

Abstract
A switch from estrogen-dependent to estrogen-independent growth is a critical step in malignant progression of breast cancer and is a major problem in endocrine therapy. However, the molecular mechanisms underlying this switch remain poorly understood. The Wilms’ tumor suppressor gene, wt1, encodes a zinc finger protein WT1 that functions as a transcription regulator. High levels of the WT1 expression have been associated with malignancy of breast cancer. The goal of this study was to investigate the function of WT1 in malignant progression of breast cancer. We found that the high passage ER-positive breast cancer MCF7H cells expressed EGFR, HER2 and WT1 at higher levels compared to the low passage MCF7L cells. MCF7H cells responded weakly to estrogen stimulation, grew rapidly in the absence of estrogen and were insensitive to anti-estrogens such as ICI 182,780 and 4-hydroxy-tamoxifen (4OH-TAM). We also established stable cell lines from the low passage MCF7L cells to constitutively express exogenous WT1 and found elevated levels of EGFR and HER2 expression, estrogen-independent growth and anti-estrogen insensitivity in WT1-transfected MCF7L cells. These results suggested WT1 promotes estrogen-independent growth and anti-estrogen resistance in ER-positive breast cancer cells presumably through activation of the signaling pathways mediated by the members of EGFR family.
Keywords: The Wilms tumor suppressor 1, human epidermal growth factor receptor 2, epidermal growth factor receptor, estrogen and anti-estrogens, breast cancer
Introduction
The roles of endogenous estrogens in breast cancer etiology have been supported by a number of epidemiological, clinical and laboratory studies (1). Estrogen signaling pathways, in particular the mitogenic pathway, mediated by the estrogen receptor-α (ER-α) is believed pivotal in development of breast cancer stimulated by estrogen (2,3). Most non-metastatic breast cancers originally contain cells that express ER-α and respond to mitogenic estrogen signaling (4). However, majority of these tumors will transit from an estrogen-dependent to an estrogen-independent state that is usually associated with an aggressive form of the disease. Many estrogen-dependent breast tumors at the time of clinical diagnosis will respond to endocrine therapy with anti-estrogens such as tamoxifen. However, after a prolonged treatment, most breast cancers eventually become resistant to endocrine therapy (5). The molecular mechanisms underlying this transition and how cell proliferation is regulated once estrogen independence is achieved are under extensive research (reviewed in refs. 6 and 7). Several models have been proposed as possible mechanisms for developing estrogen-independence and anti-estrogen resistance including: expression of variant or mutated ER-α, ligand-independent activation of ER-α by other signaling pathways, altered expression of co-factors or downstream estrogen target genes, post-receptor and pharmacological alterations (reviewed in ref. 7).
There is a growing body of evidence indicating that constitutive expression of growth factors or growth factor receptors in breast cancer cells is one of the major mechanisms for developing estrogen independence and anti-estrogen resistance. Increased autocrine and papracrine growth factor signaling pathways could bypass the requirement of mitogenic estrogen signaling during progression of breast cancer, which would also render anti-estrogen resistance. In many cases, estrogen-independent and tamoxifen-resistant tumors display enhanced expression of the epidermal growth factor receptor (EGFR) or ErbB family members, such as EGFR (HER1 or ErbB1), HER2 (ErbB2 or Neu), and high basal levels of MAP kinase activation (8 and reviewed in refs. 9–11). In clinical studies, ER-positive patients with HER2-positive tumors are less likely to benefit from endocrine therapy than patients with HER2-negative tumors (reviewed in ref. 11). However, the molecular mechanisms by which breast cancer cells acquire enhanced mitogenic signaling through the growth factor pathways are largely unknown.
The Wilms’ tumor susceptibility gene, wt1, at chromosome locus 11p13 (12–14) encodes a C2-H2-type zinc finger protein, WT1. Through alternative splicing, there are four protein isoforms of WT1 that differ by the presence of one 17 amino acid insert between its transcription regulatory domain and DNA binding domain and one 3 amino acid (KTS) insert between the third and fourth zinc fingers (reviewed in refs. 15 and 16). The different isoforms are referred to as A, B, C and D; the A isoform lacks both 17 amino acid and KTS inserts; B isoform contains the 17 amino acid insert but lacks KTS insert; C isoform lacks the 17 amino acid insert but contains KTS insert; and D isoform contains both inserts. Mutations of wt1 were found to be associated with subsets of Wilms’ tumor (reviewed in refs. 15 and 16), mesothelioma and ovarian tumor (reviewed in ref. 17), consistent with the role of WT1 as a tumor suppressor. However, high levels of the wild-type WT1 mRNA and protein have been found in leukemia (18), lung cancer (19) and breast cancer (20–22). Breast cancer patients with tumors that highly express WT1 usually have a lower 5-year disease-free survival rate than patients with tumors of low WT1 expression (22), suggesting WT1 expression is associated with aggressive phenotype of breast cancer. However, the biological function of WT1 in development of the aggressive breast tumors is currently unclear.
The MCF7 cell line, originally established from an ER-positive breast adenocarcinoma is an estrogen-dependent breast cancer cell line that responds well to anti-estrogens such as tamoxifen. In this study, we found a high passage (>75) subline of MCF7 cells that can proliferate in an estrogen-independent manner even retaining ER-α expression and are estrogen responsive. We also found these cells highly express EGFR, HER2 and WT1. The biological function of WT1 in estrogen-independent proliferation and anti-estrogen resistance was studied in these high passage MCF7 cells as well as in MCF7 cells constitutively expressing recombinant WT1.
Materials and methods
Cell culture and establishment of stable cell lines
Relatively low passage MCF7 cells (MCF7L, <35 passages) were recently obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained at 37°C in a 5% CO2 atmosphere in Improved Modified Eagle’s Medium (IMEM) supplemented with 5% fetal calf serum. These cells were very sensitive to estrogen. Relatively high passage MCF7 cells were initially obtained from Dr Thomas F. Deuel’s laboratory at the Scripps Research Institute. After continuous 50–60 passages in the above media, we noted a gradual loss of estrogen responsiveness. The subline of MCF7 used in this study, MCF7H had been cultured for >75 passages and had obtained estrogen-independent growth compared to the MCF7L cells.
