

Abstract
Autosomal dominant polycystic disease (ADPKD) is the most common form of inherited kidney disease that results renal failure. The understanding the pathogenesis of ADPKD has advanced significantly since the discovery of the two causative genes, PKD1 or PKD2. Dominantly inherited gene mutations followed by somatic second hit mutations inactivating the normal copy of the respective gene result in renal tubular cyst formation that deforms the kidney and eventually impairs its function. The respective gene products, polycystin-1 and polycystin-2, work together in a common cellular pathway. Polycystin-1, a large receptor molecule, forms a receptor-channel complex with polycystin-2, which is a cation channel belonging to the TRP family. Both polycystin proteins have been localized to the primary cilium, a non-motile microtubule based structure that extends from the apical membrane of tubular cells into the lumen. Here we discuss recent insights in the pathogenesis of ADPKD including the genetics of ADPKD, the properties of the respective polycystin proteins, the role of cilia, and some cell signaling pathways that have been implicated in the pathways related to PKD1 and PKD2.
Keywords: Polycystic kidney, Cilia, Polycystin, TRP channel, GPS cleavage
Autosomal dominant polycystic kidney disease (ADPKD; MIM 173900) is a systemic disorder characterized by age-dependent occurrence of bilateral, multiple renal cysts as well as a variety of extrarenal manifestations. The latter include cysts in the liver bile ducts, pancreatic ducts, seminal vesicles, and arachnoid membrane, as well as non-cystic manifestations, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias (1). The study of this disease through human genetics, animal models, classical cell biology and biochemical approaches has yielded remarkable insights; however, the goal of finding an effective treatment is still a work in progress. Mutations in two genes, PKD1 and PKD2, are responsible for ADPKD (2–4). Subsequently, the genes for a number of other inherited diseases that manifest with varying degrees of cyst formation (e.g., recessive polycystic kidney disease, nephronophthisis, Bardet Biedl syndrome, and several others) have been discovered (5–8). Mechanistic studies of ADPKD and the other cystic kidney diseases have identified a previously little appreciated organelle, the primary cilium and its associated basal body complex, as the central cellular compartment in the pathogenesis of this group of disorders (9). These insights have accelerated a broader understanding of the clinical diseases and have moved the field to the threshold of directed therapeutic clinical trials for ADPKD.
GENETICS OF ADPKD
Mutations in at least two genes cause the clinical presentation of ADPKD. PKD1, located on chromosome 16p13.3, accounts for 85% of families; PKD2, on chromosome 4q21, accounts for the remainder (Table 1). Families with mutations in PKD1 tend to have more severe clinical presentations, but there is marked intra-familial and inter-familial variability in the clinical manifestations of ADPKD that go beyond the germline mutation and may suggest a possible role for genetic background in disease progression (10–12). Genotype-phenotype correlations have been difficult to identify in ADPKD although there is some indication that patients with mutations in the 5' region of PKD1 tend to have a more severe disease with increased risk of intracranial aneurysms and earlier onset of ESRD compared to patients with 3' mutations (13–15). Trans-heterozygous individuals in a rare family segregating mutations in both PKD1 and PKD2 showed a more severe presentation compared to family members with single mutations (16), consistent with an additive effect of having mutations in both genes. While it is likely that complete loss of function mutations affecting either gene would result in non-viable progeny (16;17), a recent report described several individuals carrying two incompletely penetrant, hypomorphic, PKD1 mutations (18).
Table 1.
Summary of ADPKD gene and protein characteristics.
| PKD1 | PKD2 | |
|---|---|---|
| Mutations in ADPKD families | 85% | 15% |
| Mean age of onset ESRD | 53 years | 69 years |
| Gene characteristics | Chromosome 16q13.3 46 exons gene size, ~45 kb spliced transcript, ~12.9 kb |
Chromosome 4q21–23 15 exons gene size, ~50 kb spliced transcript, ~3.5 kb |
| Protein characteristics | Polycystin 1 (PC1) 4302 amino acids 11 transmembrane domains Receptor-like protein Undergoes proteolytic cleavage |
Polycystin 2 (PC2; TRPP2) 968 amino acids 6 transmemebrane domains Homology to TRP channels |
| Subcellular localization | Cilia, cell junctions, apical and basolateral plasma membrane | Cilia, endoplasmic reticulum, plasma membrane |
| Function | Receptor | Cation channel |
THE PKD GENES AND THEIR PROTEIN PRODUCTS
PKD1 and PKD2, respectively encode for proteins polycystin-1 (PC1) and polycystin-2 (PC2) (Fig. 1). Polycystin-1 (PC1) is comprised of 4302 amino acids with a large extracellular domain (3074 aa), 11 transmembrane spanning segments and a short cytoplasmic tail (197 aa) (19;20) (Fig 1). The extracellular NH2-terminal domain contains a distinct combination of protein motifs involved in protein-protein, protein carbohydrate interactions (19;21), a receptor egg jelly (REJ) domain (22) and a GPCR proteolytic site (GPS domain) (23). The region of the last five transmembrane spans of PC1 share sequence homology with polycystin-2 (PC2) (24). PC1 occurs as a full length protein but also undergoes autoproteolytic cleavage at the GPS site to yield the extracellular NH2-terminal fragment (NTF) and the intramembranous COOH-terminal fragment (CTF) which remains tethered after cleavage (13). A novel mouse model (Pkd1v/v) carrying a non-cleavable mutation in Pkd1 suggested that GPS cleavage is required to prevent cyst formation (25). The cytoplasmic tail of PC1 also has been reported to undergo cleavage events. In one proposed model the entire C-terminal tail (p200) is cleaved and translocates to the nucleus (26) and binds β-catenin preventing TCF mediated gene transcription (27). In another study, a different cleavage event occurs resulting a 112 amino acid fragment of the C-terminus that interacts with STAT6 and p100 which is thought to stimulate transcriptional activity (28).
