

Abstract
Isolated polycystic liver disease (PCLD) is an autosomal dominant disease with genetic and clinical heterogeneity. Apart from liver cysts, it exhibits few extra-hepatic manifestations and the majority of patients with this condition are asymptomatic or subclinical. However, a small fraction of these patients develop acute liver-cyst-related complications and/or massive cystic liver enlargement, causing morbidity and mortality. Currently, the management for symptomatic PCLD is centered on palliating symptoms and treating complications.
GENETICS
Polycystic liver disease was first described in the mid-19th century. Towards the end of the century polycystic liver disease was recognized as a condition of embryonal mal-development and of progressive cystic dilatation of bile ducts(1). In 1906, Moschcowits reported 91 cases of polycystic liver disease, 85 from existing literature and 6 from his own collection(2). He found that in 82 cases cysts involved both the liver and kidneys; in the remaining few cases, cysts were confined to the liver. Based on these observations, he asserted that polycystic liver disease coexists with cystic kidney disease, and that the rare cases of isolated PCLD are likely variants with delayed or yet-to-be developed polycystic kidneys.
Contrary to Moschcowit’s assertion, the notion that isolated PCLD might be a separate disease entity, independent of cystic kidneys, was proposed in the 1950’s, as cases of isolated PCLD repeatedly appeared in literature(3). A subsequent report in 1982 of two large, four-generation kindreds substantiated this assumption and revealed not only its existence but also its autosomal dominant pattern of inheritance(4).
It was not until 2003 that linkage analysis of eight Finnish families finally confirmed that isolated PCLD is a disease entity genetically distinct from polycystic kidney disease (which is caused by mutations in PKD genes) and is genetically heterogeneous(5). Such heterogeneity was verified by the identification of specific PCLD genes. Li et al and Davila et al show that isolated PCLD can be caused by mutations in either PRKCSH or SEC63(6, 7).
PRKCSH is located on the short arm of chromosome 19 (19p13.2-13.1)(7). It consists of 18 exons and encodes a 1.6 kb message that translates to a 527 amino acid protein termed hepatocystin(8). Hepatocystin is the non-catalytic β-subunit of glucosidase II. This enzyme is an endoplasmic reticulum (ER) luminal protein that is involved in the quality control of newly synthesized glycoproteins(9).
SEC63 is located on the long arm of chromosome 6 (6q21-q23)(6). It is a 21-exon gene and encodes a 3.4 kb message that translates into a 760 amino acid integral ER membrane protein, Sec63(6, 10). It is a part of the multi-component translocon that comprises the protein translocation machinery for integral membrane and secreted proteins. In mammalian systems, the translocon mediates cotranslational passage of nascent peptides into the ER and is functionally upstream of PRKCSH-dependent protein processing(11).
Mutations in either PRKCSH or SEC63 are postulated to result in defects in protein maturation and in the PCLD phenotype. Existing information does not support mutational hot spots or an apparent genotype-phenotype association(12). It is estimated that ~20 to 30% of the isolated PCLD cases are due to mutations in these two genes(6), suggesting the existence of additional yet-to-be identified gene(s) that, when mutated, can result in defects of protein maturation.
PATHOGENESIS
The cellular mechanisms by which mutations in PRKCSH or SEC63 gene ultimately lead to liver cyst formation have not been fully elucidated. The histopathological features of PCLD, however, bear striking similarities to liver cystic disease seen in autosomal dominant polycystic kidney disease (ADPKD). It has been postulated that mutations of PRKCSH or Sec63 cause aberrant maturation of newly synthesized glycoproteins(12) including polycystins. Gao H et al recently show that in zebrafish over-expression or deletion of PRKCSH causes developmental changes that are indistinguishable from those induced by the over-expression or depletion of PC2. Moreover, Abnormalities induced by PRKCSH deletion can be ameliorated by co-injection of PC2 mRNA(13). These findings support the notion of a common pathogenic pathway shared by PCLD and ADPKD. However, it remains unclear as to why the cystic phenotype in PCLD is confined to the liver, while in ADPKD both the liver and kidneys are affected.
Morphological studies of individual liver cysts reveal that liver cysts originate from biliary microhamartomas, also termed von Meyenburg’s complexes(14) that arise by proliferation of biliary ductules, and from peribiliary glands(2, 15, 16). As the cysts enlarge, they typically become detached from their origins. It is generally believed that the expansion of liver cysts is caused by concerted effects of (1) proliferation in cyst-lining epithelia, (2) fluid secretion into the cysts, (3) remodeling of the extracellular matrix surrounding the cysts, and (4) neovascularization(17). Because the common histopathological features shared by PCLD and ADPKD and the lack of orthologous PCLD animal model, studies of polycystic liver disease in non-orthologous animal models have frequently been extrapolated to PCLD.
