

Abstract
Apelin, a ligand of the G protein-coupled putative angiotensin II-like receptor (APJ-R), exerts strong vasodilating, cardiac inotropic and chronotropic actions. Its expression is highly up-regulated during heart failure. Apelin also increases cardiac conduction speed and excitability. While our knowledge of apelin cardiovascular actions is growing, our understanding of the physiological mechanisms behind the cardiac effects remains limited. We tested the effects of apelin on the cardiac sodium current (INa) using patch clamp technique on cardiac myocytes acutely dissociated from dog ventricle. We found that apelin-13 and apelin-17 increased peak INa by 39% and 61% and shifted its mid-activation potential by −6.8 ± 0.6 mV and −17 ± 1 mV respectively thus increasing channel opening at negative voltage. Apelin also slowed INa recovery from inactivation. The effects of apelin on INa amplitude were linked to activation of protein kinase C. Apelin also increased INa “window” current by up to 600% suggesting that changes in intracellular sodium may contribute to the apelin inotropic effects. Our results reveal for the first time the effects of apelin on INa. These effects are likely to modulate cardiac conduction and excitability and may have beneficial antiarrhythmic action in sodium chanelopathies such as Brugada Syndrome where INa amplitude is reduced.
Keywords: Arrhythmia, Sodium channels, Ventricle, Dog, Myocyte, Contraction, INa
1. Introduction
Apelin, an endogenous peptide, modulates vascular tonus. The active peptides; apelin-36 (A-36), apelin-17 (A-17), and apelin-13 (A-13) results from cleavage of preproapelin. Apelin is degraded by the angiotensin conversion enzyme 2 (ACE2) [1].
It binds to the angiotensin-like putative receptor (APJ-R) [2] with affinities of 0.37 nM, 2.5 nM and 20 nM for A-13, A-17 and A-36 respectively [3]. APJ-R and cardiac sodium channels [4,5] are located at the Z-lines and intercalated discs in myocytes [6] which are key areas for conduction and contractility. Because the inhibitory effect of apelin on cAMP production is abolished by pertussis toxin, APJ-R is thought to be coupled to a Gi protein [2]. Apelin [7–9] may also modulate protein kinase A and C activity.
APJ-R is found in the cardiovascular system of many species including human [10–15]. Apelin has strong inotropic effects in isolated rat heart [9]. It is one of the strongest endogenous positive inotropic agent identified so far, yielding above 70% of the increase force observed with isoproterenol. The effects at the cardiac level are both direct (i.e. increased contractile force) and indirect (i.e. vascular) [9,16–18]. Its effects are similar to phosphodiesterase III inhibitors which exhibit cAMP- but Ca2+-ATPase-dependent inotropic and vasodilating effects.
Apelin has no effect on potassium and calcium currents (Ito, IK,sus, and ICa) and its ability to increase contraction is related to activation of phospholipase C (PLC) and protein kinase C (PKC) [9,19]. The sodium-hydrogen (NHE) and sodium–calcium exchangers (NCX) seem to contribute to the apelin-driven inotropic action by regulating calcium homeostasis and intracellular pH. Whether apelin directly increases intracellular calcium [Ca2+]i or sensitizes myofilaments to calcium remains controversial [6,19,20].
Because apelin activates NHE, it was proposed that changes in cytosolic pH increases myofilaments sensitivity for calcium[6]. However, Dai et al. [20] found no effect of apelin on the myofilaments but demonstrated an increased systolic [Ca2+]i in failing cardiac myocytes. Similarly, Wang et al. [19] reported an increase in systolic [Ca2+]i but a decrease in diastolic [Ca2+]i via a PKC-dependent mechanism. Apelin also increases the activity of NCX and the sarcoplasmic reticulum Ca2+-ATPase (SERCA) [19] via unknown mechanisms. Changes in INa amplitude and kinetics may modulate intracellular Na+ concentrations and the turnover of NCX to increase intracellular calcium. Thus, modulation of INa by Apelin could play a role in its inotropic effect.