Figure 1. Structures of polycystin-1 and polycystin-2.

Thick green line represents the membrane bilayer. The protein motifs are identified in the boxed figure legend. Light blue and green cylinders represent putative transmembrane segments. Structures are not drawn to scale.
Polycystin-2 (PC2) consists of 968 amino acids with 6 transmembrane domains (29). It is a non-selective cation channel permeable to Ca2+ that belongs to the TRPP subfamily of TRP cation channels (TRPP2, reviewed in (30). The last five transmembrane spans in PC2 bear a strong TRP channel signature and the region between S5 and S6 (transmembrane segments 5 and 6) contains the putative pore region (Fig 1) (24). The cytoplasmic tail of PC2 contains a Ca2+ binding EF-hand (31);(4) and a coiled coil domain responsible for numerous protein-protein interactions (31;32);(33). Mammalian PC2 has at least one phosphorylation site in its C-tail that modulates the Ca2+ dependence of channel activity (34) and has been suggested to have a role in trafficking of PC2 between ER, Golgi and plasma membrane compartments (35). PC1 and PC2 interact through their respective C-termini (33;36;37) (Fig 1). The interaction depends on the integrity of the coiled coil domain in the C-terminus of PC1 and has led to the hypothesis that PC1 may serve as a receptor that controls the channel activity of PC2 as part of the polycystin signaling complex.
MOLECULAR MECHANISMS OF CYST FORMATION IN ADPKD
Early microdissection studies of ADPKD kidneys indicated that cysts initially appear as focal lesions in kidney tubules that otherwise appear to be normal along most of their length (38). A molecular explanation for the focal nature of cyst formation in the setting of heterozygous germline mutations came with the discovery that cyst lining cells from human ADPKD cysts have loss of heterozygosity (LOH) in the chromosomal regions of the respective PKD genes in both the kidney (39–42) and liver (43). These findings implicated a cellular recessive mechanism for cyst formation in ADPKD and suggested the possibility that the observed intrafamilial variation in disease severity may at least in part be determined by variation in the timing and number of somatic `second hit' mutations in individual family members (44). The causal relationship between `second hit' mutations and cyst formation in adult kidneys was validated in a mouse model expressing a modified Pkd2 allele (Pkd2WS25) that expresses functional PC2 but is prone to genomic rearrangement leading to somatic loss of PC2 (45).
Alternative mechanisms of cyst formation also exist and the relative importance of these in human clinical disease remains to be determined. Cyst formation resulting from trans-heterozygous somatic mutations involving PKD1 and PKD2 have also been proposed (46;47). However, trans-heterozygous mutations alone are unlikely to be sufficient for cyst formation. Individuals with bilineal inheritance of PKD1 and PKD2 mutations (16) and trans-heterozygous mice (48) show more severe polycystic kidney disease, but the overall severity is within the range consistent with additive effects of single gene mutations. Recent evidence indicates that significant reduction of functional PC1 expression below a critical threshold level is sufficient to result in cyst formation in some situations (Fig. 2) (18;49;50). A unique chimeric animal model produced by mosaic embryos combining Pkd1−/− cells with wild type cells formed kidney cysts with severity proportionate to the degree of Pkd1−/− contribution to the mosaic animal (51). Interestingly, the cysts were mosaic with both Pkd1−/− and wild type cells in the early stages, but over time, the Pkd1−/− cells replaced the wild type cells by inducing apoptosis. These data suggest the possibility that cellular loss of PC1 produces cyst formation both by expansion of the null cell mass and by induction of programmed cell death in surrounding normal cells.
Figure 2. Threshold model of cyst formation in ADPKD.

This model is based on the hypothesis that a critical level of PKD1 and PKD2 functional activity is required to form and maintain tubule structure (solid red line). In this model, a cyst forms when the combined activity of two alleles falls below the specific threshold for an individual cell. The requisite threshold may be vary based on a combination of factors such as genetic variants at modifier loci, environmental effects, the developmental stage of the kidney, or physiologic demands such as in response to injury (dashed red lines). The actual of level of activity achieved is determined by the specific combination of allelic variants in a gene, e.g., PKD1. In this example, three different wild type variant alleles (WT1–WT3) have their own unique set of polymorphisms that impact PKD1 activity with WT1 having the greatest activity. Combinations of wild type alleles with each other or with a null allele provide sufficient activity to avert cyst growth (leftmost three columns). Mutant alleles have less activity than the wild type variants. These range from complete loss of function (Null) to missense mutations with reduced activity (Miss, Miss-1, Miss-2). Individuals who inherit any of these hypomorphic, reduced function, allele haplotypes through the germline may acquire somatic mutations in individual cells (rightmost six columns) that in combination may fall below cell-specific threshold levels of activity and lead to cyst formation. In the illustration, Null/Null and Null/Miss-2 allele combinations will invariably result in cysts, whereas Miss/Miss-1 will never result in cysts with the other combinations having different effects depending on the actual cellular thresholds. This model also applies to PKD2.
