL (8.3 g, 114 mmol) of tert-butylamine was added, and the solution was heated with a Dean−Stark water separator overnight. The contents of the flask were distilled under atmospheric pressure to yield 11.8 g (78%) of clear colorless oil, bp 153−154 °C. 1H NMR: δ 7.47 (t, 1 H, JH−F = 4.7 Hz), 6.20 (q, 1 H, JH−F = 4.0 Hz), 1.23 (s, 9 H). 13C NMR: δ 145.5 (t, JC−F = 146 Hz), 120.3 (q, JC−F = 280 Hz), 117.9 (t, JC−F = 270 Hz), 78.8 (qt, JC−F = 40, 5.5 Hz), 58.9, 28.7. 19F NMR: δ −78.22 (bd, 1 F, JF−F = 146 Hz), −78.96 (dd, 1 F, JF−F = 146 Hz, JH−F = 4.7 Hz), −80.0 (bs, 3 F). HRMS (CI+): m/z calculated for C8H12ClF5NO (M + H)+ 268.0528; found 268.0525.
Preparation of 1-tert-Butyl-3-(difluoro-(1-chloro-2,2,2-trifluoroethoxy)methyl)diaziridine (4)
To a 25 mL rb flask with stir bar was added 1.08 g (4.0 mmol) of 3 and 2 mL of absolute ethanol, and the solution was cooled in an ice bath. A mixture of 0.48 g (4.2 mmol) of hydroxylamine-O-sulfonic acid (HOSA) in 2.5 mL of absolute ethanol was cooled in an ice bath, and 0.40 g (4.0 mmol) of triethylamine was added dropwise over the course of 2 min with good stirring. The resulting clear, colorless HOSA solution was added dropwise to the solution of 3 over the course of 5 min, and the resulting solution was stirred for 20 min at 0 °C. The ice bath was removed, and the mixture was allowed to stir at rt for 1 h during which time white precipitate formed. The mixture was evaporated on a rotary evaporator, and the resulting semisolid was triturated with ether (3 × 20 mL). Evaporation of the ether left 0.56 g (50%) of clear, colorless oil that was sufficiently pure for conversion to diazirine. An analytical sample was purified by silica gel chromatography using 1:10 ethyl acetate/hexane. The product was an approximately 1:1 mixture of two diastereomers as analyzed by capillary gas chromatography. 1H NMR: δ 6.14 (m, 1 H), 3.19 (m, 1 H), 2.16 (m, 1 H) 1.30 (m, 9 H). 13C NMR: δ 121.39 (t, JC−F = 271 Hz), 120.28 (q, JC−F = 280 Hz), 78.53 (m), 55.84, 50.92 (t, JC−F = 35.8 Hz), 50.89 (t, JC−F = 36.4 Hz), 25.25, 25.23. 19F NMR: δ −80.16 (m, 6 F), −83.07 (bd, 1 F, JF−F = 144 Hz), −83.55 (bd, 1 F, JF−F = 144 Hz), −83.60 (bd, 1 F, JF−F = 141 Hz), −84.47 (dd, 1 F, JF−F = 141 Hz). HRMS (CI+): m/z calculated for C8H13ClF5N2O (M + H)+ 283.0637; found 283.0625.
