

Abstract
We describe studies carried out in the DNA context to test how a common fluorescence quencher, dabcyl, interacts with oligodeoxynu-cleoside fluorophores (ODFs)—a system of stacked, electronically interacting fluorophores built on a DNA scaffold. We tested twenty different tetrameric ODF sequences containing varied combinations and orderings of pyrene (Y), benzopyrene (B), perylene (E), dimethylaminostilbene (D), and spacer (S) monomers conjugated to the 3′ end of a DNA oligomer. Hybridization of this probe sequence to a dabcyl-labeled complementary strand resulted in strong quenching of fluorescence in 85% of the twenty ODF sequences. The high efficiency of quenching was also established by their large Stern–Volmer constants (KSV) of between 2.1 × 104 and 4.3 × 105M−1, measured with a free dabcyl quencher. Interestingly, quenching of ODFs displayed strong sequence dependence. This was particularly evident in anagrams of ODF sequences; for example, the sequence BYDS had a KSV that was approximately two orders of magnitude greater than that of BSDY, which has the same dye composition. Other anagrams, for example EDSY and ESYD, also displayed different responses upon quenching by dabcyl. Analysis of spectra showed that apparent excimer and exciplex emission bands were quenched with much greater efficiency compared to monomer emission bands by at least an order of magnitude. This suggests an important role played by delocalized excited states of the π stack of fluorophores in the amplified quenching of fluorescence.
Keywords: DNA, excimer, exciplex, fluorescence quenching, fluorescent probes
Introduction
Fluorescence quenching-based strategies have become a highly useful tool for interrogating biological systems. Quencher–fluorophore pairs are in common use for studying protein interaction and recognition, and have been extremely valuable in DNA sequence reporting.[1] Various fluorogenic oligonucleotide probe formats have been developed, such as molecular beacons,[2] Scorpion primers,[3] Taqman probes[4–6] and QUAL probes.[7,8] These methods rely on the efficient quenching of the fluorophore at close proximity to the quencher either through collisional quenching and/or Förster energy transfer mechanisms. DNA hybridization leads to the removal of the quencher either by physical separation or by enzymatic or chemical reaction and fluorescence is restored, signaling the biomolecular interaction.
Fluorescence signal readouts in such approaches depend not only on the photophysical properties of fluorophores, but also on the efficiency of the interaction with quenchers. The search for better quenchers has brought about the development of multiquencher designs[9] and the use of novel materials, such as carbon nanotubes[10] and gold,[11] as quenchers. Another approach to higher-efficiency quenching involves harnessing the intrinsic properties of multichromophoric systems to produce amplified quenching. An important example of this is found in luminescent conjugated polymers, some of which have demonstrated a remarkable degree of enhanced fluorescence quenching. Small-molecule quenchers, such as methyl viologen, had been shown to efficiently quench the fluorescence of conjugated polymers with exceptionally high Stern–Volmer quenching constants.[12] Hyper-efficient quenching of cationic conjugated polymers by gold nanoparticles has also been observed.[13] Such enhanced quenching phenomena have been used in DNA detection assays[14,15] and in enzyme detection, such as with proteases.[16]
Conjugated polymers can exhibit such high sensitivity to quenching due to their multichromophoric structures.[17] The exceptional amplification of quenching is due to the efficient migration of the exciton in the extended π conjugation along the polymer backbone.[18] Thus when the quencher is brought in close proximity with the conjugated polymer through electrostatic interactions, “superquenching” occurs through a combination of high mobility of the exciton and favorable noncovalent association between the fluorescent polymer and the quencher.
While such polymeric systems can be effective in biomolecular detection assays, it would be useful in many cases if similar enhanced quenching properties were available in small, discrete molecules. This could allow for conjugation to biomolecules (e.g., antibodies and DNA) and other small molecules, and would enable many specific assays and better controlled interactions. Moreover, small molecules can be simpler to characterize, and their properties, such as solubility, cell permeability and photophysical characteristics, can be relatively straightforward to vary and control.
In recent years, the deoxyribose backbone of DNA has been employed as a scaffold for the design of multichromophoric systems. Base substitutions have been made with phenanthrenes and pyrenes,[19,20] clusters of methyl red and naphthyl red dyes[21,22] and bipyridyl and biphenyl moieties.[23] Pyrenes have also been covalently attached to the 5′ position of uridine to form regular helical π arrays along the major groove of the DNA duplex.[24,25] Besides DNA, multilabeling of other nucleic acid architectures, for example, RNA[26] and LNA,[27] have also been carried out. Noncovalent assembly of chromophores on the DNA structure has also been observed with intercalating dyes.[28]
Our laboratory has developed a novel system of discrete fluorophores built on the deoxyribose–phosphate backbone, called oligodeoxyfluorosides (ODFs), in which consecutive aromatic fluorophores, such as pyrene, perylene and benzopyrene, replace the natural DNA bases.[29,30] This DNA-like structure allows aromatic fluorophores to interact electronically in the ground state through π stacking, and results in multiple forms of energy and excitation transfer in the excited state. Excimers and exciplexes are commonly observed and result in large Stokes’ shifts of more than 200 nm.[31] By varying the length, monomers and sequence, highly diverse and tunable properties result; for example, sets of dyes with multiple emission colors have been identified that can be excited at one excitation wavelength.[32] In addition to their useful photophysical properties, ODFs have the advantages of being readily synthesized in any sequence on an automated DNA synthesizer; moreover, the anionic DNA backbone renders them water soluble.