More recently the conditional Cre-lox system has been used to bypass the embryonic lethality of null mice and inactivate genes of interest in a tissue selective or temporally controlled manner (49;50;52;53). Temporally controlled inactivation of Pkd1 has shown that the timing of gene inactivation impacts the rate of cyst growth (54;55) Inactivation of Pkd1 in the developing kidney results in rapid cyst growth while inactivation in the adult kidney results in markedly slower cyst growth (Fig. 3) (54;55). A developmentally regulated change in the gene expression profile of the kidney coincides with the differential effect of Pkd1 loss on cyst formation (55). The underlying proliferative potential of the cells as a function of developmental stage may account for part of the difference in the rate of cyst formation (53). This is supported by the finding that regenerating tubules after ischemia reperfusion injury have an increased rate of cyst formation (56–58). Factors such as the timing of gene inactivation, the degree of inactivation of the protein product and the presence of other forms of renal injury are likely to contribute to the severity of ADPKD in individuals.
Figure 3. Phenotypic response to acquired Pkd1 loss is exquisitely sensitive to developmental life stage.
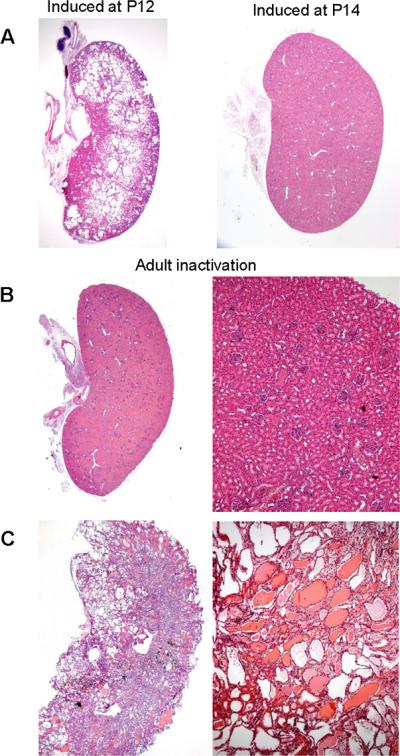
A, Kidneys of Pkd1cond/cond;tamoxifen-Cre+ mice with inactivation of Pkd1 induced at postnatal day 12 (P12) became cystic within 3 weeks (left panel), whereas if Pkd1 inactivation occurs at P14, they remained normal 3 months later (right panel) [from (55)]. B,C, Pkd1 inactivation in adult kidneys results in late-onset renal cystic disease. Kidneys from Pkd1cond/cond;tamoxifen-Cre+ mice harvested 3 months (B) or 6 months (C) after Pkd1 inactivation was induced at 6 weeks of age [from (55)].
CILIA AND ADPKD
Several lines of complementary evidence led to the emergence of cilia as central to the pathogenesis of ADPKD. The PC1 ortholog in C.elegans is localized to cilia (59). Mutations in genes associated with situs inversus, a known cilial phenotype, are associated with polycystic kidneys (60). Mutations in genes encoding intraflagellar transport (IFT) proteins required for cilia structure are also associated with polycystic kidneys (61). The subsequent demonstration of PC2 and PC1 localization to primary cilia of renal epithelia (62;63) solidified the hypothesis that cilia have a role in the pathogenesis of ADPKD. Further support of the cilia link with ADPKD came with the discovery that Pkd2−/− mice have defects in left-right axis formation (64) through a lateralized calcium signal at the left margin of the embryonic node during mammalian development (65). A prospective study selectively inactivating Kif3a, a component of the kinesin-2 motor complex required for cilia maintenance, in the kidney resulted in cyst formation further supporting the role of intact cilia in polycystic diseases (66).
While there is a clear connection between PKD and dysfunction of ciliary proteins, the precise nature of this relationship is not completely understood. There is general agreement that PC1 and PC2 are localized to primary cilia (62;63) but several other cellular locations have been identified for each protein (Fig. 4). For example, PC1 has been localized to cell-cell junctions and both apical and basolateral membranes (67). PC2 is a resident ER protein (29). Many of the other proteins localized to the primary cilium are also found in additional sub-cellular locations— e.g., kinesin-2 serves as an anterograde motor for more than just the primary cilium. In light of these complexities, the determination of the relative contributions from these various sites of action to the polycystic phenotype has remained imprecise. It is thought that cilia in renal epithelia may function as sensory organelles with the polycystins possibly acting as a receptor/channel complex and localized calcium influx acting as a second messenger. The nature of the stimuli to which this complex responds remains a matter of investigation. The cilium and PC1/PC2 have been proposed to respond to flow-dependent mechanosensory stimuli (68). This hypothesis must be tempered by the finding that inactivation of Pkd1 or an essential IFT gene, tg737 (IFT88), in adult mice does not result in cyst formation until months after deletion of either gene (54;55). The prolonged delay between gene inactivation and the onset of cystic change is difficult to understand if the role of the PC1/PC2 receptor-channel complex is to signal in response to a continuous and dynamic process like luminal flow. Another recent study suggests that PC2 utilizes TRPV4, another TRP protein, to form a mechanosensitive calcium channel in the cilium (69). Inactivation of TRPV4 in either zebrafish or mice does not result in renal cystic disease further calling into question the role of the primary cilium as a mechanosensor for flow as a signal to regulate luminal diameter. Homologs of PC1 and PC2 have been shown to form receptor channel complexes acting as sour taste receptors (70), raising the possibility that PC1/PC2 may also respond to ligand mediated events. A combined mechanism of flow-dependent delivery of a ligand remains an intriguing hypothesis.