Proliferation of biliary epithelial cells is required for the development and expansion of liver cysts. A number of signaling pathways, including cAMP-mediated activation of the mitogen activated protein kinase/extracellular regulated kinase (MAPK/ERK) pathway(18) and the mammalian target of rapammycin (mTOR)-mediated signaling cascade(19, 20), have been shown to be aberrantly activated and likely contribute to the proliferation of cystic epithelium. A recent study in PCK rats (an orthologus model of autosomal recessive PKD with prominent polycystic liver phenotype) also has shown that there is a global alteration in the expression pattern of micro RNAs (miRNAs, non-coding RNAs of ~20–22 nucleotides in length) in cystic epithelia(21). miRNAs can bind to their target mRNAs and decrease their expression via degradation or silencing(22). In this study(21), Lee et al show that of the 126 miRNAs analyzed, 76 are shared by both normal and cyst-lining epithelium, and 67 of the 76 (91%) are significantly (>2-fold) down-regulated in cystic epithelium. miR-15a is one of the down-regulated miRNAs and its down regulation results in an overexpression of the cell cycle regulator, Cdc25A, which strongly correlates with hyperproliferation of cyst-lining epithelial cells.
Fluid secretion by the cyst-lining epithelium contributes to the cyst expansion(23). The epithelial lining of liver cysts is shown to retain its secretin responsiveness and secretory capacity(24). Secretin activates cAMP-dependent signaling cascades leading to fluid secretion from cyst-lining epithelium and cyst expansion.
Remodeling of the extra cellular matrix surrounding the cysts is necessary for cyst expansion. Such remodeling is mediated by the activity of metalloproteases(25), which is highly elevated in the liver cyst epithelia(25). Cyst expansion also requires vascular supply. Studies have shown that the density of the vascular beds surrounding the liver cysts is significantly increased(26, 27), consistent with an adaptive angiogenesis and active neovascularization. Such neovascularization is promoted by a number of factors including cytokines IL-6 and IL-8 and growth factors such as vascular endothelial growth factor (VEGF)(28–30).
Compelling evidence indicates that estrogen promotes the development and progression of liver cysts(31–33). Estrogen acts both directly through estrogen receptors aberrantly expressed in cyst-lining cells in PCLD(34) and indirectly by potentiating the effects of growth factors to promote cyst-lining epithelium proliferation and secretion(35, 36). Moreover, by potentiating the effects of VEGF, estrogen enhances adaptive angiogenesis(37). Thus, estrogen can promote liver cyst growth via multiple mechanisms.
EPIDEMIOLOGY AND NATURAL HISTORY
Isolated PCLD is considered rare with an estimated incidence of < 0.01%. However, several autopsy studies have shown its occurrence only slightly lower than that of ADPKD(1, 3, 38). This discrepancy may be related to the milder and less symptomatic nature of isolated PCLD, compared to ADPKD. Therefore it seems likely that the incidence of PCLD has been underestimated.
Liver cysts in isolated PCLD usually begin to appear after affected individuals reach puberty. The rate of cyst growth and liver enlargement is heterogeneous with significant intrafamilial variability. However, for each patient, the liver cysts grow steadily with age in both number and size. Although men and women inherit PCLD equally, women present with massive and symptomatic cystic liver more frequently(39, 40), suggesting a hormonal component. Accordingly, multiparous women or women who have been on long-term oral contraceptives or estrogen replacement therapy tend to have larger-sized cystic liver and be more symptomatic(33). With a continuous increase in life expectancy, more patients with PCLD are expected to develop significant cystic liver enlargement and become symptomatic.
CLINICAL MANIFESTATIONS
The majority (> 80%) of PCLD patients are clinically asymptomatic(32). Liver synthetic function is typically preserved despite innumerable cysts(39). The only laboratory abnormalities in PCLD seen infrequently are mild elevations of γ-glutamyltransferase and alkaline phosphatase(32, 39). When symptoms arise, they are typically related to compression of neighboring organs by an enlarging liver. Cystic liver enlargement can cause abdominal distention and can result in chronic dull abdominal pain, early satiety or dyspnea. Compression of the inferior vena cava or rarely the biliary system can give rise to lower extremity edema and jaundice(41).
Acute pain may result from cyst rupture, hemorrhage or infection. Cyst rupture and hemorrhage can occur spontaneously or secondary to trauma and thought to result from stretching of the surrounding vessels. The precise frequency of their occurrence is unknown. In the majority of cases, pain is self-limited and can be managed with supportive care including analgesia.