We previously reported the highly efficient quenching properties of oligomeric pyrene excimers (not conjugated to DNA) using methyl viologen as a free quencher.[33] The quenching efficiency, with Stern–Volmer constants up to 4.7 × 106M−1 was comparable to those observed in conjugated polymers. This observation raised a number of questions. For example, are pyrene excimers, which are especially long lived, particularly prone to this quenching, or are other ODF sequences and types of excited states viable as well? Second, is methyl viologen unique as a highly effective quenching partner because of its dicationic, flat aromatic structure, or can other common quenchers exhibit such effects? Third, can ODFs be conjugated to a biomolecule, for example, DNA, and still retain their ability to be quenched by common quenchers?
Here, we investigate these issues by conjugating a variety of ODFs to a 14-mer oligonucleotide strand. The effect of conjugation to DNA on the quenching properties is investigated, as is the effect of varied chromophore sequence and composition. We also report on the use of dabcyl, a common quencher in DNA fluorogenic probes, as an efficient quencher of an assortment of sequences of ODFs. We find that the susceptibility of ODFs to efficient quenching depends upon the formation of delocalized excited states, such as excimers or exciplexes. As a result, quenching efficiency is highly dependent not only on ODF composition, but also on sequence, as demonstrated by a number of ODF sequence anagrams that have very different properties. The results underscore that polyfluorophore systems can behave as cooperative units in the excited state, and that close interactions of neighboring chromophores can have a strong influence on their properties.
Results
A representative set of twenty tetrameric sequences of ODFs was chosen from a larger random library for this study; for hybridization studies, they were appended to the 3′ terminus of a 14-mer probe DNA sequence (Table 1). The ODF sequences were chosen to represent multiple random chromophore compositions, as well as varied sequences of the same compositions. The ODF labels were synthesized from five monomers, in which the base-replacement fluorophore was a benzopyrene (B), pyrene (Y), perylene (E), dimethylaminostilbene (D) or an abasic spacer tetrahydrofur-an moiety (S). α-Glycosidic anomers, which can stack with one another in a DNA context,[31] were used for synthesis convenience in this study. Figure 1 shows the chemical structures of these monomeric fluorosides and Figure 2 shows the structures of two representative ODF labels. Note that sequences of ODFs are listed in 5′ to 3′ order in analogy to DNA.
Table 1.
DNA sequences used in this study.
| Strand ID | Sequencea |
|---|---|
| P1–P20 | 5′-TCC ACA GAA ACA TA–XXXX-3′ |
| D1 | 3′-AGG TGT CTT TGT AT-5′ |
| Q1 | 3′-AGG TGT CTT TGT AT–dabcyl-5′ |
XXXX denotes the sequence of ODF; refer to Table 2 for ODF sequences.
Figure 1.
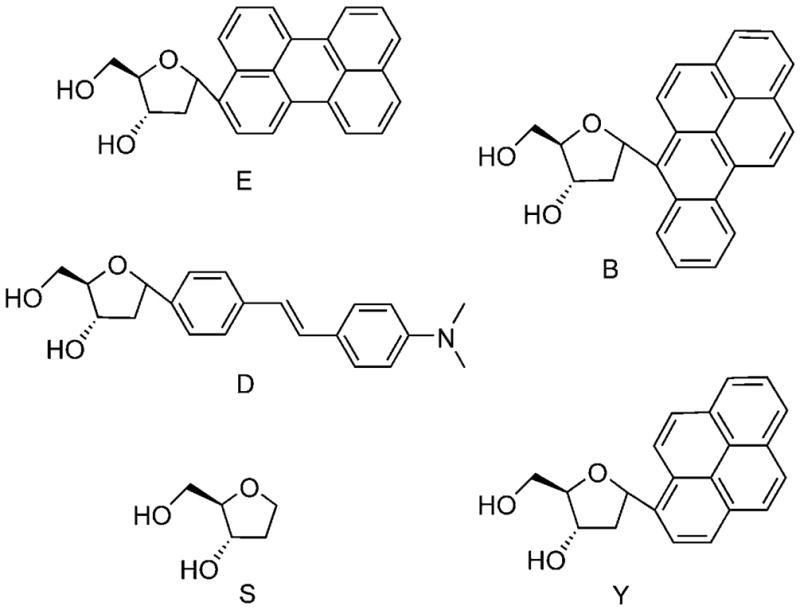
Structures of monomeric deoxyfluorosides perylene (E), benzopyrene (B), dimethylaminostilbene (D), pyrene (Y) and spacer (S).
Figure 2.

Structures of two representative oligodeoxyfluorosides from the twenty in this study. These are attached to the 3′ end of a DNA probe sequence (R).