Figure 4. Subcellular localization of “cystoproteins”.

Numerous proteins associated with cystic kidneys (“cystoproteins”) localize to the primary cilium and basal body complex but are also found in other intracellular compartments. AJ, adherens junction; BB, basal body; Cen, centriole; ER, endoplasmic reticulum; FAP, focal adhesion plaque; TJ, tight junction. [Adapted from (67) and Menezes and Germino, Methods in Cell Biology 94: 273–297, 2009].
CELLULAR PATHWAYS ASSOCIATED WITH ADPKD
There have been a number of approaches used to define the functional connection between disease gene and disease phenotype. As detailed above, a good deal of progress in identifying polycystic disease genes and suggesting the importance of cilia in these disorders has been achieved through genetic approaches. The challenges that remain, center on understanding the in vivo functions of the polycystins at the cellular and molecular level. Several factors complicate this undertaking. First, PKD is a disease of three dimensional organ structure and as such, the applicability of in vitro cell-based models remains uncertain. Without clearly defined stimuli such as ligands that are typically used in functional studies of other receptor complexes, assessing cellular responses specific to polycystins remains a challenge. Second, cilia are not readily tractable as a cellular compartment limiting the ability to discover novel pathways specifically operational in this minute cellular space. This has led to bias in investigational focus on establishing a cilia connection for a number of previously known cellular pathways. Finally, it has been challenging to convincingly define larger multiprotein complexes beyond PC1 and PC2. This difficulty stems from a number of factors including the possibility that the partners differ based on different cellular location of the polycystins and complexes specific to a minor compartment like the cilia will be hard to discover given the quantitative challenge. The fact that the most proximate signal to the PC1/PC2 complex may be calcium rather than direct protein interaction further compounds this problem. Here we present some of the cellular pathways that have been implicated in polycystin-related functions.
Intracellular calcium, secretion and cAMP
The role of PC2 as a calcium channel and of PC1 as a receptor regulating its activity has led to a focus on calcium in the cellular ADPKD phenotype. Direct assessment of any calcium effects in the cilia compartment has been limited, but studies examining total cellular calcium have provided some insights. As a member of the TRP family of ion channel proteins abundantly expressed in the ER, functional studies showed that PC2 over-expression enhances the release of Ca2+ from intracellular stores (71). PC2 associates with a number of Ca2+ channel proteins (TRPC1 and TRPV4) and homo-multimerizes with itself via its C-terminus (36;69;72;73). In keeping with its putative role in regulating cellular Ca2+ homeostasis via intracellular Ca2+ pools, PC2 interacts directly with the inositol 1,4,5-trisphosphate receptor (IP3R) (74) regulates the activity of the ryanodine receptor through direct interaction (75) and has its own activity regulated by association with syntaxin-5 (76). Overexpression of PC1 also appears to modulate the properties of intracellular Ca2+ stores by inhibiting capacitative Ca2+ entry and rate of Ca2+ re-uptake by the endoplasmic reticulum (77). While the interdependence of PC2 and PC1 in these process and the significance these effects on intracellular Ca2+ homeostasis in the pathophysiology of cystic disease remains to be determined, these studies do lend support to the hypothesis that the polycystins impact a physiologically critical second messenger pathway that may influence a broad array of cellular functions.
Several studies report that cAMP levels are elevated in cyst epithelial cells and furthermore that cAMP stimulates cyst fluid and electrolyte secretion (78;79). CFTR has been proposed as the apical chloride channel responsible for driving fluid secretion, providing a possible molecular explanation for cAMP's pro-secretory effects (80;81). A report that small-molecule inhibitors of CFTR reduced cyst growth in a murine model of PKD (82) support a role for CFTR in cyst growth. In vivo genetic studies combining CFTR and PKD mutant mice have not been reported. Whether CFTR is the sole channel responsible for fluid secretion in cysts is uncertain since CFTR does not appear to be expressed in all cysts (83). The reasons for the high cytosolic cAMP concentrations in cyst cells is not well understood. It is possible that the perturbations in cytosolic Ca2+ levels may in part account for dysregulation of cAMP in cyst cells. The polycystin proteins may directly or indirectly alter the activities of G-protein coupled receptors that signal through cAMP. Expression and activity of the V2 vasopressin receptor, for example, is elevated in a number of animal models of PKD (78;84). This fact is being exploited through the development of V2 receptor antagonists as potential therapeutic agents that can potentially slow or prevent cyst fluid accumulation by reducing cytosolic cAMP levels (84;85).