Cyst infection is rare but serious with a reported mortality rate of 2%(42). Affected patients typically present with fever, chills and right upper quadrant pain. However, some can present with non-specific generalized malaise, vague abdominal discomfort, and fever of unknown origin. Signs of cyst infection include leukocytosis, positive blood culture, and changes on imaging studies, i.e., thickened, irregular cyst walls, and hyperdense cyst content with air-fluid levels(43, 44). Indium-labeled white blood cell scan can be helpful in providing supportive information and localizing the infection. Recently, 18F-FDG Positron emission tomography (PET)/computed tomography (CT) imaging technique, in which leukocytes labeled with 18F-FDG (a nonspecific tracer of increased glucose consumption), has emerged as a promising alternative for detecting and localizing infected liver cysts(45, 46). This method integrates metabolic (18F-FDG PET) and structural (CT) data and demonstrates sensitivity similar to that of 111In-labeled leukocyte scan in detecting infection, but with a lower specificity(47, 48). Thus, clinical correlation is imperative in the interpretation of the test results. Cyst infection should be managed in a timely fashion to reduce the risk of life-threatening complications. A combination of systemic antibiotics and cyst-drainage provides the best outcome(49).
Extrahepatic abnormalities in PCLD have been described. In our series of 146 individuals from 35 families in which at least one individual was known to have isolated polycystic liver disease, there was an increased incidence of cardiac valve abnormalities (mitral valve prolapse and incompetence) and of intracranial aneurysms, in up to 30% and 6% of affected individuals, respectively(32). However, the significance of these results is limited by the relatively small number of patients and families in the study. Although the occurrence of intracranial aneurysm in PCLD has also been described previously by Schievink et al(50) and recently by Hoevernaren et al (5.3% incidence) in their series of 19 PCLD patients(51), further study is required to ascertain these occurrences; indiscriminate screening is not recommended at present.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
PCLD is diagnosed by imaging studies, including ultrasound, CT and magnetic resonance imaging (MRI). Ultrasound is generally preferred because of its low cost, easy accessibility, and absence of radiation exposure. However, CT and MRI are more sensitive and accurate in detecting the presence and size of liver cysts.
The differential diagnosis of PCLD includes simple liver cysts and liver cysts resulting from other systemic diseases such as ADPKD (Figure 1). Simple liver cyst is a benign condition occurring with age in normal individuals. Our group has proposed ultrasonographic diagnostic criteria that can be used to differentiate simple liver cysts from PCLD in those who are at 50% risk of inheriting PCLD (at risk individuals)(32). In at risk individuals of < 40 years of age, the presence of any liver cysts is considered to be diagnostic of PCLD. In those at risk individuals, > 40 years of age, 4 or more liver cysts are considered affected with PCLD. These criteria were validated in the two largest PCLD families by comparing the imaging results with gene-linkage analysis(52). These criteria, however, need to be further vetted in a larger cohort of PCLD patients.
Figure 1.
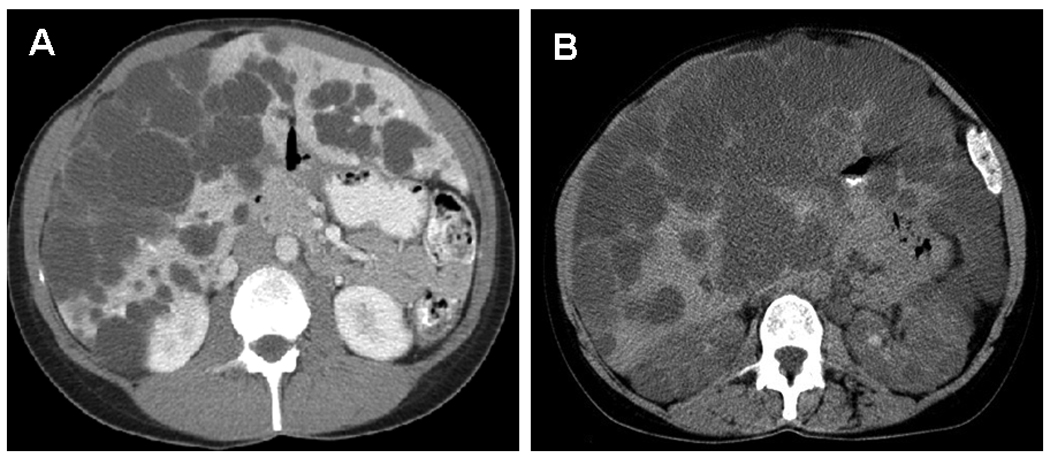
Transverse CT scan images from a patient with PCLD (1A) and a patient with ADPKD (1B). In PCLD, numerous cysts are confined in the liver, but in ADPKD cysts involve both the liver and kidneys.