Preparation of Azi-isoflurane (1)
A 25 mL rb flask with stir bar was filled with 0.18 g (0.64 mmol) 4 and 0.55 g of dichloroethane. The solution was cooled in an ice bath with stirring, and 0.12 g (0.67 mmol) of N-bromosuccinimide was added in one portion. After the mixture was stirred for 10 min, the ice bath was removed, and the solution was allowed to stir for 1 h at rt. The volatiles were transferred to a U-trap cooled to −78 °C under continuous pumping with a vacuum pump. Purification of the solution by preparative gas chromatography was accomplished using a 10 ft × 0.25 in. column packed with 10% Carbowax 20 M on Chromasorb W. GC collection conditions were as follows: injector 75 °C; column 50 °C; detector 70 °C; helium flow rate = 120 mL/min. The order of elution was tert-butylbromide, 1, and dichloroethane. Product 1 was collected in a U-trap cooled to −78 C °C. 1H NMR: δ 6.05 (qd, 1 H, JH−F = 4.0, 1.0 Hz), 1.68 (t, 1 H, JH−F = 4.3 Hz). 13C NMR: δ 120.4 (q, JC−F = 280 Hz), 120.2 (t, JC−F = 269 Hz), 78.6 (qt, JC−F = 41.1, 5.5 Hz), 20.7 (t, JC−F = 42 Hz). 19F NMR: δ −75.42 (bd, 1 F, JF−F = 147 Hz), −76.40 (dd, 1 F, JF−F = 147 Hz, JH−F = 4.5 Hz), −80.11 (m, 3 F). HRMS (CI+): m/z calculated for C4H2ClF4N2O (M − F)+ 204.9792; found 204.9794.
Physical Properties. Water Solubility and Hydrophobicity
Density was measured directly in tared, sealed vials. Maximal water solubility was measured by vigorous mixing of excess azi-isoflurane in water, centrifugation at 1000g for 10 min, and then measuring absorbance at 300 nm (after using methanolic solutions to calculate an extinction coefficient, 126 M−1 at 300 nm). This water solution (9.9 mL) was then loaded into 10 mL gastight Hamilton syringes. Absorbance at 300 nm of an aliquot was recorded to allow calculation of maximal water solubility, and then exactly 0.1 mL of octanol was added to the syringe, and the two phase system was mixed by rotation for an hour. The octanol−water mixture was allowed to completely separate for another hour, and then absorbance of the water phase was measured again. Molar partition was calculated by multiplying the difference in water absorbance by the ratio of water to octanol volume and dividing by the ending water absorbance.
Electronic structure calculations at the ab initio B3LYP/6-311+G(d,p) geometry optimized level (50) reveal that there are three lowest energy conformations for azi-isoflurane. These three lowest energy conformations are related to each other by rotation about the C−C bond connecting the CF2 group and the diazirine ring. The total energy of each of these three conformations differed by less than 120 cal. The calculated dipole moment for the separate conformations was 2.16, 1.26, and 1.17 D, respectively. The dipole moment for azi-isoflurane is predicted to be 1.5 D assuming that each conformation contributes equally to the average molecular dipole moment at room temperature. A similar calculation on isoflurane (51) predicts that the average molecular dipole moment for this molecule is 1.67 D at room temperature.
Isothermal Titration Calorimetry
The thermodynamic parameters for the binding of azi-isoflurane to HSAF at 20 °C were determined by ITC using a Microcal, Inc. VP ITC (Northampton, MA; http://www.microcalorimetry.com/). The ITC consists of a matched pair of sample and reference vessels (1.43 mL) enclosed in an adiabatic enclosure and a rotating stirrer-syringe for titrating aliquots of the ligand solution into the sample vessel. The sample cell contained 0.01 mM HSAF, and the reference cell contained water. Saturated photolabel (1.1 mM azi-isoflurane) was loaded in the syringe (volume = 0.28 mL) for injecting into the sample. Four separate titrations were performed, including three controls, ligand into buffer, buffer into protein, and buffer into buffer, which were then used to correct the experimental titration, ligand into protein. Origin 5.0 (Microcal Software, Inc., Northampton, MA) was used to fit thermodynamic parameters (single binding site class) to the heat profiles.