Mitogen activated protein kinase/extracellular regulated kinase
The mitogen activated protein kinase/extracellular regulated kinase (MAPK/ERK) cascade couples extracellular signals received by a variety of surface receptors, through the activation of small G-proteins and the involvement of a variety of adaptors, to the successive phosphorylation of Raf, MEK and MAP kinase/ERK. Activated MAPK/ERK can modulate protein translation and can enter the nucleus to regulate the activities of transcription factors and the cell cycle. Activation of the MAPK/ERK pathway occurs in cell culture based models of ADPKD (86;87) as well as in vivo mouse models of the disease (53). ERK activation observed in cultured ADPKD patient cyst-derived cells has been attributed to cAMP-dependent activation of B-Raf (86;87). Inhibition of the MAPK/ERK cascade has slowed cyst formation in a murine cystic model based on a gene for human nephronophthisis (88) but the MEK1/2 inhibitor U0126 failed to alter the course of ADPKD in a conditional Pkd1 gene inactivation model (53). In light of the observations that the MAPK/ERK cascade is active in a variety of cell and animal models of PKD, the MAPK/ERK cascade kinases may be potential targets for future therapies in ADPKD.
mTOR
The mTOR (mammalian target of rapamycin) protein is a kinase whose activation leads to increased protein translation and cell growth. The mTOR pathway is stimulated by cell surface receptors that signal through PI3 kinase to activate the AKT kinase. Activated AKT phosphorylates the tuberous sclerosis complex (TSC), composed of the TSC1 and TSC2 proteins (hamartin and tuberin). It is interesting to note that the TSC2/tuberin gene lies close to the PKD1 gene on chromosome 16 and occasionally results in a contiguous gene deletion syndrome manifesting severe ADPKD and features of TSC as well. Support for link between the mTOR pathway and ADPKD is provided by studies demonstrating that downstream effectors of the mTOR pathway are inappropriately activated in cyst lining cells (89). Administration of rapamycin in rodent models of PKD has slowed cyst development suggesting the possibility that inappropriate activation of the mTOR pathway is associated with or in part responsible for the excessive proliferation of renal epithelial cells that characterizes PKD (89–91). Recent evidence suggests that PC-1 inhibits the mTOR pathway in a Tsc2-dependent manner and thereby regulates cell size as well (92). Taken together, these data suggest that under normal circumstances the PC1 exerts an inhibitory influence on the strength of mTOR signaling. This pathway represents another potential target for therapy in ADPKD.
Wnt
Oriented cell division (OCD) is controlled by the planar cell polarity (PCP) pathway through non-canonical Wnt signaling (93). Several lines of evidence support the role of oriented cell division in shaping tissues (94) and a role for loss of OCD in cyst formation has been proposed (Fig. 5) (95;96). The elongating nephron of the developing kidney shows significant cellular proliferation yet the tubules grow in a longitudinal direction without an appreciable increase in cross section (95). The mitotic spindles during this tubular elongation phase appear to orient along the long axis of the tubule and cell division takes place in this orientation. Loss of OCD occurs prior to cyst formation in two cystic animal models, Hnf1β deficient mice and the pck rat (orthologous ARPKD model (95) Consistent with these findings, disruption of the PCP-related protocadherin Fat4 results in tubular cysts in kidney development, a process further exacerbated by reduction in the dose of the core PCP gene, Vangl2 (97). Recent data suggests a more complex view of PCP processes in the kidney. During tubular condensation in the earliest stages of kidney development, convergent extension movements, not OCD, seem to drive establishment of tissue polarity (98). A mouse model with a hypomorphic ARPKD gene mutation shows similar loss of OCD but never develops cysts, suggesting that loss of OCD is not sufficient for cyst formation (99). Conversely, Pkd1 and Pkd2 mouse models that develop cystic kidneys have normal OCD in pre-cystic tubules but lose this property as the tubules start to dilate (99). Loss of OCD is a marker for dysregulated PCP activity and some cystic disease genes (e.g., Pkhd1) may impact PCP phenotypes, but loss of OCD may not be a direct cause of cystic expansion. Further studies will likely serve to resolve some of these issues.
Figure 5. Planar cell polarity and tubular morphogenesis.

Planar cell polarity may help to establish normal tubular architecture through directional cell division (top left) and convergent extension/directional cell migration (bottom left). Disruption of these processes could potentially result in cyst formation (right). [Adapted from (96) and Menezes and Germino, Methods in Cell Biology 94: 273–297, 2009].
While PCP uses components of the non-canonical Wnt signaling pathway, the role of the canonical Wnt pathway is more compelling in ADPKD. The canonical Wnt pathway functions through β-catenin and controls cell proliferation and differentiation during development (100). The canonical Wnt pathway actively regulates the availability of β-catenin for nuclear translocation. Evidence for a role of canonical Wnt signaling in PKD comes from studies showing that transgenic expression of constitutively active β-catenin or kidney-specific inactivation of the APC gene results in cyst formation (98;101;102). The direct connection between ADPKD and canonical Wnt signaling remains speculative since conflicting reports state in one case that the C-terminus of PC1 acts as an activator of β-catenin transcription (103) whereas a more recent study implicates the C-terminal tail of PC1 as an inhibitor of β-catenin-TCF mediated transcription (27). Taken together, it appears that Wnt signaling constitutes a plausible downstream effecter mechanism for some aspects of PKD and that the balance between canonical and non-canonical signaling may be a factor (104) but it is unlikely that this mechanism uniformly underlies all manifestations of these diseases.