As discussed elsewhere in this issue, cystic liver disease is the most common extrarenal manifestation of ADPKD. MRI imaging analysis performed as part of the CRISP study demonstrated that the prevalence of liver cysts increases with age, occurring in 58, 85, and 94% of affected individuals ages 15–24, 25–34, and 35–46(53). In addition, the severity of symptomatic cystic liver disease in ADPKD patients appears to parallel the severity of cystic kidney enlargement and kidney dysfunction(40, 54). The natural history of ADPKD and PCLD, however, is different primarily because individuals with PCLD do not develop renal failure. A recent study by Hoevenaren et al(51) also suggested that isolated PCLD patients tended to have fewer associated comorbidities, a greater number of liver cysts and larger cyst volume on presentation than those in ADPKD. Since ADPKD patients tend to be diagnosed at earlier disease stage, they have smaller liver cyst volume on presentation than patients with PCLD, who are typically diagnosed later in their disease course.
It should be noted that the extent of the liver cysts in PCLD varies from a few cysts to massive cystic liver enlargement. When the cysts are liver predominant but also present in the kidneys, the number of liver cysts is small, and a family history of PCLD or ADPKD is absent, it may be impossible to distinguish the two disease entities. Under such circumstances, ongoing clinical follow-up is helpful.
Recently, a study by Waanders et al shows that serum concentration of carbohydrate antigen 19-9 (CA19-9), secreted by cyst lining epithelium, correlates with the liver volume in PCLD(55). The authors suggest that serum CA19-9 could potentially be considered as a biomarker for monitoring liver cyst burden. However, large variations in serum CA19-9 levels exist among PCLD patients, and a small percentage of the Caucasian population (Lea–b– genotype) does not express CA 19-9. Thus, further study is required to evaluate the utility of serum CA19-9 in PCLD.
DNA testing for individual PCLD patients has limited utility at present because not all mutations responsible for PCLD have been identified. The two genes, PRKCSH and Sec63, identified thus far cover only a fraction of PCLD patients.
TREATMENT
No specific medical regimen has been established to prevent or retard cystic liver enlargement, although several agents have shown promise and are undergoing clinical studies. Today, the mainstay management for PCLD is interventional and centers on palliating symptoms.
Interventions for symptomatic PCLD include percutaneous cyst aspiration followed by sclerotherapy, cyst fenestration, hepatic resection with fenestration, selective hepatic artery embolization, stenting of obstructed bile ducts and large abdominal veins and rarely, liver transplantation. Choice of specific treatment depends on the number, size, and location of the cysts and on local expertise.
Percutaneous cyst aspiration followed by sclerotherapy is feasible in treating one or several large cysts that cause symptoms. Sclerotherapy using 95–99% alcohol or acidic solutions of tetracycline or minocycline ablates the fluid-producing cyst epithelia and attenuates the cyst reexpansion. With this method, ~70 to 90% treated cysts can be successfully obliterated. For cysts > 8 cm in diameter, repeat treatments are often necessary to achieve a sustained effect(56–59). Cysts eventually recur in nearly 80% of patients, and retreatment for recurrent symptoms is required in 50% of patients(60). Overall, this method is minimally invasive and rarely associated with complications.
Cyst fenestration(61), laparoscopically or by open laparotomy, involves unroofing and excising the cystic area down to the interface of the liver parenchyma. The unroofed cyst-lining epithelium is then treated by argon laser beam coagulation and electrocoagulation. Patients with a small number of large, superficial cysts are the most suitable candidates. Symptom relief can be achieved in 30 – 80%(60, 62–65). Laparoscopic fenestration is associated with short hospital stays and without mortality, but with recurring symptoms in ~50% necessitating repeat laparoscopic cyst aspiration. Approximately 10% require surgical conversion and 30% suffered from perioperative complications. Open laparotomy is associated with prolonged hospitalization, morbidity including bleeding, infection, bile leak and ascites (0–69%) and mortality (3%)(66).
Hepatic resection with fenestration is effective in reducing liver mass. Large superficial and deep seated cysts in the remaining liver segments can be fenestrated during the operation. Suitable candidates are those with massive hepatomegaly due to innumerable small- to medium-sized cysts with preservation of at least two contiguous liver segments with adequate hepatic venous drainage. The mortality and morbidity rates are highly dependent on the degree of expertise in the team of caregivers. In the largest series of 124 patients from Mayo Clinic, rates of morbidity and mortality were 63% and 3%, respectively(67). The most common morbidities include bile leak and ascites. The latter may require stenting of the inferior vena cava or hepatic veins, or more rarely placement of a LaVeen shunt.
Selective hepatic artery embolization has been reported from a single center(68, 69). This method targets hepatic regions with no discernible normal parenchyma. It appears to be safe with an average hepatic mass reduction by 23%. The major side effects are post procedural pain and fever, which typically resolve within a week. This method is best suited for patients who are poor candidates for surgery.
Stenting of a compressed hepatic vein or inferior vena cava can reestablish patency and relieve obstruction-associated symptoms, such as intractable ascites and lower extremity edema(70, 71).