To establish cells expressing recombinant WT1, MCF7L cells were plated at a density of 1×105 cells per 60-mm dish and transfected 24 h later with a WT1 expression vector driven by the cytomagalovirus (CMV) promoter in the mammalian expression vector pCB6+ as described elsewhere (23), using the FuGene 6 transfection reagent (Roche Applied Sciences, Indianapolis, IN). The empty expression vector was also transfected into MCF7L cells to serve as a control. Forty-eight hours after transfection, the cells were re-plated and selected with 500 μg/ml of G418 (Invitrogen Corporation, Carlsbad, CA) for two weeks. The medium was changed every 3 days until colonies appeared. We established a number of clonal cell lines that highly expressed WT1, three of which are described in detail in this study (MCF7/WTl-2, -3 and -4). More than 20 individual clones from cells transfected with the empty expression vector were pooled and used as control MCF7/V cells.
To examine growth of WT1 transfected cell in normal medium, cells maintained in IMEM with 5% fetal calf serum were seeded at 1×104 cells per well in 6-well plates and counted every other day for 8 days using a hemacytometer. Five wells were examined for each time point and experiments were repeated three times.
To examine cell growth in the presence or absence of E2 and different drugs, cells maintained for 3 days in phenol red-free IMEM plus 5% dextran-charcoal-stripped fetal calf serum (HyClone, Logan, UT) were treated with 1 nM of 17β-estradiol (E2), or DMSO vehicle as a control. Anti-estrogens 4-hydroxytamoxifen (1 μM, Sigma, St. Louis, MO) and ICI 182,780 (1 μM, Sigma) were also included in the experiments. The cells were seeded at 1×104 cells per dish in 60-mm dishes and counted after 8 to12 days using a hemacytometer. Three dishes were used for each experiment and experiments were repeated three times. Alternatively, cells seeded at 1×104 cells per well in 6-well plates were counted every other day for 8 days using a hemacytometer. Five wells were examined for each time point and experiments were repeated three times.
Western blot analysis
Cells were washed with phosphate-buffered saline (PBS) and lysed with lysis buffer [50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.25 mM EDTA pH 8.0, 0.1% sodium dodecyl sulfate (SDS), 1% Triton® X-100, 50 mM NaF and the protease inhibitor cocktail from Sigma]. After adjustment to the same total protein content, cell lysates were analyzed by Western blot analysis. Twenty micrograms of cell lysates were boiled for 5 min in SDS gel loading buffer and separated on a 12 or 10% SDS-PAGE gel. After electrophoresis, the proteins were transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA). The membranes were probed with different primary antibodies, incubated with appropriate HRP-conjugated secondary antibodies and visualized with enhanced chemiluminescence (ECL) detection reagents (Amersham Pharmacia Biotech. Piscataway, NJ). The same membranes were stripped and reprobed with an antibody against β-actin (I-19) (Santa Cruz Biotechnology, Santa Cruz, CA) to confirm equal loading.
Anti-phospho-AKT (Ser473) rabbit mAb, polyclonal anti-AKT antibody, anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (197G2) rabbit mAb, anti-p44/42 MAPK (Erk1/2) (137F5) rabbit mAb, anti-EGF receptor (15F8) rabbit mAb and anti-phospho-ER-α (Ser118) antibody were purchased from Cell Signaling Technology (Beverly, MA). Anti-cyclin D1, c-Myc and HER2/Neu antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz). Anti-ER-α antibody (Ab-15) was obtained from Lab Vision Products (Fremont, CA). Polyclonal anti-WT1 antibody was from Invitrogen Corporation.
Luciferase assay
Cells were transfected using FuGene 6 transfection reagent (Roche Applied Science) with a plasmid encoding the firfly luciferase gene driven by a promoter bearing two-tandem estrogen response elements, p2X ERE-Luc reporter plasmid (a kind gift from Dr Katarine Pettersson at Karolinska Institute, Sweden) and a cytomegalovirus-driven Renilla luciferase plasmid, pRL-CMV (Promega, San Louis Obispo, CA) to establish transfection efficiency. Cells were treated with 1 μmol/l 4OH-TAM and 5 μmol/l lapatinib (GW572016, LC Laboratories, Woburn, MA) in the absence or presence of 1 nM of E2 for 12 h. Forty-eight hours after transfection, cell extracts were prepared and luciferase activity was determined and normalized using the Dual-Luciferase Assay System (Promega) and a TD 20/20 Luminometer (Turner BioSystems, Inc. Sunnyvale, CA) as instructed by the manufacturer.
Statistical analysis
Data were summarized as the mean ± standard error (SE) using GraphPad InStat software program. Tukey-Kramer Multiple Comparisons Test was also used and the significance was accepted at p<0.05.
Results
High passage MCF7 cells (MCF7H) exhibited estrogen-independent and anti-estrogen insensitive growth
During our research, we observed a progressive development of estrogen-independent growth in a MCF7 cell subline (MCF7H) of long-term passage (>75) when compared to our low passage (<35) MCF7L subline. In the absence of estrogen, MCF7L cells proliferated at a very slow rate while were stimulated to grow rapidly by 1 nM 17β-estradiol (E2) (Fig. 1A), consistent with the fact that ER-positive MCF7 cells are estrogen-dependent cells that require the presence of estrogen for their optimum proliferation. MCF7H cells proliferated rapidly in the absence of estrogen compared to MCFL cells. Further growth of MCF7H cells was weakly stimulated by E2 (Fig. 1B), suggesting that MCF7H cells exhibit estrogen-independent growth but are still estrogen responsive (Fig. 1B). Compared to MCF7L, MCF7H exhibited less sensitivity to anti-estrogens such as ICI 182,780 (ICI) and 4-hydroxytamixifen (4OH-TAM), one of the active metabolites of tamoxifen (Fig. 1B).
Figure 1.

High passage MCF7 cells (MCF7H) exhibit estrogen-independent and anti-estrogen insensitive growth. (A). Growth rate of MCF7H and MCF7L cells. Cells maintained in steroid-free medium were seeded at 1×104 cells per well in 6-well plates, incubated in medium containing DMSO (V) or 1 nM of 17β-estradiol (E2) and counted every other day. Data presented are means of three independent experiments; bars, SE. (B) Cells were seeded at 1×104 cells per well in 60-mm dishes, incubated in medium containing DMSO (V), 1 nM of 17β-estradiol (E2), 1 μM 4OH-TAM (4OH) or 1 μM ICI 182,780 (ICI) and counted in 12 days. Column, means of three independent experiments; bars, SE. a and b, P<0.01 vs. cells in the absence E2; c and d, P<0.01 vs. MCF7L cells treated with anti-estrogens.