Apelin increases the frequency of spontaneous activation and the speed of conduction [6,16]. We therefore tested if these changes in cardiac excitability were linked to modulation of INa in cardiomyocytes freshly isolated from dog left ventricle. Our results show that A-13 and A17 increase INa by 39% and 61% and shift its mid-activation potential by −6.8 mV and −8.6 mV, respectively. This resulted in a significant increase in INa window current. Neither A-13 nor A-17 modified the steady state availability (inactivation, h∞) of the sodium channels. Recovery from inactivation was slowed by apelin and activation of PKC but not PKA was involved in the modulation of INa. Our results suggest that the apelin-induced increase in excitability is due to activation of INa at more negative potentials and that a larger window current may contribute to its inotropic effect.
2. Materials and methods
2.1. Cell dissociation
All methods and care of the dogs conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and were approved by the institutional animal ethics review committee of the University de Sherbrooke. Myocytes were isolated by enzymatic dissociation as previously described [21]. Briefly, adult mongrel dogs weighing between 30 and 35 kg were sedated with acepromazine (Atravet, 0.25 mg/kg I.M.), a phenothiazine tranquilizer, for 15 min, and then anaesthetized with heparin (5000 U) and sodium pento-barbital (25 mg/kg I.V) and their hearts quickly removed and placed in Gerrit Isenberg’s Kraft-Bruhe solution. A wedge consisting of the left ventricular free wall supplied by the left anterior descending coronary artery was excised. The coronary artery was cannulated and flushed for 5 min at a rate of 5 ml/min with Ca-free Tyrode solution supplemented with EGTA 2 mM and 0.1% BSA. Perfusion was then switched to Tyrode solution at 33 °C containing 0.1 mM Ca2+ and 230 U/ml collagenase (CLS 2, Worthington, Freehold, NJ) and recirculated for 25–45 min. Dissociated cells were stored in Gerrit Isenberg’s Kraft-Bruhe solution ( in mM): 100 Potassium glutamate, 10 Potassium aspartate, 25 KCl, 10 KH2PO4, 2 MgSO4, 20 Taurine, 5 Creatine, 0.5 EGTA, 20 Glucose, 10 HEPES, 2% BSA, supplemented with 0.2 mM CaCl2.
2.2. Electrophysiology
2.2.1. Patch clamp
Myocytes were placed in a chamber mounted on the stage of an inverted microscope (Nikon Diaphot, Tokyo, Japan) and superfused with a low sodium solution containing (mM): 120 Choline-Cl, 5 NaCl, 5 NaOH, 2.8 Na-Acetate, 4 KOH, 0.5 CaCl2, 1.5 MgCl2, 10 HEPES, 10 Glucose (pH 7.4 with NaOH) to insure adequate voltage control. Tetraethylammonium chloride (5 mM) was added to the external solution to block TEA-sensitive native potassium currents and CoCl2 (1 mM), 4-AP (2 mM) and BaCl2 (0.5 mM) were used to block ICaL, ITo and IK1 currents, respectively. Membrane currents were measured in the whole-cell configuration of the patch-clamp technique as previously described [22,23]. All recordings were made at room temperature (22 °C) using an Axopatch 200B amplifier (Axon instruments, Union City CA). Patch pipettes pulled from Corning 7052 glass (Model PP-89, Narashige, Japan) had resistance between 1.5 and 2 MΩ when filled with a solution containing (in mM) 15 NaCl, 5 KCl, 120 Cs-Aspartate, 1 MgCl2, 4 Na2-ATP, 10 EGTA and 10 HEPES (pH 7.3 with CsOH). All solutions were adjusted at 300 mOsm with sucrose. Currents were filtered with a four pole Bessel filter at 5 kHz and digitized at 10–50 kHz. Data acquisition and analysis were performed using pCLAMP programs V9.2 (Axon Instruments), EXCEL (Microsoft) and ORIGIN 6.1 (Microcal Software, Northampton, MA) softwares. Whole cell capacitance and series resistance compensation (85%) were optimized to minimize the duration of the capacitive artefact and reduce voltage errors. Only recordings from cells displaying more than six well controlled peak INa measurements between the threshold and the maximum current were analyzed. This is a well established criterion to demonstrate voltage control during INa recordings. Access resistance varied between 500 and 750 kΩ.