Fluorescence properties of ODFs
Absorption and emission spectra of the twenty ODF labels conjugated to single-stranded DNA were measured in pH 7.0 hybridization buffer (see the Experimental Section). Lists of the absorption and emission bands are given in Table 2, and emission spectra are shown in the Supporting Information. Quantum yields for fluorescence were measured in the same buffer; these are also given in Table 2. In general, the absorption spectra were simple combinations of the added spectra of the monomer components consistent with ground-state stacking of the chromophores (data not shown), although in many cases there was moderate line broadening and a decrease in peak/valley ratio, as reported previously.[34] However, emission spectra of the ODF labels were in nearly all cases quite different from the combined monomer emissions. All cases except one (P6) showed broad bands between 450–600 nm; this is consistent with excited state dimer (exciplex and excimer) states (see the Supporting Information). The maxima of these bands varied from 460 to 530 nm, and some cases showed multiple maxima (as many as three) in this range. The one exception (P6, sequence BSDY) is a blue–violet fluorophore with no longer-wavelength bands.
Table 2.
Sequences and photophysical data for twenty composition- and sequence-varied ODFs appended to the 3′ end of the DNA probe sequence. a
| Strand ID | ODF sequence (5′ → 3′) | λmaxabs [nm] | λmaxem [nm] | Φem | Qeff [%] | KSV [M−1] b exciplex | KSV [M−1] b monomer | |||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | D | E | B | Y | 257, 347, 399, 448 | 375, 395, 464, 489 | 0.229 | 81 | 1.3×105 | neg. c |
| P2 | E | D | B | Y | 258, 349, 403, 452 | 461, 490 | 0.188 | 87 | 2.9×105 | 3.5×104 |
| P3 | D | Y | B | E | 256, 349, 403, 442 | 462, 492 | 0.130 | 77 | 2.5×105 | neg. c |
| P4 | B | E | B | Y | 258, 347, 401, 450 | 492 | 0.194 | 85 | 5.7×104 | neg. c |
| P5 | E | Y | B | E | 257, 347, 401, 451 | 465, 493 | 0.092 | 84 | 1.4×105 | neg. c |
| P6 | B | S | D | Y | 264, 326, 342, 401 | 375, 395 | 0.428 | 7 | n.a. d | 8.3×102 |
| P7 | B | Y | D | S | 258, 346, 349, 402 | 375, 416, 467 | 0.045 | 73 | 4.9×104 | neg. c |
| P8 | E | D | S | Y | 264, 342, 451 | 375, 395, 459 | 0.443 | 1 | 4.3×105 | 4.3×104 |
| P9 | E | S | Y | D | 257, 346, 349, 451 | 460, 488 | 0.191 | 84 | 3.5×105 | neg. c |
| P10 | D | S | E | Y | 257, 343, 349, 451 | 375, 463, 490 | 0.303 | 74 | 1.9×105 | 2.5×104 |
| P11 | B | Y | S | S | 261, 349, 403 | 375, 395, 416, 470 | 0.205 | 85 | 2.5×104 | 8.0×103 |
| P12 | E | S | B | S | 258, 402, 451 | 470, 500 | 0.219 | 88 | 1.5×105 | 2.0×104 |
| P13 | E | D | S | S | 257, 355, 450 | 461, 490 | 0.161 | 86 | 1.5×105 | neg. c |
| P14 | E | E | D | E | 254, 355, 449 | 374, 454, 478 | 0.062 | 51 | 8.4×104 | 1.5×103 |
| P15 | E | Y | Y | E | 256, 347, 451 | 464, 490 | 0.130 | 88 | 2.1×104 | neg. c |
| P16 | E | E | Y | Y | 257, 347, 451 | 490 | 0.141 | 77 | 6.2×104 | neg. c |
| P17 | E | S | E | Y | 256, 347, 451 | 375, 395, 494, 530 | 0.148 | 75 | 2.3×105 | neg. c |
| P18 | E | Y | S | S | 257, 346, 451 | 461, 490 | 0.421 | 92 | 7.4×104 | 9.1×103 |
| P19 | Y | Y | E | E | 254, 349, 441 | 462, 489 | 0.110 | 77 | 1.1×105 | 4.4×104 |
| P20 | Y | S | S | Y | 264, 332, 350 | 378, 395, 490 | 0.025 | 86 | n.a. d | 6.9×103 |
See the Experimental Section for conditions.
KSV values measured with free dabcylamide quencher (1) in water.
neg.: cases in which KSV was zero or slightly negative; see the Supporting Information for plots.
n.a.: cases in which no long-wavelength (e.g., excimer or exciplex) band was present.
In general, the twenty DNA-conjugated ODFs were efficient fluorophores, with quantum yields for emission as high as 44% (Table 2). The quantum yields varied with chromophore composition and sequence; for example, P7 (BYDS) gave a lower quantum yield of 4.5%, while the sequence P8 (EDSY), which is different by only one monomer, has a quantum yield of 44.3%. An even more striking comparison was observed between P7 (BYDS, Φfl = 4.5%) and its sequence-rearranged anagram, P6 (BSDY, Φfl = 42.8%), in which quantum efficiencies varied strongly without a change in composition.
In addition to quantum efficiency, wavelengths of emission varied substantially with sequence as well (Table 2), an effect documented in an earlier set of pyrene–perylene ODFs[34] as well as more recently in a library of unconjugated tetramer ODFs.[32] Examples in the present study include P1 (labeled with DEBY), which is a whitish multiemissive fluorophore, as compared with its anagram P3 (DYBE), a green emitter. A second comparison is sequence P8 (labeled with EDSY, a blue–violet dye) versus sequence P9 (ESYD, a green–yellow fluorophore).