MCF7H cells expressed high levels of EGFR, HER2 and WT1
It is well known that constitutive or enhanced expression of the EGFR family members such as EGFR and HER2 render estrogen-independent growth and anti-estrogen resistance (reviewed in refs. 9–11). Western blot analysis revealed that expression levels of HER2 and EGFR were increased in MCF7H cells compared to MCF7L cells (Fig. 2), indicating high passage MCF7H cells gained expression of two members of the EGFR family, EGFR and HER2. The basal levels of the phosphorylated ERK1/2 and AKT were also elevated in MCF7H cells, suggesting that the enhanced levels of EGFR and HER2 expression also resulted in an enhanced activation of the MAPK/ERK and the PI3K/AKT signaling pathways. In MCF7H cells, the levels of ER-α phosphorylation at Ser118 were also increased while the expression levels of ER-α were not significantly changed compared to MCF7L cells (Fig. 2). Finally, we observed that the expression levels of the Wilms’ tumor suppressor WT1 were greatly increased in MCF7H cells compared to MCF7L cells (Fig. 2).
Figure 2.

High passage MCF7H cells express increased levels of EGFR, HER2 and WT1. Western blot analysis of the cell lysates from MCF7H cells and MCF7L cells.
MCF7H cells exhibited ligand-independent ER activation and enhanced transcriptional activity simulated by estrogen
To evaluate the effects of high levels of HER2 and EGFR expression on ER-transcription activity and ligand responsiveness, we performed transient transfection assays. MCFL and MCF7H cells were transfected with an ER-regulated luciferase reporter gene (p2X ERE-Luc) and the internal control vector, pRL-CMV and evaluated for changes in the levels of basal (without treatment) and estrogen treatment induced transcription activity of endogenous ER-α. The transfected cells were cultured in a medium with dextran-charcoal-stripped serum to reduce steroid. In the absence of estrogen, the basal luciferase activity levels in MCF7H cells were >30-fold higher than those in MCF7L cells (Fig. 3). This suggests that the cross-talk between the signaling pathways mediated by the members of the EGFR family and ER enhances the basal transcription activity of ER-α in a ligand-independent manner. In MCF7H cells, treatment of 1 nM E2 resulted in a >2-fold increase of ERE-reporter activity relative to the high basal levels observed in the untreated MCF7H cells, whereas a ~5- to 6-fold increase in reporter activity relative to the basal levels in E2β-treated MCF7L cells (Fig. 3).
Figure 3.
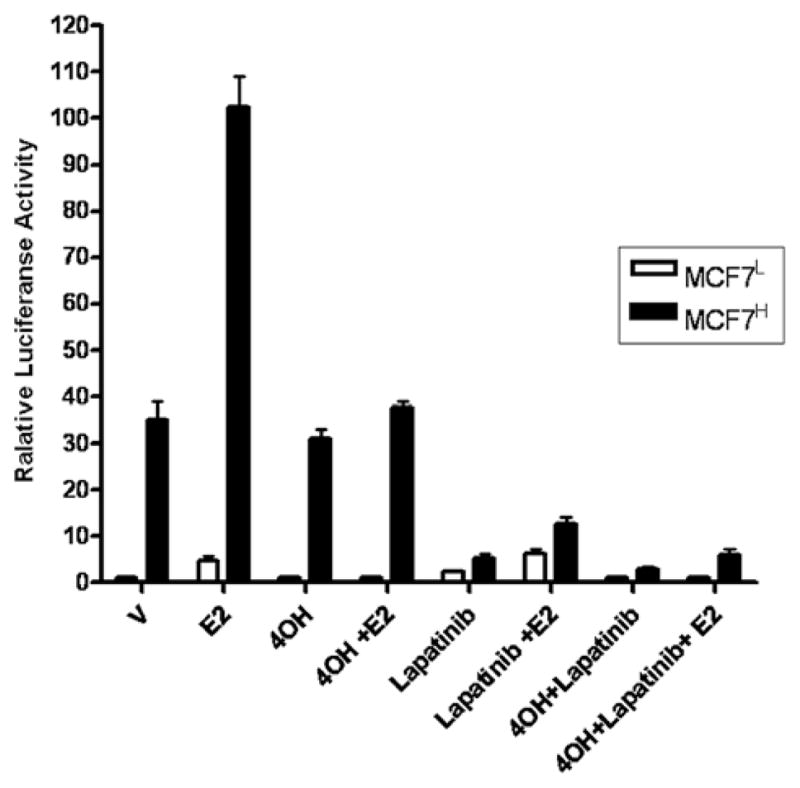
Lapatinib and 4-hydroxytamoxifen inhibit ER-mediated transcription activity in MCF7H cells. MCF7H cells and MCF7L cells were transfected with p2X ERE-Luc reporter plasmid for 36 h before treatment with DMSO (V), 1 μM 4OH-TAM (4OH), 5 μM Lapatinib or both drugs together in the absence or presence of 1 nM of E2 for an additional 12 h. Data are shown as means ± SE from three independent experiments. The value of ERE luciferase activity in vehicle-treated MCF7L cells is set as 1.
Anti-estrogen 4OH-TAM treatment inhibited E2-induced ER-α activity in both MCF7L and MCF7H cells, but failed to inhibit the ligand-independent activation of ER-α activity in MCF7H cells (Fig. 3). However, lapatinib (GW572016), an effective inhibitor for both EGFR and HER2 (24), strongly inhibited the ligand-independent ER activity in MCF7H cells and was without effect on the estrogen-induced ER activity (Fig. 3). Combination of both 4OH-TAM and lapatinib abrogated the basal and estrogen-induced ER activity in MCF7H cells. These results indicated that the transcriptional activity of ER in the presence and absence of estrogen is dramatically enhanced in the high passage MCF7H cells through the cross-talk between the signaling mediated by the members of the EGFR family and ER-α.