Liver transplantation is limited by organ shortage, perioperative risks and lifelong immunosuppression. Liver transplantation for PCLD patients, given their typically preserved liver function, is reserved for severely symptomatic patients who are not suitable for other treatment modalities. Review of United Network for Organ Sharing (UNOS) data indicates that in patients receiving liver transplantation for polycystic liver disease early post operative mortality within 6 months is ~20%. Long-term outcome in those survived beyond 6 months is excellent(60). One study showed that 1-, 3-, and 5-year survival rate was 78.1, 71.7, and 68.7%, respectively(72).
A recent study by Schnelldorfer et al summarized the Mayo Clinic experience in operative treatment of symptomatic polycystic liver disease, in 141 patients (including 24 PCLD patients). During the period of 1985 to 2006, 124 patients underwent partial hepatectomy with cyst fenestration, 10 cyst fenestration alone (2 laparoscopic), and 7 liver transplantation (65). Overall operative morbidity and mortality was 58% and 4%, respectively, with major complications in 30%. Overall 5- and 10-year survival was 90% and 78%, respectively. The 5-year survival after partial hepatectomy with cyst fenestration, cyst fenestration alone, and liver transplantation was 92%, 90%, and 60%, respectively. Although some degree of cystic liver-related symptoms recurred in 73% of patients in this study, performance status was normalized or improved in 75%, and 73% returned to work full-time.
Non-interventional management options are limited at present. Avoidance of estrogen should be considered in patients with progressive polycystic liver enlargement. This is based on both the existing research evidence showing liver-cyst growth effects of estrogen and the clinical observation of more symptomatic, rapid liver-cyst growth and more frequent requirement of interventions associated with high-level estrogen exposure(31, 33, 51, 73).
Caffeine is known to increase cellular cAMP, which acts as a second messenger to stimulate cyst growth by promoting the secretion of electrolytes and water from the cyst-lining cholangiocytes and by stimulating cholangiocyte proliferation(74, 75). Excessive consumption of caffeinated drinks might induce cAMP mediated effects and may be discouraged, although clinical or epidemiological study is lacking.
Several investigational agents are currently undergoing clinical trials. Octreotide and lanreotide belong to a group of long-acting synthetic somatostatin analogs that inhibit secretin-induced, cAMP-mediated secretion in biliary and cyst-lining epithelium. Octreotide has been shown to inhibit hepatic cyst growth in PCK rats, an orthologous autosomal recessive PKD model with prominent cystic liver enlargement(76). In a recent 8 case series (7 ADPKD, 1 PCLD), octreotide treatment (100 mcg thrice daily subcutaneous injection for 70 to 180 days) resulted in a 3.0% median reduction of liver volume(77). A pilot prospective trial in patients with polycystic liver disease using monthly octreotide treatment is currently in progress. Keimpema et al recently reported their results of lanreotide for 54 patients with polycystic liver disease. It was a prospective, randomized, placebo-controlled trial included 22 PCLD patients; lanreotide (120 mg) or placebo was administered subcutaneously every 4 weeks. After a total of 24 weeks, the liver volume reduced by 2.9% in lanreotide group but increased by 1.6% in placebo group (P < 0.01). Patients with ADPKD or PCLD showed a similar responses to lanreotide(78). These results are encouraging, although longer and larger studies are needed to determine the long-term tolerability, efficacy and the impact on the symptoms and complications associated with polycystic liver disease.
mTOR inhibitors, sirolimus and everolimus, are immunosuppressive agents and exerts an antiproliferative effect. A retrospective study in ADPKD patients after renal transplantation showed that sirolimus-containing immunosuppressive regimen was associated with a significant reduction in polycystic liver volume, suggesting a role for sirolimus in the treatment severe polycystic liver disease(19). Prospective clinical trials of sirolimus and everolimus in ADPKD are currently in progress. The results of these studies on polycystic liver disease may be applicable to PCLD considering the nearly identical clinical and histopathological characteristics of polycystic liver disease in ADPKD and PCLD, although direct study in patients with PCLD would be ideal.
SUMMARY
Isolated PCLD is a group of genetically and phenotypically heterogeneous diseases. Although the majority of PCLD patients are asymptomatic, a small fraction of them develop polycystic liver related complications that can cause significant morbidity and mortality. To date, management for PCLD rests primarily on operative measures to correct complications. A number of candidate medications aiming to deter cyst growth are undergoing clinical trials. The results of these trials are eagerly awaited.
ACKNOWLEDGMENTS
Supported in part by DK073567 and Mayo Foundation Career Development Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure Statement: None.