Dabcyl quenching of ODF strands upon hybridization
Dabcyl (4-((4′-dimethylamino)phenylazo)benzoic acid) is a common quencher widely used in a variety of biomolecular applications[35] and nucleic acid probes.[36–38] Thus we chose to explore its quenching effects on ODFs upon hybridization, to explore the utility of varied ODF–dabcyl fluorophore quencher pairs as reporters of DNA hybridization. Dabcyl was attached to the 5′ end of the complementary DNA strand (Q1; Table 1); thus upon hybridization to each of the 3′-ODF-labeled strands (P1–P20), dabcyl was brought into proximity with the various labels. The fluorescence spectrum of each of the ODFs was measured before and after the addition of Q1. The percentage quenching of fluorescence was determined by the relative fluorescence intensities at the maximum emission wavelength before and after the addition of Q1. We used a one molar excess equivalent to ensure that all ODF-labeled DNAs were hybridized with a quencher.
The experiments revealed that efficient quenching by dabcyl occurred in 17 of the 20 ODFs tested (Table 2). These sequences had more than 70% of their fluorescence quenched upon the addition of the dabcyl-conjugated complement Q1. Two instances of this effect are shown in Figure 3 with labeled probes P4 (BEBY) and P15 (EYYE), which yielded 85 and 88% quenching, respectively.
Figure 3.
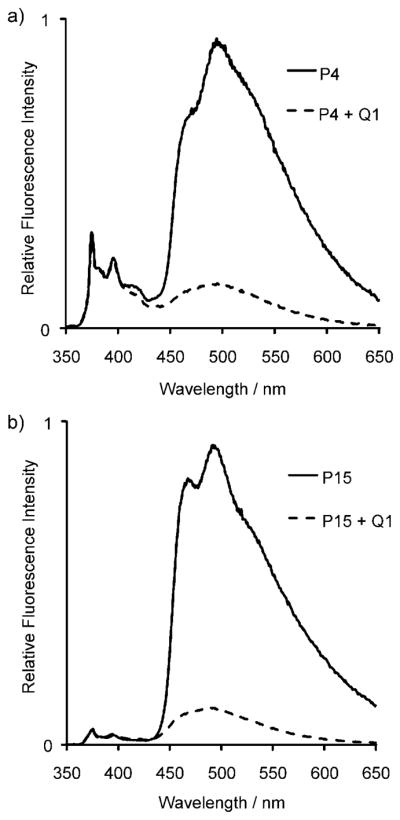
Fluorescence emission spectral changes upon hybridization of ODF-labeled probes: a) P4, and b) P15 with dabcyl-conjugated complement, Q1 (λex = 330 nm).
Interestingly, inspection of emission spectra before and after hybridization with Q1 (see the Supporting Information and Figure 3) revealed that quenching was nearly always much more efficient with longer wavelength apparent exciplex emission bands compared to the monomer emission band in cases when both were present. The majority of labeled sequences showed this effect; some examples include dramatic monomer versus excimer differences, such as in sequences P4 (BEBY; Figure 3a), P1 (DEBY), P7 (BYDS), and P10 (DSEY), all of which showed strong quenching of long-wavelength bands between 450–600 nm, but essentially no quenching of shorter-wavelength bands. Several other cases showed similarly strong quenching of long-wavelength bands (see, for example, Figure 3b) but had very little in the way of short-wavelength emission for comparison, apparently due to highly efficient excimer–exciplex formation with nearly complete lack of monomer emission.
Three ODF labels showed poor quenching by dabcyl on the complementary strand. Both sequence P6 (BSDY) and P8 (EDSY) exhibited predominantly the pyrene monomer emission, which showed almost no quenching. Probe P14 (EEDE) showed 51% quenching at the emission wavelengths between 450 and 500 nm.
Hybridization of ODF-labeled strands with DNA alone
To evaluate whether quenching of the fluorescence of ODFs was due to dabcyl and not just the complementary DNA strand, all twenty ODF-labeled sequences were hybridized to the natural complementary oligomer, D1 (Table 1), which lacked the quencher group. The effects of hybridization to D1 varied from enhancement of fluorescence to a small degree of quenching, and several of the ODFs displayed no change at all (see spectra in the Supporting Information). The two largest changes in ODF emission were with sequences P16 (EEYY) and P3 (DYBE). The fluorescence of EEYY was quenched by 33% (see the Supporting Information), while the fluorescence of DYBE was enhanced by 47%. Most others exhibited less than 20% change in emission. Taken together, the effects of the complementary DNA strand alone were generally small; thus in the majority of cases, the quenching effects observed in the dabcyl hybridization experiments came entirely (or nearly so) from the presence of the dabcyl group on the opposite DNA strand.
Stern–Volmer relation
To quantify the intrinsic sensitivity of DNA-conjugated ODFs to dabcyl, a small water soluble dabcylamide compound (1) was synthesized from the free
acid of dabcyl through its acid chloride derivative. Synthesis details can be found in the Supporting Information. Quenching efficiency was quantified by using the Stern–Volmer relation:
where I0 and I are the steady state fluorescence intensities in the absence and presence of quencher 1. The Stern–Volmer constant (KSV) was derived from the slope of linear fits to the data from each complex constrained to intersect at y=1. The Stern–Volmer constants of all ODFs attached to DNA are given in Table 2.