Regulation of cyclin D1 and c-Myc expression by estrogen in MCFL cells is switched to the MAPK/ERK pathway in MCF7H cells
We then examined the expression levels of two estrogen-inducible genes, cyclin D1 and c-Myc, both of which are also known to contribute to estrogen-stimulated cell growth (25). In the absence of estrogen, both cyclin D1 and c-Myc were expressed at very low levels in MCF7L cells while estrogen treatment strongly induced expression of both cyclin D1 and c-Myc in MCF7L cells (Fig. 4A). However, the basal levels of cyclin D1 and c-Myc expression were strongly enhanced in MCF7H cells in the absence of estrogen and estrogen treatment had no further effect on their expression (Fig. 4A), indicating that in the absence of estrogen, MCF7H cells express high levels of cyclin D1 and c-Myc that could not be induced further by estrogen. It was reported that MCF7 cells become estrogen hypersensitive after a long-term culture under estrogen-deprived condition (26). It is thus possible that the high passage MCF7H cells might become estrogen hypersensitive and trace amount of remaining estrogen in the dextran-charcoal stripped serum may maintain sustained expression of both genes observed in MCF7H cells. To examine this possibility, we included anti-estrogens in our experiments. In MCF7L cells cultured in a steroid-reduced medium, c-Myc expression was significantly reduced (Fig. 4B), suggesting c-Myc expression is mainly dependent on estrogen signaling in estrogen sensitive cells. Expression levels of cyclin D1 were also reduced in MCF7L cells in the absence of estrogen, which was not further reduced with anti-estrogens such as 1 μM ICI 182,780 or 4OH-TAM but was totally diminished by the MEK inhibitor UO126 (Fig. 4B), indicting that cyclin D1 expression was partially dependent on estrogen signaling and also relies on the MAPK/ERK pathway in estrogen-sensitive cells. In the MCF7H cells, however, estrogen withdrawal and inclusion of both anti-estrogens had no effects on the sustained expression of cyclin D1 and c-Myc and only the MEK inhibitor UO126 was able to abrogate their expression (Fig. 4B), indicating that in MCF7H cells, expression of cyclin D1 and c-Myc was regulated predominantly by a mechanism that is dependent on the MAPK/ERK signaling pathway and thus bypassed the requirement for estrogen signaling.
Figure 4.
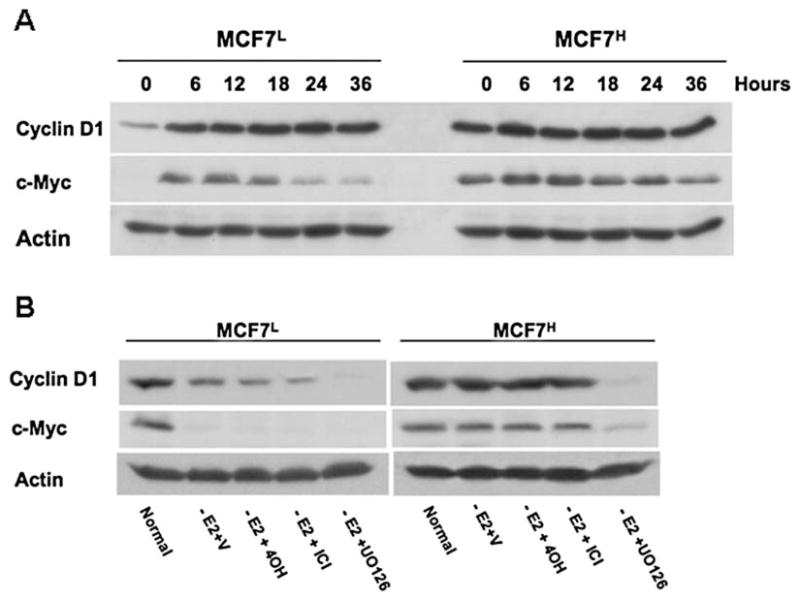
Cyclin D1 and c-Myc expression is regulated by the MAPK/ERK pathway in MCF7H cells. (A). Western blot analysis of the lysates from MCF7H cells and MCF7L cells treated with 1 nM of E2 for different period of time as indicated using anti-cyclin D1 and c-Myc antibodies. (B). Western blot analysis of the lysates from MCF7H cells and MCF7L cells cultured in normal medium containing 5% FCS or in medium containing 5% dextran-charcoal-stripped FCS containing DMSO vehicle (V), 1 μM 4OH-TAM (4OH), 1 μM ICI 182,780 (ICI) or 10 μM UO126 for 12 h.
Forced expression of WT1 induced levels of EGFR and HER2 expression
Previously, it was reported that the Wilms’ tumor suppressor WT1 binds to its cognate binding sequences in the promoter region of the EGFR gene activating its expression (27). Recently, the HER2 signaling pathway was found to increase the expression of WT1 protein in breast cancer cells (28). To determine if WT1 is the cause or result of the elevated expression of HER2 and EGFR in MCF7H cells, we transfected MCF7L cells that express undetectable levels of endogenous WT1 with a CMV promoter-driven expression vector containing the WT1 cDNA. We established three clonal cell lines that highly expressed WT1 (MCF7/WTl-2, -3 and -4). We also generated a cell line from mixture of >20 clones transfected with the empty expression vector (MCF7/V). Western blot analysis using the antibody against the WT1 confirmed that the WT1 protein (~52 kDa) was highly expressed in three transfected cell lines but not in parental (MCF7/P) and control MCF7L cells transfected with empty vector (MCF7/V) (Fig. 5A). The expression levels of EGFR and HER2 were dramatically elevated in WT1-transfected MCF7L cells compared to the parental (MCF7/P) and control (MCF7/V) cells (Fig. 5A). ER-α expression was without significant change in WT1-transfected cells but the levels of ER-α phosphorylation at Ser118 were increased (Fig. 5A). Accordingly, the basal levels of the activated ERK1/2 and expression levels of cyclin D1 and c-Myc were all elevated in WT1-tranfected cells (Fig. 5B). These results strongly suggested that WT1 expression induces the enhanced expression of EGFR and HER2, sustained activation of the MAPK/ERK signaling and increased expression of cyclin D1 and c-Myc observed in MCF7H cells.
Figure 5.