CONCLUDING REMARKS
The discovery of the genes and their respective proteins that are associated with ADPKD has revolutionized the field of PKD biology. They have provided the means for determining how mutations cause disease, yielded the tools for studying the proteins' functions in cell culture systems, and led to the development of preclinical models useful for understanding the pathophysiology of disease and for testing potential therapies. Important questions remain, however. We must yet explain the molecular details that link ciliary dysfunction to renal cystic disease. We must establish an unambiguous “chain of evidence” that directly links PC1 and PC2 through their various downstream signaling pathways to the cellular processes that regulate tubular morphology. We must determine whether the PC1/PC2 complex really functions as a mechanosensor or whether it is modulated by more classical ligand binding events. If it is a mechanosensor, is it primarily responding to flow at the primary cilium or is it sensing forces between cells or cells and matrix as well? Or is the complex a sensor for something else? Are the instances of divergence in PC1 and PC2 phenotypes, as in left-right axis determination, indicative of promiscuity in functional interacting partners for either protein? We must also work backwards from the cystic phenotype through the many pathogenic steps that result from mutation of PKD genes to identify potential therapeutic targets. What is the nature of the relationship between proliferation and cyst formation and is this a step that can be safely targeted in humans? What accounts for the delayed cystic transformation that results from loss of Pkd1 in adult tissues?
We have made great progress since the first identification of PKD1 15 years ago and the ground work is laid for important discoveries in the future. These studies hold great promise that the future will bring profound mechanistic understanding and effective treatment.
Acknowledgements
This work was supported by grants from NIH/NIDDK including DK51041, DK54053 and DK57328 (S.S.) and the Intramural Research Program of the NIH, NIDDK (G.G.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no financial disclosures or conflicts-of-interest relevant to this manuscript to declare.
Reference List
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.The European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;78:725. [PubMed] [Google Scholar]
- 3.The International Polycystic Kidney Disease Consortium Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell. 1995;81:289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 4.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 5.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 7.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guay-Woodford LM. Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr. Nephrol. 2006;21:1369–1376. doi: 10.1007/s00467-006-0164-9. [DOI] [PubMed] [Google Scholar]
- 10.Hateboer N, Lazarou LP, Williams AJ, Holmans P, Ravine D. Familial phenotype differences in PKD11. Kidney Int. 1999;56:34–40. doi: 10.1046/j.1523-1755.1999.00541.x. [DOI] [PubMed] [Google Scholar]
- 11.Milutinovic J, Rust PF, Fialkow PJ, Agodoa LY, Phillips LA, Rudd TG, Sutherland S. Intrafamilial phenotypic expression of autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1992;19:465–472. doi: 10.1016/s0272-6386(12)80956-5. [DOI] [PubMed] [Google Scholar]
- 12.Persu A, Duyme M, Pirson Y, Lens XM, Messiaen T, Breuning MH, Chauveau D, Levy M, Grunfeld JP, Devuyst O. Comparison between siblings and twins supports a role for modifier genes in ADPKD. Kidney Int. 2004;66:2132–2136. doi: 10.1111/j.1523-1755.2004.66003.x. [DOI] [PubMed] [Google Scholar]
- 13.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossetti S, Burton S, Strmecki L, Pond GR, San Millan JL, Zerres K, Barratt TM, Ozen S, Torres VE, Bergstralh EJ, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J. Am. Soc. Nephrol. 2002;13:1230–1237. doi: 10.1097/01.asn.0000013300.11876.37. [DOI] [PubMed] [Google Scholar]
- 15.Watnick T, Phakdeekitcharoen B, Johnson A, Gandolph M, Wang M, Briefel G, Klinger KW, Kimberling W, Gabow P, Germino GG. Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am. J. Hum. Genet. 1999;65:1561–1571. doi: 10.1086/302657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pei Y, Paterson AD, Wang KR, He N, Hefferton D, Watnick T, Germino GG, Parfrey P, Somlo S, George-Hyslop P. Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am. J. Hum. Genet. 2001;68:355–363. doi: 10.1086/318188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paterson AD, Wang KR, Lupea D, George-Hyslop P, Pei Y. Recurrent fetal loss associated with bilineal inheritance of type 1 autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 2002;40:16–20. doi: 10.1053/ajkd.2002.33908. [DOI] [PubMed] [Google Scholar]
- 18.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van't Hoff WG, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–855. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 20.Nims N, Vassmer D, Maser RL. Transmembrane domain analysis of polycystin-1, the product of the polycystic kidney disease-1 (PKD1) gene: evidence for 11 membrane-spanning domains. Biochemistry. 2003;42:13035–13048. doi: 10.1021/bi035074c. [DOI] [PubMed] [Google Scholar]
- 21.Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, Wilson RK, Chissoe S, Pepin K, Bateman A, Chothia C, et al. Comparative analysis of the polycystic kidney disease 1 (PKD1) gene reveals an integral membrane glycoprotein with multiple evolutionary conserved domains. Hum. Mol. Genet. 1997;6:1483–1489. doi: 10.1093/hmg/6.9.1483. [DOI] [PubMed] [Google Scholar]