Oocyte Expression and K+ Channel Electrophysiology
The Shaw2 F335A mutant channel was used in this study because it expresses more robustly than the corresponding wild-type in Xenopus oocytes and exhibits unaltered biophysical and pharmacological properties (9,21). To harvest oocytes, Xenopus laevis frogs were handled according to a protocol approved by the IACUC of Thomas Jefferson University. The mRNA was synthesized in vitro and microinjected into individual oocytes as described previously (21,22). According to established procedures, the two-electrode voltage-clamp method was used to record the resulting whole-oocyte currents in normal extracellular bath solution (22). Dilutions of azi-isoflurane and isoflurane were prepared fresh before each experiment at 1 and 2 mM final concentrations in a gastight glass screw-top vial (penetrable Tuf-Bond Teflon septa; Thermo Scientific, Rockford, Ill.). Dissolved compounds were delivered through Teflon tubing directly into the recording chamber via a 50 mL Hamilton gastight syringe and syringe pump. Generally, macroscopic currents were low-pass filtered at 0.5−1 kHz and digitized at 1−2 kHz. The program pClamp 8−9 (Axon Instruments and Molecular Devices, Sunnyvale, CA) was used for acquisition, data reduction, and initial analysis of the recorded currents. Leak current was subtracted off-line by assuming a linear leak. All recordings were obtained at room temperature (23 ± 1 °C).
Tadpole Studies
Xenopus tadpoles were used to examine the anesthetic potency of both azi-isoflurane and isoflurane. Briefly, groups of 10 tadpoles were placed in 20 mL sealed glass vials containing pond water and increasing concentrations of the compounds, presolubilized by vigorous shaking/sonication of pondwater with aliquots of neat compound. Volumes were adjusted to minimize gas volume in the vials (∼5%). After a 5 min exposure, tadpoles were transferred to Petri dishes and scored for the presence of a startle reflex. The pond water was then changed and recovery documented for 24 h. Control experiments verified an absence of effect of the manipulations on tadpole activity in the absence of added compound. Percent mobile at each concentration were fitted to variable slope Hill plots.
Photolabeling and Nano-LC/nanospray/LTQ
Photolysis rates were determined in azi-isoflurane/buffer solutions by measuring the loss of absorbance at 300 nm over time of exposure to 300 nm light in quartz cuvettes at a distance of 6 mm from bulb surface. Small aliquots of pH 7 phosphate buffer containing 1 mg/mL HSAF and integrin LFA I domains with and without saturated azi-isoflurane were placed in a 1 mm path length quartz cuvette and exposed to 300 nm light (Rayonet RPR-3000 lamp; emission from 280 to 320 nm) at 2 mm distance for 15 min. Control samples received only UV irradiation. The UV-treated proteins were then passed through an HPLC C-18 analytical column to separate the labeled apoferritin H and L subunits, which were then resuspended in 0.1% TFA. Mass spectrometry equipment and software were available in the Proteomics Core Facility at the University of Pennsylvania. Small aliquots of UV-treated sample L chain were trypsinized and injected into a 10 cm C18 capillary column to separate the digested peptides. Eksigent NanoLC proteomics experiments were run at 200 nL/min for 60 min with gradient elution. Nanospray was used to spray the separated peptides into LTQ (Thermo Electron). Xcalibur is used to acquire the raw data, and modified (photolabeled) peptides were identified using Sequest.
Acknowledgments
The authors thank Michael Hall for assistance with synthesis, and mass spectrometry analysis was provided by Proteomics Core Facility, University of Pennsylvania, supported by grants P30CA016520 and ES013508-04.
Supporting Information Available
Original 1H, 19F, and 13C NMR spectra for compounds 1, 3, and 4; structures, total energies, and dipole moments of the three lowest energy conformations of 1 optimized at the B3LYP/6-311G* level; and original mass spectra showing adduction of 1 with proteins. This information is available free of charge via the Internet at http://pubs.acs.org/.