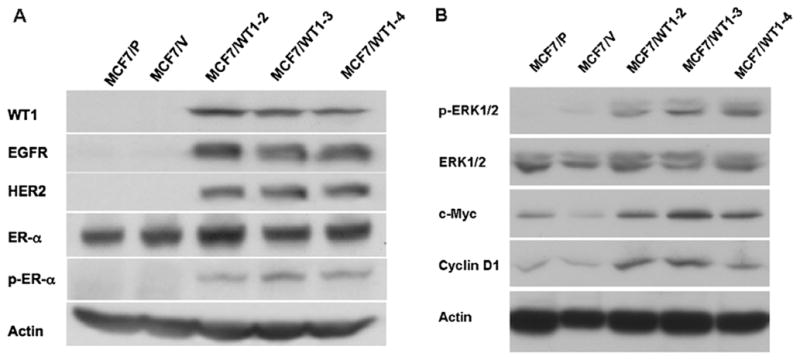
Constitutive expression of recombinant WT1 in MCF7L cells induces protein levels of EGFR and HER2. (A and B) Western blot analysis of the lysates from parental MCF7L cells (MCF7/P), MCF7L cells transfected with the empty expression vector (MCF7/V) and clonal cell lines from MCF7L cells transfected with WT1 expression vector (MCF7/WT1-2, -3 and -4).
WT1 transfected MCF7L cells become estrogen-independent and tamoxifen insensitive
The WT1-expressing MCF7L cells and the MCF7/V cells were maintained under optimal growth condition in the presence of estrogen. The growth rate of each cell line was determined by counting the number of cells every other day. The cell lines expressing high levels of exogenous WTl (MCF7/WTl-2, -3 and -4) had ~5-fold higher growth rate than the control MCF7/V cells (Fig. 6A). These data indicated that the high levels of WT1 expression dramatically enhanced the growth rate of transfected MCF7L cells. In the absence of estrogen, WT1-transfected cells also proliferated at a more rapid rate (~8-fold) than the control MCF7/V cells (Fig. 6B), indicating that WT1 expressing cells grew in an estrogen-independent manner. In addition, WT1-transfected MCF7L cells also exhibited less sensitivity to anti-estrogens such as ICI 182,780 and 4OH-TAM when compared to the control MCF7/V cells (Fig. 6B). These results suggested that acquired WT1 expression may act as a switch of estrogen-dependence to estrogen-independence though activation of EGFR and HER2 expression.
Figure 6.
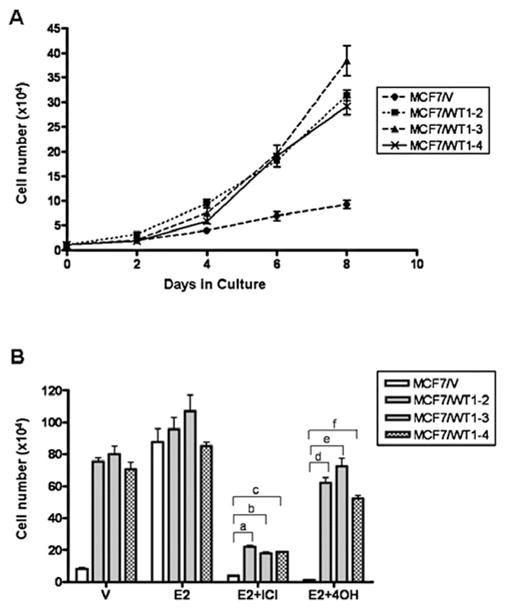
WT1-transfected MCF7L cells exhibit estrogen-independent and anti-estrogen insensitive growth (A). Growth rate of MCF7L cells transfected with the empty expression vector (MCF7/V) and clonal cell lines from MCF7L transfected with WT1 expression vector (MCF7/WT1-2, -3 and -4). Cells maintained in normal medium containing 5% FCS were seeded at 1×104 cells per well in 6-well plates and counted every other day. Data are shown as means ± SE from three separate experiments (B). Cells were seeded at 1×104 cells per 60-mm dishes, incubated in medium containing DMSO (V), 1 nM of 17β-estradiol (E2), 1 μM 4OH-TAM (4OH) or 1 μM ICI 182,780 (ICI) and counted at day 12. Column, means of three independent experiments; bars, SE. a–c, P<0.01 vs. MCF7/V cells; d–f, P<0.01 vs. MCF7/V cells.
Discussion
Etiological, clinical and experimental data demonstrate that the mitogenic estrogen signaling plays an important role in both the initiation and progression of breast cancer (1). The majority of human breast tumors is thought to initiate as estrogen-dependent lesions. However, with the malignant progression of the disease, most breast tumors will develop into estrogen-independent but retain estrogen receptor expression and are still estrogen responsive before they eventually develop into estrogen-independent and unresponsive phenotypes usually accompanied by lack of ER expression.
The switch of breast tumor from an estrogen-dependent to an estrogen-independent phenotype is also associated with a progression from a benign form to a more aggressive form of the disease that represents a major clinical problem. Endocrine therapy such as anti-estrogen tamoxifen that binds and antagonizes ER has been used to treat both early and advanced ER-positive breast cancer patients for almost three decades. Unfortunately, however, ~50% of patients with advanced primary breast tumors that are ER-positive do not respond to first line treatment with tamoxifen and thus have de novo resistance to endocrine therapy (29). Of those that initially respond to endocrine therapy, most eventually will develop estrogen independence and acquire resistance to endocrine therapy. Further understanding of the molecular mechanisms underlying the development of endocrine resistance will allow us to overcome these obstacles.
In the present study, we found that the high passage breast cancer MCF7 cells (MCF7H) exhibit estrogen-independent growth and are less sensitive to anti-estrogens such as ICI 182,780 and 4OH-TAM compared to the low passage MCF7 cells, MCF7L. The MCF7H cells also express increased levels of both EGFR and HER2, members of the ErbB family and exhibited sustained phosphorylation levels of the MAPK/ERK and the AKT. Along the same lines, the expression levels of the downstream target genes such as cyclin D1 and c-Myc were also increased compared to MCF7L cells. In addition, we observed that phosphorylation level of ER-α Ser118 residue is increased in MCF7H cells while the expression levels of ER-α were not significantly changed compared to MCF7L cells. The basal (in the absence of estrogen) transcription activity of ER in MCF7H cells was significantly higher than that in MCF7L cells and E2 stimulated stronger ERE-dependent transcription in MCF7H cells compared to MCF7L cells, which was efficiently blocked by anti-estrogen 4OH-TAM. Consistent with these observations, previous studies have shown that that peptide growth factor signaling pathways can activate ER-α in absence of its ligand through phosphorylation of ER-α by the MAPK/ERK pathway (30–32). Our data further established an important role for the cross-talk between the growth factor pathways and ER-dependent transcription activity in development of estrogen-independence and anti-estrogen resistance in breast cancer cells.