The data show that the Stern–Volmer constants for the ODFs are large. Interestingly, in all cases in which it was present, the apparent exciplex emission bands were much more efficiently quenched than the monomer emission. More specifically, the long-wavelength, broad bands (~430–600 nm) were strongly quenched by increasing amounts of dabcylamide, while the monomer emission bands typical of B, Y, D monomers (330–440 nm) were only slightly affected or were unaffected. Table 2 shows KSV values listed for both sets of bands. The KSV values of the entire set of long-wave-length bands ranged from 2.1 × 104M−1 in P15 (EYYE) to 4.3 × 105M−1 in P8 (EDSY). Monomeric emission bands, in contrast to this, gave smaller KSV values of 8.3 × 102 to 4.4 × 104M−1, typically one order of magnitude less efficient than that of the excimer–exciplex band in a given sequence. Half (10 of 20) of the ODF sequences tested did not show any quenching of the emission of the monomer band. Some sequences even showed an enhancement of emission; for example, sequence P5 (EYBE) gave a 55% increase in monomer emission with the addition of 5 μM dabcylamide, while at the same time the exciplex emission was decreased by 39%. The formation of the excimer or exciplex band was dependent on buffer conditions, as observed in the case of P20 (YSSY), in which the excimer band was missing under the conditions used for determining KSV values.
Notably, quenching of ODFs was highly sequence dependent even when monomer composition was the same. This could be readily observed in anagrams of ODF sequences. For example, the quenching was two orders of magnitude higher in P7 (BYDS), with KSV of 4.9 × 104M−1, than with P6 (BSDY), for which the KSV was 8.3 × 102M−1. As with the above experiments with dabcyl-conjugated complementary DNA, this correlated with the presence or absence of long wavelength exciplex bands, as the exciplex band (463 nm) was large in P7 (BYDS), but no exciplex band was present in P6 (BSDY). In a second comparison, P19 (YYEE) and P15 (EYYE) showed considerably better excimer–exciplex quenching in the former as compared with the latter. Interestingly, the monomer bands also showed large differences, with P19 yielding strong quenching (KSV = 4.4 × 104M−1) for the pyrene–perylene monomer emissions, while P15 showed no quenching of these bands. A similar effect was seen in anagrams P9 and P10 (Table 2 and the Supporting Information), as well as in P2 as compared with its anagrams P1 and P3.
Examination of data for anagrams P8, P9 and P10 showed cases of strong sequence dependence as well. With D and S between E and Y, there was less excited state delocalization as seen by the much smaller exciplex band in P8 (EDSY) compared to P9 (ESYD) and P10 (DSEY). The dominant emission band in P8 (EDSY) was the monomer emission band at 375 and 395 nm while the dominant band in P9 (ESYD) and P10 (DSEY) was the exciplex band at 460 and 490 nm. The exciplex emission band was better quenched by an order of magnitude greater than the quenching of the monomeric emission band. Fluorescence emission spectra of P9 and P10 in the quenching experiments are shown in Figure 4, together with their respective Stern–Volmer plots.
Figure 4.

Sequence dependence of fluorescence and of quenching in ODF fluorophores that have the same dye composition but with different ordering. Shown are the fluorescence emission spectra of: a) P8 (sequence EDSY), b) P9 (ESYD), and c) P10 (DSEY) with varied amounts of quencher 1 added. Their respective Stern–Volmer plots for short- and long-wavelength emission bands are shown below the respective emission spectrum; note the large response of long-wavelength bands as compared with short-wavelength bands. Additional examples are available in the Supporting Information file.
The sequence dependence of quenching as observed in anagrams of ODFs could be observed in hybridization experiments carried out in cuvettes. Changes were apparent by visual inspection as seen in Figure 5. P8 and P9 are sequence anagrams containing the same constituents as discussed earlier. Quenching of P8 (EDSY) by Q1 produced a color change from blue to violet, consistent with loss of the long wavelength emission upon quenching. P9 (ESYD), which possessed a more yellowish tone due to a dominant emission at 450–550 nm, yielded a different color change upon quenching.
Figure 5.

Varied fluorescence and quenching in anagrams of ODF-labeled oligonucleotide strands in buffer, observed by visual inspection. Images of buffered solutions of: a) P8 (EDSY), and b) P9 (ESYD), before (left) and after (right) hybridization with Q1. Cuvettes were placed on a UV transilluminator (354 nm).
Discussion
In our study, a common quencher, dabcyl, was tested for its effectiveness in quenching the fluorescence of various multi-chromophoric ODF sequences. Large Stern–Volmer constants were found with a free quencher, demonstrating strong inherent ability of the ODFs to be quenched. For a DNA-conjugated dabcyl moiety, the results have shown that quenching is effective in 85% of the twenty ODFs tested. In all cases, the long wavelength emission band, which was apparently due to exciplex or excimer emission, was quenched more efficiently than the monomeric emission band.