REFERENCES
- 1.Melnick PJ. Polycystic liver; analysis of seventy cases. AMA Arch Pathol. 1955;59(2):162–172. [PubMed] [Google Scholar]
- 2.Moschcowitz E. Non-parasitic cysts (congenital) of the liver, with a study of aberrant bile ducts. The American Journal of the Medical Sciences. 1906;131(4):674–699. [Google Scholar]
- 3.Feldman M. Polycystic disease of the liver. Am J Gastroenterol. 1958;29(1):83–86. [PubMed] [Google Scholar]
- 4.Berrebi G, Erickson RP, Marks BW. Autosomal dominant polycystic liver disease: a second family. Clin Genet. 1982;21(5):342–347. doi: 10.1111/j.1399-0004.1982.tb01381.x. [DOI] [PubMed] [Google Scholar]
- 5.Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kaariainen H, Hockerstedt K. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38(1):39–43. doi: 10.1016/s0168-8278(02)00348-3. [DOI] [PubMed] [Google Scholar]
- 6.Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36(6):575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 7.Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72(3):691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33(3):345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 9.Trombetta ES, Fleming KG, Helenius A. Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry (Mosc) 2001;40(35):10717–10722. doi: 10.1021/bi010629u. [DOI] [PubMed] [Google Scholar]
- 10.Skowronek MH, Rotter M, Haas IG. Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biol Chem. 1999;380(9):1133–1138. doi: 10.1515/BC.1999.142. [DOI] [PubMed] [Google Scholar]
- 11.Schnell DJ, Hebert DN. Protein translocons: multifunctional mediators of protein translocation across membranes. Cell. 2003;112(4):491–505. doi: 10.1016/s0092-8674(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 12.Drenth JP, Martina JA, van de Kerkhof R, Bonifacino JS, Jansen JB. Polycystic liver disease is a disorder of cotranslational protein processing. Trends in molecular medicine. 2005;11(1):37–42. doi: 10.1016/j.molmed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Gao H, Wang Y, Wegierski T, et al. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum Mol Genet. 19(1):16–24. doi: 10.1093/hmg/ddp463. [DOI] [PubMed] [Google Scholar]
- 14.von Meyenburg H. Uber die cystenleber. Beitr Path Anat. 1918;64 477A/532. [Google Scholar]
- 15.Bistritz L, Tamboli C, Bigam D, Bain VG. Polycystic liver disease: experience at a teaching hospital. Am J Gastroenterol. 2005;100(10):2212–2217. doi: 10.1111/j.1572-0241.2005.50258.x. [DOI] [PubMed] [Google Scholar]
- 16.Perrone RD, Grubman SA, Rogers LC, et al. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol. 1995;269(3 Pt 1):G335–G345. doi: 10.1152/ajpgi.1995.269.3.G335. [DOI] [PubMed] [Google Scholar]
- 17.Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Current opinion in gastroenterology. 2009;25(3):265–271. doi: 10.1097/MOG.0b013e328328f4ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63(6):1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 19.Qian Q, Du H, King BF, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19(3):631–638. doi: 10.1681/ASN.2007050626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103(14):5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SO, Masyuk T, Splinter P, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118(11):3714–3724. doi: 10.1172/JCI34922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 23.Ye M, Grantham JJ. The secretion of fluid by renal cysts from patients with autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329(5):310–313. doi: 10.1056/NEJM199307293290503. [DOI] [PubMed] [Google Scholar]
- 24.Everson GT, Emmett M, Brown WR, Redmond P, Thickman D. Functional similarities of hepatic cystic and biliary epithelium: studies of fluid constituents and in vivo secretion in response to secretin. Hepatology. 1990;11(4):557–565. doi: 10.1002/hep.1840110406. [DOI] [PubMed] [Google Scholar]
- 25.Murray SL, Grubman SA, Perrone RD, et al. Matrix metalloproteinase activity in human intrahepatic biliary epithelial cell lines from patients with autosomal dominant polycystic kidney disease. Connect Tissue Res. 1996;33(4):249–256. doi: 10.3109/03008209609028882. [DOI] [PubMed] [Google Scholar]
- 26.Ubara Y. New therapeutic option for autosomal dominant polycystic kidney disease patients with enlarged kidney and liver. Ther Apher Dial. 2006;10(4):333–341. doi: 10.1111/j.1744-9987.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- 27.Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60(1):37–45. doi: 10.1046/j.1523-1755.2001.00768.x. [DOI] [PubMed] [Google Scholar]
- 28.Nichols MT, Gidey E, Matzakos T, et al. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40(4):836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 29.Fabris L, Cadamuro M, Fiorotto R, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43(5):1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 30.Amura CR, Brodsky KS, Groff R, Gattone VH, Voelkel NF, Doctor RB. VEGF receptor inhibition blocks liver cyst growth in pkd2(WS25/-) mice. Am J Physiol Cell Physiol. 2007;293(1):C419–C428. doi: 10.1152/ajpcell.00038.2007. [DOI] [PubMed] [Google Scholar]