Cyclin D1 is a transcription target of the estrogen signaling (33) and in some studies its overexpression in ER-positive breast cancer has been associated with poorer prognosis (34). Cyclin D1 is also a downstream target of HER2/Neu and is essential for HER2/Neu to induce mammary tumorigenesis in transgenic mice (35). Likewise, c-Myc proto-oncogene is also a downstream target gene of estrogen signaling and a downstream effector of the peptide growth factor signaling pathways (36). In the low passage MCF7L cells, expression of both cyclin D1 and c-Myc are mainly subjected to estrogen regulation whereas their expression was switched to regulation by the MAPK/ERK pathway in MCF7H cells. Unlike the observation that estrogen stimulated much higher ER-transcription activity in the transient transfection experiment with the ERE-luciferase reporter in MCF7H cells, we did not observe estrogen stimulation of cyclin D1 and c-Myc expression in MCF7H cells. This suggests that in MCF7H cells, stimulatory activities mediated by the growth factor signaling override the effects of estrogen signaling.
Previously, several laboratories reported that long-term estrogen-deprived (LTED) breast cancer cells develop estrogen hypersensitivity and increased expression of the MAPK/ERK which still can be inhibited with the pure anti-estrogen ICI 182,780 (37) and expression of constitutively activated MEK1 showed hypersensitivity to estrogen without diminishing sensitivity to the inhibitory effects of anti-estrogens (38). Thus, it is possible that ER primed by the increased MAPK/ERK activity in MCF7H cells may utilize the residual estrogen in the dextran-charcoal stripped serum to promote cell growth. However, in MCF7H cells, the sustained expression of cyclin D1 and c-Myc in the absence of estrogen and in the presence of the pure anti-estrogen ICI 182,780 excludes the involvement of ER-mediated transcription since all known ERs are sensitive to the inhibitory effect of ICI 182,780. We observed down-regulation of ER-α in MCFH cells treated with ICI 182,780 (data not shown). In addition, we observed that the expression of c-Myc and cyclin D1 was completely abolished by the MEK inhibitor UO126 in MCF7H cells, suggesting the growth factor signaling pathways acting through the MAPK/ERK pathway contribute predominantly to the sustained expression of cyclin D1 and c-Myc and estrogen-independent proliferation of MCF7H cells.
We also observed that growth of MCF7H cells were inhibited by both ICI 182,780 and 4-OH-TAM although less sensitive compared to MCF7L cells, which seems contradictory to the result that ICI 182,780 failed to block cyclin D1 and c-Myc expression. In anti-estrogen sensitive cancers, anti-estrogens reduce tumor size and increase overall survival by inducing a G0-G1 cell cycle arrest and inducing apoptotic cell death (reviewed in ref. 39). Tamoxifen has been shown to induce apoptosis in breast cancer cells through the p38 pathway (40, 41) and interferon regulatory factor-1 mediates the proapoptotic, but not cell cycle arrest effects of ICI 182,780 (42). Thus, anti-estrogens may still induce apoptotic cell death while loss of cell cycle arrest effects in MCFH cells.
It is now well established that in ER-positive primary breast cancers, overexpression of both EGFR and HER2 is associated with resistance to endocrine therapy (9–11). However, the mechanisms whereby ER-positive breast cancer cells progressed to gradually gain EGFR and HER2 expression are largely unknown. Here, our data demonstrated that the Wilms’ tumor gene product WT1 plays an important role in the up-regulation of EGFR and HER2 expression in breast cancer cells. WT1 was initially discovered as a tumor suppressor gene of the genitourinary system. The biological function of WT1 outside of the genitourinary system remains controversial. Mutations of wt1 gene were reported in different types of cancer, such as mesothelioma, ovarian and prostate cancer and acute myeloid leukemia (16, 17). However, accumulating evidence indicates a potentially oncogenic role of WT1 in leukemia and breast cancer (reviewed in ref. 43). WT1 expression was found in primary breast tumors (20–22) and high levels of WT1 expression were shown to predict a poor prognosis in breast cancer patients (22), consistent with a putative oncogenic role of WT1. WT1 is a dual transcription regulator and functions to activate or suppress gene transcription depending on cell and gene context (23, 27, 44–46), which may provide an explanation of WT1 function as both a tumor suppressor and oncogene. Previously, Zapata-Benavides et al (47) found that the expression levels of WT1 correlated with the proliferation of several lines of breast cancer cells and down-regulation of WT1 expression using WT1 antisense oligonuleotides led to growth inhibition of breast cancer cells, suggesting a growth promoting role of WT in breast cancer. In this study, we found constitutive expression of WT1 in breast cancer cells conferred increased expression of EGFR and HER2, suggesting that WT1 directly or indirectly regulates their expression. It has been reported that WT1 activates the EGFR promoter activity in neuron cells (27). It is thus possible that WT1 may activate EGFR expression directly in breast cancer cells. The molecular mechanism underlying enhanced HER2 expression in WT1-transfected cells is not clear. The HER2 promoter sequence is G+C rich. The 300 bp upstream of the mRNA start site has 59% G+C content (48). It is also worth noting that a pentanucleotide sequence GGAGG appears eight times in the region upstream of the mRNA start site in the HER2 promoter region (48). In the EGFR promoter, the complement of this sequence, CCTCCTCC (the ‘TCC’ motif), appears four times upstream to the first ATG, which was required for WT1-mediated transcription activation of the EGFR gene (27). It is reasonable to speculate that the same ‘TCC’ motif in the HER2 promoter may be used for the WT1 binding and activation of HER2 expression. It was also reported that WT1 expression can be increased by HER2 signaling pathway through AKT (28). It is thus possible that the HER2 signaling and WT1 may form a positive feed back to maintain high levels of both expressions.