Contact quenching is likely to play a central role in this quenching effect since the quencher, dabcyl, was in close proximity to ODFs upon hybridization and the lifetimes of many ODFs are long (in the 6–120 ns range).[34] In addition, the absorption spectrum of dabcyl (λmaxabs = 468 nm) overlaps with the emission spectra of many of the ODFs; thus energy transfer might also play a role in the quenching effect. The dabcylamide quencher (1) used in our measurements of KSV has only one cationic charge compared to the dicationic methyl viologen used in an earlier study of the quenching of nonconjugated ODFs.[33] Thus static quenching by the formation of an electrostatically favorable complex between ODF and the quencher is likely to be less efficient when using dabcyl. However, the Stern–Volmer constants were still comparable in both quenchers, with KSV values between 104 and 105M−1 for tetramer-length ODFs. This establishes that multiple excimer and exciplex species (beyond pyrene alone[33]) can be highly efficiently quenched. Since the quenching with DNA-conjugated dabcyl was not as efficient as with the free quencher, we surmise that constraints on dye location and motion due to the tether could have had a negative effect; future tests with different tether lengths and structures can shed light on this.
We hypothesize that the efficient inherent quenchability of ODFs is due to the presence of delocalized excited states, which increases the likelihood of productive contact with the quencher. In addition, we proposed recently[33] that if excimer–exciplex states exist in longer oligomers, the exciton might migrate by dynamic alignment of the adjacent chromophores. Thus the effects of delocalization and mobility of the excited state might be similar in ODFs and conjugated polymers. Association or collision with a quencher anywhere in the delocalized domain rapidly brings about quenching, as the exciton migrates rapidly to the site of the quencher. In the ODF system, this mobile exciton mechanism explains our observation of enhanced quenching of exciplex bands over monomer emission. ODF molecules that have no exciplex emission, and thus do not have this delocalization mechanism available, are poorly quenched. For example, the sequence BSDY shows only emissions similar to those of the pyrene and benzopyrene component monomers, and its KSV value with dabcyl was 8.3 × 102M−1. Other sequences with monomer bands had KSV values between 1.5 × 103 and 4.4 × 104M−1, similar to values for classical monomeric fluorophores, such as fluorescein, which we found to have a KSV value of 3.1 × 103M−1 (see the Supporting Information). When conjugated to a 14-mer oligonucleotide at the 5′ end, fluorescein had a higher KSV value of 1.6 × 104M−1; this suggests that the polyanionic DNA might increase affinity of the quencher for the fluorophore-tagged molecule as a whole. A similar effect is seen in quenching of conjugated anionic polymers by methyl viologen,[12,39] which binds to the polymers and thus favors contact quenching. However, there is an additional favorable effect of the delocalized state in the current ODFs; for example, the sequence EDBY shows clear long wavelength exciplex-like bands (461, 490 nm) and exhibits KSV = 2.9 × 105M−1.
Our hypothesis suggests that ODF sequences that have more efficient delocalization, which might be judged by higher excimer–monomer emission ratios, should be better quenched in general than those with poor delocalization. In addition, ODF structures of increasingly greater length should further enhance quenching efficiency as well. In the present cases we limited length to four, but recent studies with multipyrene ODFs with lengths of up to eight did support this prediction.[33] In the future, it will be of great interest to study the detailed mechanism of exciton mobility in this and other π stacked excimer–exciplex systems.[28,40] Time-resolved fluorescence and absorption studies would no doubt lend useful insights into this.
Recently, the quenching properties of a DNA–multichromophore system upon hybridization was also studied by Häner and co-workers, who effectively quenched pyrene excimer fluorescence by a non-nucleosidic perylene diimide moiety upon DNA duplex formation.[41] Both the pyrene and perylene diimide non-nucleosidic base surrogates were incorporated in the middle of respective DNA strands, and DNA hybridization brought them into contact; this resulted in efficient quenching of the pyrene excimer emission. Although that work did not test other emissive dyes, it does confirm that pyrene excimers can be effectively quenched by hybridization in a geometry different from the current one.
The finding of efficient quenching of different excimers and exciplexes beyond pyrene excimer alone is likely to have useful applications. For example, it might be possible to generate multiple differently colored quencher–dye pairs for probing applications, with the added advantage of being excited with the same single excitation wavelength. This multicolor property in ODFs allows for direct visualization of multiple species, even in moving systems, without the use of multiple filter sets and camera exposures.[32] The ease of synthesis of combinatorial libraries of these ODFs suggests some simple future strategies for screening of various common quenchers–dye pairs that might be more effective than the current ones.
Additional studies would be useful to shed light on the quenching mechanisms of dabcyl, methyl viologen and other quenchers in polychromophore systems. Moreover, further studies into the mechanisms of excited state mobility would be valuable, both for lending basic insight, and also for design of better fluorescent labels, probes and reporters in the future.
Experimental Section
Fluoroside phosphoramidites (E, Y, D, S, B)
Syntheses of the monomer 1′-α-2′-deoxyriboside 5′-dimethoxytrityl-3′-phosphoramidite derivatives of pyrene (Y), perylene (E), benzopyrene (B) and dimethylaminostilbene (D) were carried out as previously described.[29,30,42] The abasic tetrahy-drofuran spacer S was obtained commercially (dSpacer CE phosphoramidite from Glen Research Corporation).