- 22.Moy GW, Mendoza LM, Schulz JR, Swanson WJ, Glabe CG, Vacquier VD. The sea urchin sperm receptor for egg jelly is a modular protein with extensive homology to the human polycystic kidney disease protein, PKD1. J. Cell Biol. 1996;133:809–817. doi: 10.1083/jcb.133.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponting CP, Hofmann K, Bork P. A latrophilin/CL-1-like GPS domain in polycystin-1. Curr. Biol. 1999;9:R585–R588. doi: 10.1016/s0960-9822(99)80379-0. [DOI] [PubMed] [Google Scholar]
- 24.Li A, Tian X, Sung SW, Somlo S. Identification of two novel polycystic kidney disease-1-like genes in human and mouse genomes. Genomics. 2003;81:596–608. doi: 10.1016/s0888-7543(03)00048-x. [DOI] [PubMed] [Google Scholar]
- 25.Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, Menezes LF, Xu H, Germino GG, Zuo J, et al. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18688–18693. doi: 10.1073/pnas.0708217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lal M, Song X, Pluznick JL, Di G, V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum. Mol. Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R, Somlo S. Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 1999;274:28557–28565. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- 30.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 31.Celic A, Petri ET, Demeler B, Ehrlich BE, Boggon TJ. Domain mapping of the polycystin-2 C-terminal tail using de novo molecular modeling and biophysical analysis. J. Biol. Chem. 2008;283:28305–28312. doi: 10.1074/jbc.M802743200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casuscelli J, Schmidt S, DeGray B, Petri ET, Celic A, Folta-Stogniew E, Ehrlich BE, Boggon TJ. Analysis of the cytoplasmic interaction between polycystin-1 and polycystin-2. Am. J. Physiol Renal Physiol. 2009;297:F1310–F1315. doi: 10.1152/ajprenal.00412.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Ulbrich MH, Li MH, Buraei Z, Chen XZ, Ong AC, Tong L, Isacoff EY, Yang J. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proc. Natl. Acad. Sci. U. S. A. 2009;106:11558–11563. doi: 10.1073/pnas.0903684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai Y, Anyatonwu G, Okuhara D, Lee KB, Yu Z, Onoe T, Mei CL, Qian Q, Geng L, Wiztgall R, et al. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J. Biol. Chem. 2004;279:19987–19995. doi: 10.1074/jbc.M312031200. [DOI] [PubMed] [Google Scholar]
- 35.Kottgen M, Benzing T, Simmen T, Tauber R, Buchholz B, Feliciangeli S, Huber TB, Schermer B, Kramer-Zucker A, Hopker K, et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 2005;24:705–716. doi: 10.1038/sj.emboj.7600566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 37.Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baert L. Hereditary polycystic kidney disease (adult form): a microdissection study of two cases at an early stage of the disease. Kidney Int. 1978;13:519–525. doi: 10.1038/ki.1978.75. [DOI] [PubMed] [Google Scholar]
- 39.Brasier JL, Henske EP. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J. Clin. Invest. 1997;99:194–199. doi: 10.1172/JCI119147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pei Y, Watnick T, He N, Wang K, Liang Y, Parfrey P, Germino G, George-Hyslop P. Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 41.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 42.Torra R, Badenas C, San Millan JL, Perez-Oller L, Estivill X, Darnell A. A loss-of-function model for cystogenesis in human autosomal dominant polycystic kidney disease type 2. Am. J. Hum. Genet. 1999;65:345–352. doi: 10.1086/302501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watnick TJ, Torres VE, Gandolph MA, Qian F, Onuchic LF, Klinger KW, Landes G, Germino GG. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell. 1998;2:247–251. doi: 10.1016/s1097-2765(00)80135-5. [DOI] [PubMed] [Google Scholar]
- 44.Qian F, Germino GG. "Mistakes happen": somatic mutation and disease. Am. J. Hum. Genet. 1997;61:1000–1005. doi: 10.1086/301618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H, Jr., Kucherlapati R, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 46.Koptides M, Mean R, Demetriou K, Pierides A, Deltas CC. Genetic evidence for a trans-heterozygous model for cystogenesis in autosomal dominant polycystic kidney disease. Hum. Mol. Genet. 2000;9:447–452. doi: 10.1093/hmg/9.3.447. [DOI] [PubMed] [Google Scholar]
- 47.Watnick T, He N, Wang K, Liang Y, Parfrey P, Hefferton D, George-Hyslop P, Germino G, Pei Y. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat. Genet. 2000;25:143–144. doi: 10.1038/75981. [DOI] [PubMed] [Google Scholar]
- 48.Wu G, Tian X, Nishimura S, Markowitz GS, D'Agati V, Park JH, Yao L, Li L, Geng L, Zhao H, et al. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum. Mol. Genet. 2002;11:1845–1854. doi: 10.1093/hmg/11.16.1845. [DOI] [PubMed] [Google Scholar]
- 49.Jiang ST, Chiou YY, Wang E, Lin HK, Lin YT, Chi YC, Wang CK, Tang MJ, Li H. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am. J. Pathol. 2006;168:205–220. doi: 10.2353/ajpath.2006.050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, DeRuiter MC, Breuning MH, de HE, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 2004;13:3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 51.Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, Mochizuki T. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J. Clin. Invest. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 53.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davenport JR, Watts AJ, Roper VC, Croyle MJ, van GT, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat. Med. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM, Fonseca II, Germino GG, Onuchic LF. Pkd1 Haploinsufficiency Increases Renal Damage and Induces Microcyst Formation following Ischemia/Reperfusion. J. Am. Soc. Nephrol. 2009 doi: 10.1681/ASN.2008040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J. Am. Soc. Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature. 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 60.Mochizuki T, Saijoh Y, Tsuchiya K, Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H, Nakatsuji N, et al. Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature. 1998;395:177–181. doi: 10.1038/26006. [DOI] [PubMed] [Google Scholar]