- 31.Alvaro D, Mancino MG, Onori P, et al. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12(22):3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37(1):164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 33.Sherstha R, McKinley C, Russ P, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26(5):1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 34.Alvaro D, Invernizzi P, Onori P, et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. 2004;41(6):905–912. doi: 10.1016/j.jhep.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 35.Alvaro D, Metalli VD, Alpini G, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43(5):875–883. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Gigliozzi A, Alpini G, Baroni GS, et al. Nerve growth factor modulates the proliferative capacity of the intrahepatic biliary epithelium in experimental cholestasis. Gastroenterology. 2004;127(4):1198–1209. doi: 10.1053/j.gastro.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 37.Mancino A, Mancino MG, Glaser SS, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. 2008 doi: 10.1016/j.dld.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986;30(1):29–37. doi: 10.1111/j.1399-0004.1986.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 39.Everson GT, Scherzinger A, Berger-Leff N, et al. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8(6):1627–1634. doi: 10.1002/hep.1840080626. [DOI] [PubMed] [Google Scholar]
- 40.Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11(6):1033–1037. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 41.Dmitrewski J, Olliff S, Buckels JA. Obstructive jaundice associated with polycystic liver disease. HPB Surg. 1996;10(2):117–120. doi: 10.1155/1996/83547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abascal J, Moya M, Martin F. Infection of hepatic cysts in polycystic disease. World J Surg. 1984;8(3):424–425. doi: 10.1007/BF01655097. [DOI] [PubMed] [Google Scholar]
- 43.Levine E, Cook LT, Grantham JJ. Liver cysts in autosomaldominant polycystic kidney disease: clinical and computed tomographic study. AJR Am J Roentgenol. 1985;145(2):229–233. doi: 10.2214/ajr.145.2.229. [DOI] [PubMed] [Google Scholar]
- 44.Levine E, Grantham JJ. High-density renal cysts in autosomal dominant polycystic kidney disease demonstrated by CT. Radiology. 1985;154(2):477–482. doi: 10.1148/radiology.154.2.3966136. [DOI] [PubMed] [Google Scholar]
- 45.Dumarey N, Egrise D, Blocklet D, et al. Imaging infection with 18FFDG- labeled leukocyte PET/CT: initial experience in 21 patients. J Nucl Med. 2006;47(4):625–632. [PubMed] [Google Scholar]
- 46.Keidar Z, Gurman-Balbir A, Gaitini D, Israel O. Fever of unknown origin: the role of 18F-FDG PET/CT. J Nucl Med. 2008;49(12):1980–1985. doi: 10.2967/jnumed.108.054692. [DOI] [PubMed] [Google Scholar]
- 47.Bleeker-Rovers CP, Corstens FH, Van Der Meer JW, Oyen WJ. Fever of unknown origin: prospective comparison of diagnostic value of (18)F-FDG PET and (111)In-granulocyte scintigraphy. European journal of nuclear medicine and molecular imaging. 2004;31(9):1342–1343. doi: 10.1007/s00259-004-1634-6. author reply 1344. [DOI] [PubMed] [Google Scholar]
- 48.Kjaer A, Lebech AM, Eigtved A, Hojgaard L. Fever of unknown origin: prospective comparison of diagnostic value of 18F-FDG PET and 111In-granulocyte scintigraphy. European journal of nuclear medicine and molecular imaging. 2004;31(5):622–626. doi: 10.1007/s00259-003-1425-5. [DOI] [PubMed] [Google Scholar]
- 49.Telenti A, Torres VE, Gross JB, Jr, Van Scoy RE, Brown ML, Hattery RR. Hepatic cyst infection in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65(7):933–942. doi: 10.1016/s0025-6196(12)65154-4. [DOI] [PubMed] [Google Scholar]
- 50.Schievink WI, Spetzler RF. Screening for intracranial aneurysms in patients with isolated polycystic liver disease. J Neurosurg. 1998;89(5):719–721. doi: 10.3171/jns.1998.89.5.0719. [DOI] [PubMed] [Google Scholar]
- 51.Hoevenaren IA, Wester R, Schrier RW, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28(2):264–270. doi: 10.1111/j.1478-3231.2007.01595.x. [DOI] [PubMed] [Google Scholar]
- 52.Reynolds DM, Falk CT, Li A, et al. Identification of a locus for autosomal dominant polycystic liver disease, on chromosome 19p13.2-13.1. Am J Hum Genet. 2000;67(6):1598–1604. doi: 10.1086/316904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bae KT, Zhu F, Chapman AB, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1(1):64–69. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 54.Harris RA, Gray DW, Britton BJ, Toogood GJ, Morris PJ. Hepatic cystic disease in an adult polycystic kidney disease transplant population. Aust N Z J Surg. 1996;66(3):166–168. doi: 10.1111/j.1445-2197.1996.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 55.Waanders E, van Keimpema L, Brouwer JT, et al. Carbohydrate antigen 19-9 is extremely elevated in polycystic liver disease. Liver Int. 2009 doi: 10.1111/j.1478-3231.2009.02055.x. [DOI] [PubMed] [Google Scholar]