Synthesis of ODF-labeled oligodeoxynucleotides
Oligodeoxynucleotides were synthesized by using an Applied Biosystems 394 DNA/RNA synthesizer on a 1 μmole scale and possessed a 3′-phosphate group. Coupling employed standard β-cyanoethyl phosphoramidite chemistry, but with extended coupling time (600 s) for fluoroside phosphoramidites. ODFs were synthesized directly on the CPG beads and the DNA sequence was then built on its 5′ end. All oligomers were deprotected by using the ultramild deprotection method with potassium carbonate solution (0.05M; 55°C, 4 h), then purified by reverse-phase HPLC. The recovered material was quantified by absorbance at 260 nm with molar extinction coefficients determined by the nearest-neighbor method. Molar extinction coefficients for ODFs were estimated by adding the measured value of the molar extinction coefficient of the fluorosides (at 260 nm) to the calculated value for the natural DNA fragments. ODF-labeled oligomers were characterized by MALDI-TOF mass spectrometry (data are given in the Supporting Information). Dabcyl was purchased as the 5′-dabcyl phosphoramidite derivative from Glen Research Corporation and attached to the 5′ end of the complementary DNA sequence by using the manufacturer’s methods. Purification was achieved by using reverse-phase HPLC.
Optical methods
Absorption measurements were carried out by using a Varian Cary 100 UV-Visible Spectrophotometer. Steady-state fluorescence measurements were carried out by using a HORIBA Jobin Yvon Fluorolog 3 fluorescence spectrometer. Hybridization experiments were performed with 2 μM of each ODF-labeled oligonucleotide strand with 2.0 molar equivalents of its complementary strand (unless otherwise noted) to ensure complete hybridization. Samples were buffered at pH 7.0 with NaCl (100 mM), MgCl2 (10 mM), and Na·PIPES (10 mM); buffers were not deoxygenated. The excitation wavelength used for ODF-labeled strands was 330 nm. To prevent aggregation and reabsorption of light during quantum yield measurements, samples were diluted to solutions with absorption of less than 0.1 at wavelengths longer than λex. 9,10-Diphenylanthracene (Φ=0.90)[43] in cyclohexane was used as the reference for quantum yield calculations. Quantum yields were calculated by using the following equation:[44]
where Φ and ΦR are the quantum yields of the unknown and reference compounds, respectively; n and nR are the refractive indices of water and cyclohexane, respectively; A and AR denote the absorbances at 330 nm for the unknown and reference samples, and ∫F and ∫FR are the integrals of fluorescence emission intensities of the unknown and the reference samples, respectively.
Supplementary Material
Acknowledgments
We thank the US National Institutes of Health (GM067201) for support. Y.N.T. acknowledges an A*STAR NSS scholarship. J.N.W. acknowledges an NIH postdoctoral fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.200901607.
References
- 1.Marras SA. Mol Biotechnol. 2008;38:247–255. doi: 10.1007/s12033-007-9012-9. [DOI] [PubMed] [Google Scholar]
- 2.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 3.Whitcombe D, Theaker J, Guy SP, Brown T, Little S. Nat Bio-technol. 1999;17:804–807. doi: 10.1038/11751. [DOI] [PubMed] [Google Scholar]
- 4.Nurmi J, Wikman T, Karp M, Lovgren T. Anal Chem. 2002;74:3525–3532. doi: 10.1021/ac020093y. [DOI] [PubMed] [Google Scholar]
- 5.Holland PM, Abramson RD, Watson R, Gelfand DH. Proc Natl Acad Sci USA. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solinas A, Thelwell N, Brown T. Chem Commun. 2002:2272–2273. doi: 10.1039/b207254h. [DOI] [PubMed] [Google Scholar]
- 7.Sando S, Kool ET. J Am Chem Soc. 2002;124:2096–2097. doi: 10.1021/ja017328s. [DOI] [PubMed] [Google Scholar]
- 8.Sando S, Abe H, Kool ET. J Am Chem Soc. 2004;126:1081–1087. doi: 10.1021/ja038665z. [DOI] [PubMed] [Google Scholar]
- 9.Yang CJ, Lin H, Tan W. J Am Chem Soc. 2005;127:12772–12773. doi: 10.1021/ja053482t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang R, Jin J, Chen Y, Shao N, Kang H, Xiao Z, Tang Z, Wu Y, Zhu Z, Tan W. J Am Chem Soc. 2008;130:8351–8358. doi: 10.1021/ja800604z. [DOI] [PubMed] [Google Scholar]