Previously, it has been reported that different isoforms of WT1 have distinct effects on mammary epithelial cells (49). Recently, Han et al reported that WT1 down-regulated ER-α expression and mediated anti-estrogen resistance (50). In our study here, we did not observe significant changes of ER-α expression in WT1-transfected MCF7 cells. In the previous study (50), WT1 isoforms B and D were used, both of which contain the 17 amino acid insert. In this study, we used WT1 A isoform, which may provide an explanation for the distinct effects of WT1 on ER-α expression observed. Previously, it was found that the 17 amino acid domain harbors a transcription suppression domain and WT1 isoforms containing this domain have stronger transcription suppression activity (51). These results suggested the ratios of WT1 isoforms in mammary epithelial cells are involved in regulation of cellular responses to estrogen and anti-estrogens.
In summary, our results demonstrate that the Wilms’ tumor suppressor WT1 induces estrogen-independent growth and anti-estrogen insensitivity in ER-positive breast cancer MCF7 cells presumably through activation of EGFR and HER2 expression. These findings are consistent with the oncogenic role of WT1 in breast cancer development and suggest that examination of WT1 expression together with EGFR and HER2 assessment might be useful as a new diagnosis marker for selecting the appropriate treatment and as a prognostic factor in breast cancer patients.
Acknowledgments
This study was funded by the Nebraska Tobacco Settlement Biomedical Research Program Award (LB-595) to (Z.Y. Wang) and NIH grant DK070016 (Z.Y. Wang).
Abbreviations
- E2
17β-estrodial
- AKT
v-akt murine thymoma viral oncogene homolog
- WT1
The Wilms tumor suppressor 1
- ER
estrogen receptor
- EGFR
epidermal growth factor receptor
- DMSO
dimethyl sulfoxide
- MAPK
mitogen activated protein kinase
- ERE
estrogen response element
- ERK
extracellular signal-regulated kinase
- HER2
human epidermal growth factor receptor 2
- MEK
MAPK kinase
- PI3K
phosphatidylinositol-3-kinase
- 4OH-TAM
4-hydroxy-tamoxifen
- CMV
cytomagalovirus
- mAb
monoclonal antibody
- ECL
enhanced chemiluminescence
- ICI
ICI 182,780
References
- 1.Vorherr H. Breast Cancer. Urban & Schwarzenberg Press; Baltimore: 1980. [Google Scholar]
- 2.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 3.Klings C. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott JA, McGure WL. In: Endocrine-Dependent Tumor. Voight KD, Knabbe C, editors. Raven Press Ltd; New York: 1991. pp. 179–196. [Google Scholar]
- 5.Katzenellenbogen B. Antiestrogen resistance: mechanisms by which breast cancer cells undermine the effectiveness of endocrine therapy. J Natl Cancer Inst. 1991;83:1434–1435. doi: 10.1093/jnci/83.20.1434. [DOI] [PubMed] [Google Scholar]
- 6.Wiebe VJ, Osborne CK, Fuqua SA, DeGregorio MW. Tamoxifen resistance in breast cancer. Crit Rev Oncol Hematol. 1993;14:173–188. doi: 10.1016/1040-8428(93)90008-r. [DOI] [PubMed] [Google Scholar]
- 7.Murphy LC. Mechanisms of hormone independence in human breast cancer. In Vivo. 1998;12:95–106. [PubMed] [Google Scholar]
- 8.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 9.Nicholson RI, Staka C, Boyns F, Hutcheson IR, Gee JMW. Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endocr Relat Cancer. 2004;11:623–641. doi: 10.1677/erc.1.00778. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson RI, Hutcheson IR, Harper ME, Knowlden JM, Barrow D, McClelland RA, Jones HE, Wakeling AE, Gee JMW. Modulation of epidermal growth factor receptor In endocrine-resistant, oestrogen receptor-positive breast cancer. Endocr Relat Cancer. 2001;8:175–182. doi: 10.1677/erc.0.0080175. [DOI] [PubMed] [Google Scholar]
- 11.Grazia A, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonetta L, Kuehn SE, Huang A, Law DJ, Kalikin LM, Koi M, Reeve AE, Brownstein BH, Yeger H, Williams BR. Wilms tumor locus on 11p13 defined by multiple CpG island-associated transcripts. Science. 1990;250:994–997. doi: 10.1126/science.2173146. [DOI] [PubMed] [Google Scholar]
- 13.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 14.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343:774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 15.Coppes MJ, Campbell CE, Williams BR. The role of WT1 in Wilms tumorigenesis. FASEB J. 1993;7:886–895. doi: 10.1096/fasebj.7.10.8393819. [DOI] [PubMed] [Google Scholar]
- 16.Lee SB, Haber DA. Wilms tumor and the WT1 gene. Exp Cell Res. 2001;264:74–99. doi: 10.1006/excr.2000.5131. [DOI] [PubMed] [Google Scholar]
- 17.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997;9:209–225. doi: 10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, Miyake S, Tamaki H, Oji Y, Yamagami T, Tatekawa T, Soma T, Kishimoto T, Sugiyama H. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- 19.Oji Y, Miyoshi S, Maeda H, Hayashi S, Tamaki H, Nakatsuka S, Yao M, Takahashi E, Nakano Y, Hirabayashi H, Shintani Y, Oka Y, Tsuboi A, Hosen N, Asada M, Fujioka T, Murakami M, Kanato K, Motomura M, Kim EH, Kawakami M, Ikegame K, Ogawa H, Aozasa K, Kawase I, Sugiyama H. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int J Cancer. 2002;100:297–303. doi: 10.1002/ijc.10476. [DOI] [PubMed] [Google Scholar]
- 20.Silberstein GB, Horn KV, Strickland P, Roberts CT, Jr, Daniel CW. Altered expression of the WT1 Wilms’ tumor suppressor gene in human breast cancer. Proc Natl Acad Sci USA. 1997;94:8132–8137. doi: 10.1073/pnas.94.15.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loeb DM, Evron E, Patel CB, Sharma PM, Niranjan B, Buluwela L, Weitzman SA, Korz D, Sukumar S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001;61:921–925. [PubMed] [Google Scholar]
- 22.Miyoshi Y, Ando A, Egawa C, Taguchi T, Tamaki Y, Tamaki H, Sugiyama H, Noguchi S. High expression of Wilms’ tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin Cancer Res. 2002;8:1167–1171. [PubMed] [Google Scholar]