To avoid artefactual measurements due to time dependent voltage shifts in steady state inactivation (h∞) in whole cell configuration, we routinely measured our first h∞ curve after 8–10 min and at the end of the experiment i.e. roughly 20 to 30 min thereafter. Variability in V0.5, was typically 1 to 2 mV between the 10 min and final measurements. Control data were taken in non-treated cells that were under voltage clamp for 8 min before application of Apelin.
2.2.2. Action potential measurements
Free running epicardial preparations (strips 1 × 0.5 × 0.1 cm) were isolated from left ventricle (LV). The preparations, isolated using a dermatome (Davol Simon Dermatome, Cranston, RI, USA), were placed in a tissue bath (volume 5 ml, flow rate 12 ml/min) and allowed to equilibrate for at least 3 hours while superfused with oxygenated Tyrode’s solution (36.5 ± 0.5 °C, pH = 7.35) and stimulated at a basic cycle length (BCL) of 500 ms using field or point stimulation (rectangular stimuli 1– to 3-ms duration, 2–3 times diastolic threshold intensity). The composition of the Tyrode’s solution was (in mM): NaCl 129, KCl 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, and d-glucose 5.5. Action potentials were recorded during stimulations at a cycle length of 800 ms using standard glass microelectrodes filled with 2.7 M KCl (10 to 20 MΩ DC resistance) connected to a high input-impedance amplification system (World Precision Instruments, Sarasota, FL, USA). The signals were displayed on oscilloscopes, amplified, digitized and analyzed (Spike 2, Cambridge Electronic Design, Cambridge, England). Apelin was superfused for 20 min before recordings.
2.3. Isolation of proteins
Separation of sarcolemmal and endosomal membrane fractions was performed according to established methods [24]. Briefly, fresh heart tissue was minced in a high salt solution (2 M NaCl, 20 mM HEPES, pH 7.4) and incubated for 30 min at 4 °C to depolymerize the myofilaments. Tissue was then rinsed and homogenized in a buffer containing (mM): 20 HEPES, 250 sucrose, 2 EDTA, 1 MgCl2, pH 7.4. Fractionation was performed by centrifugations for 10 min at 2000g followed by 10 min at 5000g, 30 min at 200,00g and finally 60 min at 100,000g. The 5000g fraction containing mainly sarcolemmal membrane proteins was used for Western blots. Concentrations were determined using the BCA Protein Assay (Pierce, USA).
2.4. Immunoblots
Immunoblots were performed as previously described [23] in non-denaturing (non-reducing) conditions. Primary rabbit anti-APJ-R antibody (1:2000; Neuromics, US) was detected with an HRP-conjugated goat-anti rabbit antibody (1:3000; Bio-Rad, USA) and visualized with the Western Lightning Chemiluminescence Reagent Plus (PerkinElmer, USA). Blots were exposed to X-ray films and the intensity of each band was measured by densitometry (Quantity One software from Bio-Rad). To determine their specificity, APJ-R antibodies were pre-absorbed overnight at 4 °C against their antigen before immunoblotting or immunocytochemistry.
2.5. Immunocytochemistry
Cells in low-calcium Tyrode containing (in mM): 130 NaCl, 4 KCl, 1.2 MgSO4, 10 HEPES, 11.1 d-glucose were placed on glass slides and fixed in a solution containing: 5% ethanol, 25% acetone, 70% formaldehyde/ZnCl2, pH 6 for 15 min at 4 °C and then permeabilized for 10 min in a Ca2+-free Tyrode solution containing Saponin (0.25% w/v) and CHAPS (0.5% w/v). Primary antibody (1:200) was applied overnight at 4 °C and detected with a goat anti-rabbit antibody (1:1000) conjugated to Alexa 488 dye (Molecular Probes, USA). Propidium iodide (PI) was used to stain nucleotide rich regions (nucleus). Cells mounted with Pro-Long antifade mounting media (Molecular Probe) were visualized on a Olympus Fluoview (Olympus, Japan) confocal microscope, as previously described [4,5].