- 11.Dubertret B, Calame M, Libchaber AJ. Nat Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, McBranch DW, Wang HL, Helgeson R, Wudl F, Whitten DG. Proc Natl Acad Sci USA. 1999;96:12287–12292. doi: 10.1073/pnas.96.22.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan C, Wang S, Hong JW, Bazan GC, Plaxco KW, Heeger AJ. Proc Natl Acad Sci USA. 2003;100:6297–6301. doi: 10.1073/pnas.1132025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kushon SA, Ley KD, Bradford K, Jones RM, McBranch D, Whitten D. Langmuir. 2002;18:7245–7249. [Google Scholar]
- 15.Gaylord BS, Wang S, Heeger AJ, Bazan GC. J Am Chem Soc. 2001;123:6417–6418. doi: 10.1021/ja010373f. [DOI] [PubMed] [Google Scholar]
- 16.Kumaraswamy S, Bergstedt T, Shi X, Rininsland F, Kushon S, Xia W, Ley K, Achyuthan K, McBranch D, Whitten D. Proc Natl Acad Sci USA. 2004;101:7511–7515. doi: 10.1073/pnas.0402367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Q, Swager TM. J Am Chem Soc. 1995;117:12593–12602. [Google Scholar]
- 18.Swager TM. Acc Chem Res. 1998;31:201–207. [Google Scholar]
- 19.Damian Ackermann RH. Helv Chim Acta. 2004;87:2790–2804. [Google Scholar]
- 20.Malinovskii VL, Samain F, Häner R. Angew Chem. 2007;119:4548–4551. doi: 10.1002/anie.200700891. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:4464–4467. doi: 10.1002/anie.200700891. [DOI] [PubMed] [Google Scholar]
- 21.Kashida H, Tanaka M, Baba S, Sakamoto T, Kawai G, Asanuma H, Komiyama M. Chem Eur J. 2006;12:777–784. doi: 10.1002/chem.200500111. [DOI] [PubMed] [Google Scholar]
- 22.Kashida H, Asanuma H, Komiyama M. Angew Chem. 2004;116:6684–6687. doi: 10.1002/anie.200460870. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:6522–6525. doi: 10.1002/anie.200460870. [DOI] [PubMed] [Google Scholar]
- 23.Brotschi C, Mathis G, Leumann CJ. Chem Eur J. 2005;11:1911–1923. doi: 10.1002/chem.200400858. [DOI] [PubMed] [Google Scholar]
- 24.Mayer-Enthart E, Wagner C, Barbaric J, Wagenknecht HA. Tetrahedron. 2007;63:3434–3439. [Google Scholar]
- 25.Mayer-Enthart E, Wagenknecht HA. Angew Chem. 2006;118:3451–3453. doi: 10.1002/anie.200504210. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:3372–3375. doi: 10.1002/anie.200504210. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Murakami Y, Sasa K, Hayashi H, Yamana K. J Am Chem Soc. 2008;130:6904–6905. doi: 10.1021/ja801054t. [DOI] [PubMed] [Google Scholar]
- 27.Hrdlicka PJ, Babu BR, Sorensen MD, Harrit N, Wengel J. J Am Chem Soc. 2005;127:13293–13299. doi: 10.1021/ja052887a. [DOI] [PubMed] [Google Scholar]
- 28.Benvin AL, Creeger Y, Fisher GW, Ballou B, Waggoner AS, Armitage BA. J Am Chem Soc. 2007;129:2025–2034. doi: 10.1021/ja066354t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren RXF, Chaudhuri NC, Paris PL, Rumney S, Kool ET. J Am Chem Soc. 1996;118:7671–7678. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J, Strassler C, Tahmassebi D, Kool ET. J Am Chem Soc. 2002;124:11590–11591. doi: 10.1021/ja027197a. [DOI] [PubMed] [Google Scholar]
- 31.Cuppoletti A, Cho Y, Park JS, Strassler C, Kool ET. Bioconjugate Chem. 2005;16:528–534. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]
- 32.Teo YN, Wilson JN, Kool ET. J Am Chem Soc. 2009;131:3923–3933. doi: 10.1021/ja805502k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson JN, Teo YN, Kool ET. J Am Chem Soc. 2007;129:15426–15427. doi: 10.1021/ja075968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson JN, Gao J, Kool ET. Tetrahedron. 2007;63:3427–3433. doi: 10.1016/j.tet.2006.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adamczyk M, Moore JA, Shreder K. Org Lett. 2001;3:1797–1800. doi: 10.1021/ol015843p. [DOI] [PubMed] [Google Scholar]
- 36.Tyagi S, Marras SA, Kramer FR. Nat Biotechnol. 2000;18:1191–1196. doi: 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- 37.Tyagi S, Bratu DP, Kramer FR. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 38.Li JJ, Fang X, Tan W. Biochem Biophys Res Commun. 2002;292:31–40. doi: 10.1006/bbrc.2002.6581. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Wang D, Miller EK, Moses D, Bazan GC, Heeger AJ. Macromolecules. 2000;33:5153–5158. [Google Scholar]
- 40.Trkulja I, Häner R. Bioconjugate Chem. 2007;18:289–292. doi: 10.1021/bc0603283. [DOI] [PubMed] [Google Scholar]
- 41.Bouquin N, Malinovskii VL, Häner R. Chem Commun. 2008:1974–1976. doi: 10.1039/b802193g. [DOI] [PubMed] [Google Scholar]
- 42.Gao J, Watanabe S, Kool ET. J Am Chem Soc. 2004;126:12748–12749. doi: 10.1021/ja046910o. [DOI] [PubMed] [Google Scholar]
- 43.Hamai S, Hirayama F. J Phys Chem. 1983;87:83–89. [Google Scholar]
- 44.Williams ATR, Winfield SA, Miller JN. Analyst. 1983;108:1067–1071. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.