- 61.Lehman JM, Michaud EJ, Schoeb TR, Aydin-Son Y, Miller M, Yoder BK. The Oak Ridge Polycystic Kidney mouse: modeling ciliopathies of mice and men. Dev. Dyn. 2008;237:1960–1971. doi: 10.1002/dvdy.21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 2002;12:R378–R380. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 63.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 64.Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr. Biol. 2002;12:938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 65.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 66.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watnick T, Germino G. From cilia to cyst. Nat. Genet. 2003;34:355–356. doi: 10.1038/ng0803-355. [DOI] [PubMed] [Google Scholar]
- 68.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 69.Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang AL, Chen X, Hoon MA, Chandrashekar J, Guo W, Trankner D, Ryba NJ, Zuker CS. The cells and logic for mammalian sour taste detection. Nature. 2006;442:934–938. doi: 10.1038/nature05084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 72.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang P, Luo Y, Chasan B, Gonzalez-Perrett S, Montalbetti N, Timpanaro GA, Cantero MR, Ramos AJ, Goldmann WH, Zhou J, et al. The multimeric structure of polycystin-2 (TRPP2): structural-functional correlates of homo- and hetero-multimers with TRPC1. Hum. Mol. Genet. 2009;18:1238–1251. doi: 10.1093/hmg/ddp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J. Biol. Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 75.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc. Natl. Acad. Sci. U. S. A. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geng L, Boehmerle W, Maeda Y, Okuhara DY, Tian X, Yu Z, Choe CU, Anyatonwu GI, Ehrlich BE, Somlo S. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hooper KM, Boletta A, Germino GG, Hu Q, Ziegelstein RC, Sutters M. Expression of polycystin-1 enhances endoplasmic reticulum calcium uptake and decreases capacitative calcium entry in ATP-stimulated MDCK cells. Am. J. Physiol Renal Physiol. 2005;289:F521–F530. doi: 10.1152/ajprenal.00355.2004. [DOI] [PubMed] [Google Scholar]
- 78.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Jr., Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 80.Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10206–10211. doi: 10.1073/pnas.93.19.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sullivan LP, Wallace DP, Grantham JJ. Epithelial transport in polycystic kidney disease. Physiol Rev. 1998;78:1165–1191. doi: 10.1152/physrev.1998.78.4.1165. [DOI] [PubMed] [Google Scholar]
- 82.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lebeau C, Hanaoka K, Moore-Hoon ML, Guggino WB, Beauwens R, Devuyst O. Basolateral chloride transporters in autosomal dominant polycystic kidney disease. Pflugers Arch. 2002;444:722–731. doi: 10.1007/s00424-002-0880-3. [DOI] [PubMed] [Google Scholar]
- 84.Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 85.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004;10:363–364. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 86.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 87.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 88.Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, Awazu M. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J. Am. Soc. Nephrol. 2006;17:1604–1614. doi: 10.1681/ASN.2004090800. [DOI] [PubMed] [Google Scholar]
- 89.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 2005;16:46–51. doi: 10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- 91.Wahl PR, Serra AL, Le HM, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD) Nephrol. Dial. Transplant. 2006;21:598–604. doi: 10.1093/ndt/gfi181. [DOI] [PubMed] [Google Scholar]
- 92.Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gong Y, Mo C, Fraser SE. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature. 2004;430:689–693. doi: 10.1038/nature02796. [DOI] [PubMed] [Google Scholar]
- 94.Ahringer J. Control of cell polarity and mitotic spindle positioning in animal cells. Curr. Opin. Cell Biol. 2003;15:73–81. doi: 10.1016/s0955-0674(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 95.Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 96.Germino GG. Linking cilia to Wnts. Nat. Genet. 2005;37:455–457. doi: 10.1038/ng0505-455. [DOI] [PubMed] [Google Scholar]
- 97.Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- 98.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moon RT. Wnt/beta-catenin pathway. Sci. STKE. 2005;2005:cm1. doi: 10.1126/stke.2712005cm1. [DOI] [PubMed] [Google Scholar]
- 101.Qian CN, Knol J, Igarashi P, Lin F, Zylstra U, Teh BT, Williams BO. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J. Biol. Chem. 2005;280:3938–3945. doi: 10.1074/jbc.M410697200. [DOI] [PubMed] [Google Scholar]
- 102.Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, Perret C. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 103.Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 104.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]