- 23.Wang ZY, Qiu QQ, Deuel TF. The Wilms’ tumor gene product WT1 activates or suppresses transcription through separate functional domains. J Biol Chem. 1993;268:9172–9175. [PubMed] [Google Scholar]
- 24.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 25.Rochefort H. Oestrogen- and anti-oestrogen-regulated genes in human breast cancer. Ciba Found Symp. 1998;191:254–265. doi: 10.1002/9780470514757.ch15. [DOI] [PubMed] [Google Scholar]
- 26.Masamura S, Santner SJ, Heitjan F, Santen RJ. Estrogen deprivation causes E2 hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995;80:2918–2925. doi: 10.1210/jcem.80.10.7559875. [DOI] [PubMed] [Google Scholar]
- 27.Liu XW, Gong LJ, Guo LY, Katagiri Y, Jiang H, Wang ZY, Johnson AC, Guroff G. The Wilms’ tumor gene product WT1 mediates the down-regulation of the rat epidermal growth factor receptor by nerve growth factor in PC12 cells. J Biol Chem. 1998;273:27047–27050. doi: 10.1074/jbc.M008776200. [DOI] [PubMed] [Google Scholar]
- 28.Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene. 2005;24:1648–1652. doi: 10.1038/sj.onc.1208345. [DOI] [PubMed] [Google Scholar]
- 29.Taylor R, Powles T, Humphreys J, Bettelheim R, Dowsett M, Casey A, Neville A, Coombes R. Effects of endocrine therapy on steroid-receptor content of breast cancer. Br J Cancer. 1982;45:80–84. doi: 10.1038/bjc.1982.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchyama S, Sasaki H, Masushige S, Gotoh Y, Hishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 31.Bunone G, Briand P, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 32.Ingar-Trowbridge DM, Nelson KG, Bidwell MC, Curtis SW, Wasburn TF, McLachlan JA, Korach KS. Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci USA. 1992;89:4658–4662. doi: 10.1073/pnas.89.10.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prall OWJ, Sarcevic B, Musgrove EA, Watts CKW, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272:10882–10894. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt EV. Happenstance, circumstance or enemy action: cyclin D1 in breast, eye and brain. BioEssays. 1996;18:6–8. doi: 10.1002/bies.950180104. [DOI] [PubMed] [Google Scholar]
- 35.Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, Haines GK, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000;20:672–683. doi: 10.1128/mcb.20.2.672-683.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143–164. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]
- 37.Santen RJ, Song RX, Zhang Z, Yue W, Kumar R. Adaptive hypersensitivity to estrogen: mechanism for sequential responses to hormonal therapy in breast cancer. Clin Cancer Res. 2004;10:337S–345S. doi: 10.1158/1078-0432.ccr-031207. [DOI] [PubMed] [Google Scholar]
- 38.Atanaskova Natasha, Keshamouni VG, Krueger JS, Schwartz JA, Fred M, Reddy KB. MAP kinase/estrogen receptor crosstalk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene. 2002;21:4000–4008. doi: 10.1038/sj.onc.1205506. [DOI] [PubMed] [Google Scholar]
- 39.Clark R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2005;53:25–71. [PubMed] [Google Scholar]
- 40.Zhang CC, Shapiro DJ. Activation of the p38 mitogen activated protein kinase pathway by estrogen or by 4-hydroxy-tamoxifen is coupled to estrogen receptor-induces apoptosis. J Biol Chem. 2000;275:475–489. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- 41.Obrero M, Yu DV, Shapiro DJ. Estrogen receptor-dependent and estrogen receptor-independent pathways for Tamoxifen and 4-Hydroxytamoxifen-induced programmed cell death. J Biol Chem. 2002;277:45695–45703. doi: 10.1074/jbc.M208092200. [DOI] [PubMed] [Google Scholar]
- 42.Bouker KB, Skaar TC, Fernandez DR, O’Brien KA, Riggins RB, Cao DH, Clarks R. Interferon regulatory factor-1 mediates the proapoptotic but not cell cycle arrest effects of the steroidal antiestrogen ICI 182, 780 (Faslodex, Fulvestrant) Cancer Res. 2004;64:4030–4039. doi: 10.1158/0008-5472.CAN-03-3602. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Han Y, Saiz FS, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 44.Han Y, San-Marina S, Liu J, Minden MD. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004;23:6933–6941. doi: 10.1038/sj.onc.1207609. [DOI] [PubMed] [Google Scholar]
- 45.Hewitt SM, Hamada S, McDonnell TJ, Rauscher FJ, III, Saunders GF. Regulation of the proto-oncogenes bcl-2 and c-myc by the Wilms’ tumor suppressor gene WT1. Cancer Res. 1995;55:5386–5389. [PubMed] [Google Scholar]
- 46.Englert C, Hou X, Maheswaran S, Bennett P, Ngwu C, Re GG, Garvin AJ, Rosner MR, Haber DA. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zapata-Benavides P, Tuna M, Lopez-Berestein G, Tari AM. Downregulation of Wilms’ tumor 1 protein inhibits breast cancer proliferation. Biochem Biophys Res Commun. 2002;295:784–790. doi: 10.1016/s0006-291x(02)00751-9. [DOI] [PubMed] [Google Scholar]
- 48.Tal M, King CR, Kraus MH, Ullrich A, Schlessinger J, Givollt D. Human HER2 (neu) promoter: evidence for multiple mechanisms for transcriptional initiation. Mol Cell Biol. 1987;7:2597–2601. doi: 10.1128/mcb.7.7.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simpson LA, Thompson KA, Loeb DM. Isoforms of Wilms’ tumor suppressor (WT1) have distinct effects on mammary epithelial cells. Oncogene. 2007;26:3423–3430. doi: 10.1038/sj.onc.1210127. [DOI] [PubMed] [Google Scholar]
- 50.Han YQ, Lin Y, Suarez-Saiz F, San-Matina S, Cui J, Minden MD. Wilms’ tumor 1 suppressor gene mediates anti-estrogen resistance via down-regulation of estrogen receptor-α expression in breast cancer cells. Mol Cancer Res. 2008;6:1347–1355. doi: 10.1158/1541-7786.MCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 51.Wang ZY, Qiu QQ, Deuel TF. ‘Alternatively spliced products of the Wilms’ tumor suppressor gene, wt1, have altered DNA binding specificity and regulated transcription in different ways’. Oncogene. 1995;10:415–422. [PubMed] [Google Scholar]