- 56.Tikkakoski T, Makela JT, Leinonen S, et al. Treatment of symptomatic congenital hepatic cysts with single-session percutaneous drainage and ethanol sclerosis: technique and outcome. J Vasc Interv Radiol. 1996;7(2):235–239. doi: 10.1016/s1051-0443(96)70767-4. [DOI] [PubMed] [Google Scholar]
- 57.Blonski WC, Campbell MS, Faust T, Metz DC. Successful aspiration and ethanol sclerosis of a large, symptomatic, simple liver cyst: case presentation and review of the literature. World J Gastroenterol. 2006;12(18):2949–2954. doi: 10.3748/wjg.v12.i18.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshida H, Onda M, Tajiri T, et al. Long-term results of multiple minocycline hydrochloride injections for the treatment of symptomatic solitary hepatic cyst. J Gastroenterol Hepatol. 2003;18(5):595–598. doi: 10.1046/j.1440-1746.2003.03025.x. [DOI] [PubMed] [Google Scholar]
- 59.van Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci. 2008;53(8):2251–2257. doi: 10.1007/s10620-007-0121-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13(38):5052–5059. doi: 10.3748/wjg.v13.i38.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin TY, Chen CC, Wang SM. Treatment of non-parasitic cystic disease of the liver: a new approach to therapy with polycystic liver. Ann Surg. 1968;168(5):921–927. doi: 10.1097/00000658-196811000-00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farges O, Bismuth H. Fenestration in the management of polycystic liver disease. World J Surg. 1995;19(1):25–30. doi: 10.1007/BF00316975. [DOI] [PubMed] [Google Scholar]
- 63.Konstadoulakis MM, Gomatos IP, Albanopoulos K, Alexakis N, Leandros E. Laparoscopic fenestration for the treatment of patients with severe adult polycystic liver disease. Am J Surg. 2005;189(1):71–75. doi: 10.1016/j.amjsurg.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 64.Martin IJ, McKinley AJ, Currie EJ, Holmes P, Garden OJ. Tailoring the management of nonparasitic liver cysts. Ann Surg. 1998;228(2):167–172. doi: 10.1097/00000658-199808000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagorney DM. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2009;250(1):112–118. doi: 10.1097/SLA.0b013e3181ad83dc. [DOI] [PubMed] [Google Scholar]
- 66.Robinson TN, Stiegmann GV, Everson GT. Laparoscopic palliation of polycystic liver disease. Surg Endosc. 2005;19(1):130–132. doi: 10.1007/s00464-004-8813-1. [DOI] [PubMed] [Google Scholar]
- 67.Que F, Nagorney DM, Gross JB, Jr, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108(2):487–494. doi: 10.1016/0016-5085(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 68.Takei R, Ubara Y, Hoshino J, et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2007;49(6):744–752. doi: 10.1053/j.ajkd.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 69.Ubara Y, Takei R, Hoshino J, et al. Intravascular embolization therapy in a patient with an enlarged polycystic liver. Am J Kidney Dis. 2004;43(4):733–738. doi: 10.1053/j.ajkd.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 70.Grams J, Teh SH, Torres VE, Andrews JC, Nagorney DM : Inferior vena cava stenting: a safe and effective treatment for intractable ascites in patients with polycystic liver disease. J Gastrointest Surg. 2007;11(8):985–990. doi: 10.1007/s11605-007-0182-3. [DOI] [PubMed] [Google Scholar]
- 71.Mudge DW, Taylor J, Bannister KM. Hepatic vein stenting for recurrent ascites in polycystic liver and kidney disease. Nephrol Dial Transplant. 2005;20(11):2566–2568. doi: 10.1093/ndt/gfi051. [DOI] [PubMed] [Google Scholar]
- 72.US Scientific Registry of Transplant: 2004 OPTN / SRTR Annual Report. US Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients. U.S. government-sponsored work. 2004 wwwustransplantorg.
- 73.Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172(2):321–332. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Banales JM, Masyuk TV, Bogert PS, et al. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173(6):1637–1646. doi: 10.2353/ajpath.2008.080125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49(1):160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology. 2007;132(3):1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 77.van Keimpema L, Drenth JP. Effect of octreotide on polycystic liver volume. Liver Int. 2009 doi: 10.1111/j.1478-3231.2009.02123.x. [DOI] [PubMed] [Google Scholar]
- 78.van Keimpema L, Nevens F, Vanslembrouck R, et al. Lanreotide Reduces the Volume of Polycystic Liver: A Randomized, Double-Blind, Placebo-Controlled Trial. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]