Major funding was from NIH Grants P01GM55876 (R.G.E.), R01AI063421 (M.S.), and R01AA010615 (M.C.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eckenhoff R. G. (2001) Mol. Interventions 1, 258–268. [PubMed] [Google Scholar]
- Eckenhoff R. G.; Johansson J. S. (1997) Pharmacol. Rev. 49, 343–367. [PubMed] [Google Scholar]
- Eckenhoff R. G.; Shuman H. (1993) Anesthesiology 79, 96–106. [DOI] [PubMed] [Google Scholar]
- Eger E. I.; Raines D. E.; Shafer S. L.; Hemmings H. C. Jr.; Sonner J. M. (2008) Anesth. Analg. 107, 832–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Xie Z. (2009) Curr. Alzheimer Res. 6, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt P. F. Jr.; Wang C.; Weihrauch D.; Bienengraeber M. W.; Kersten J. R.; Pagel P. S.; Warltier D. C. (2006) Curr. Opin. Anaesthesiol. 19, 397–403. [DOI] [PubMed] [Google Scholar]
- Xi J.; Liu R.; Rossi M. J.; Yang J.; Loll P. J.; Dailey W. P.; Eckenhoff R. G. (2006) ACS Chem. Biol. 1, 377–384. [DOI] [PubMed] [Google Scholar]
- Huang C. G.; Rozov L. A.; Halpern D. F.; Vernice G. G. (1993) J. Org. Chem. 58, 7382–7387. [Google Scholar]
- Shahidullah M.; Harris T.; Germann M. W.; Covarrubias M. (2003) Biochemistry 42, 11243–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M., Bhattacharya A. A., Harris T., Kaplan B., and Germann M. W. (2005) in Basic and Systemic Mechanisms of Anesthesia (Mashimo T., Ogli K., Uchida I., Eds.) pp 55−60, Elsevier B.V., Amsterdam. [Google Scholar]
- Franks N. P. (2006) Br. J. Pharmacol. 147(Suppl 1), S72–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Loll P. J.; Eckenhoff R. G. (2005) FASEB J. 19, 567–576. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Astrof N. S.; Liu J. H.; Wang J. H.; Shimaoka M. (2009) FASEB J. 23, 2735–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H. H. (1899) Arch. Exp. Pathol. Pharmakol. (Naunyn Schmiedebergs) 42, 109–137. [Google Scholar]
- Overton C. E. (1901) Studien über die Narkose, Zugleich ein Beitrag zur allgemeinen Pharmakologie, Jena, Fischer . [Google Scholar]
- Butts C. A.; Xi J.; Brannigan G.; Saad A. A.; Venkatachalan S. P.; Pearce R. A.; Klein M. L.; Eckenhoff R. G.; Dmochowski I. J. (2009) Proc. Natl. Acad. Sci. U.S.A 106, 6501–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M.; Xiao T.; Liu J. H.; Yang Y.; Dong Y.; Jun C. D.; McCormack A.; Zhang R.; Joachimiak A.; Takagi J.; Wang J. H.; Springer T. A. (2003) Cell 112, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki K.; Astrof N. S.; Bracken C.; Yoo R.; Silkworth W.; Soriano S. G.; Shimaoka M. (2008) FASEB J. 22, 4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara D. C.; Dangott L. J.; Eckenhoff R. G.; Cohen J. B. (2003) Biochemistry 42, 13457–13467. [DOI] [PubMed] [Google Scholar]
- Eckenhoff R. G.; Petersen C. E.; Ha C. E.; Bhagavan N. V. (2000) J. Biol. Chem. 275, 30439–30444. [DOI] [PubMed] [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.. Gaussian 03, revision C.02; Gaussian, Inc.: Wallingford, CT, 2004. [Google Scholar]
- Biedermann P. U.; Cheeseman J. R.; Frisch M. J.; Schurig V.; Gutman I.; Agranat I. (2003) J. Org. Chem. 64, 3878–3884. [Google Scholar]
- Harris T.; Graber A. R.; Covarrubias M. (2003) Am. J. Physiol. Cell. Physiol. 285, C788–C796. [DOI] [PubMed] [Google Scholar]
- Harris T.; Shahidullah M.; Ellingson J. S.; Covarrubias M. (2000) J. Biol. Chem. 275, 4928–4936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.