2.6. Data analysis and statistics
Data are expressed as Mean ± SEM. Difference was considered statistically significant for a p value < 0.05 using ANOVA statistical analysis, a Student’s t test for paired values and a Student’s F-test for differences between fit to data. Steady-state inactivation was fitted to a Boltzmann distribution, I / Imax = 1 / (1 + exp((V − V0.5) / k)) where V0.5 and k are the midpoint and the slope factor, respectively, and V is the membrane potential. Sodium window currents (IW) were calculated from their I/V relationships and the fit to steady state inactivation (h∞) and activation (m∞) curves using the classical Hodgkin and Huxley formulation: where ENa represents the reversal potential for INa, Vm is the membrane potential where current is to be calculated, and GMax is the maximum conductance obtained from the slope of the linear portion of the I/V relationship.
3. Results
3.1. APJ-R cardiac expression and localization
APJ-R was present in isolated dog cardiomyocytes from the left ventricle (LV, Fig. 1). Immunoblots on proteins from dog LV showed a band at 42 kDa consistent with the molecular weight of the monomeric form of the receptor [15] (Fig. 1A). Control experiments using APJ-R antibodies pre-absorbed against their antigen overnight at 4 °C did not reveal the expected bands. Specific bands, which might reflect multimeric species of APJ-R, were detected at approximately 80 and 160 kDa in the dog ventricle. Immunoblots of whole rat brain proteins used as control showed the expected immunoreactive band of the monomer.
Fig. 1.
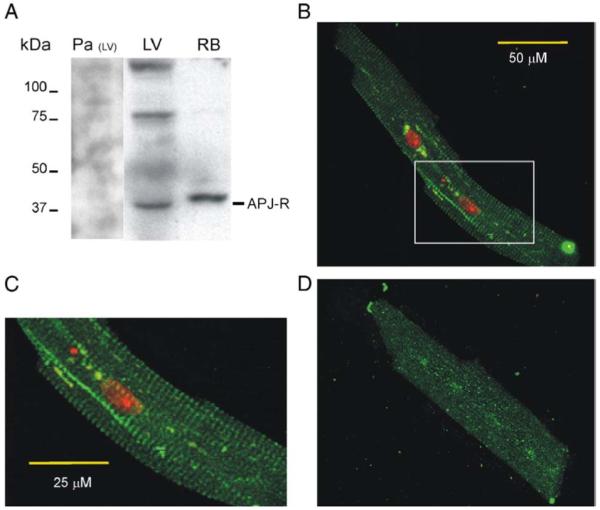
Expression of APJ receptors in dog cardiac left ventricle (LV). (A) Identification of endogenously expressed APJ-R by Western blot in both dog cardiac left ventricle (LV) and rat brain (RB) used as control. Pre-absorbed antibodies (Pa) did not reveal the specific bands observed in LV. (B, C). Cellular distribution of APJ-R in isolated LV myocytes. Staining with rat APJ-R antibodies in green and cellular nucleus by propidium iodide in red. (C) Magnified image of the inset highlighted in B. (D) Control photomicrograph of the APJ-R antibody pre-absorbed against its antigen. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The distribution of APJ-R in isolated dog cardiomyocytes was assessed by immunofluorescence confocal microscopy. Strong labelling was observed at the Z-line (Fig. 1B, C). By contrast, sections incubated with antiserum pre-absorbed with the immunogenic peptide were devoid of specific labelling (Fig. 1D). We previously reported a similar localization for the cardiac sodium channels [23].
3.2. Sodium channel activation under apelin stimulation
Patch clamp experiments on isolated cardiomyocytes showed that both active fragments A-13 and A-17 at a concentration of 100 nM gradually increased INa amplitude to a maximum level after a 20 min perfusion period (Fig. 2). Similar effects were obtained in myocytes under voltage clamp acutely exposed to Apelin or pre-incubated for 20 min before patch clamp measurements. Current-voltage (I/V) analysis showed that A-13 and A-17 increased INa by 39% (−90 ± 13 pA/pF) and 61% (−104 ± 14 pA/pF) compared to untreated cardiomyocytes (−64.8 ± 3.8 pA/pF) (Fig. 3A). Maximum conductance (slope of the linear portion of I/V) was not altered by Apelin with respective values of 2.5 ± 0.2 pS/pF, 2.6 ± 0.3 pS/pF and 2.7 ± 0.1 pS/pF for Control, A-13 and A-17 thus suggesting that changes in gating rather than recruitment of new channels is responsible for the increase in peak current.
Fig. 2.
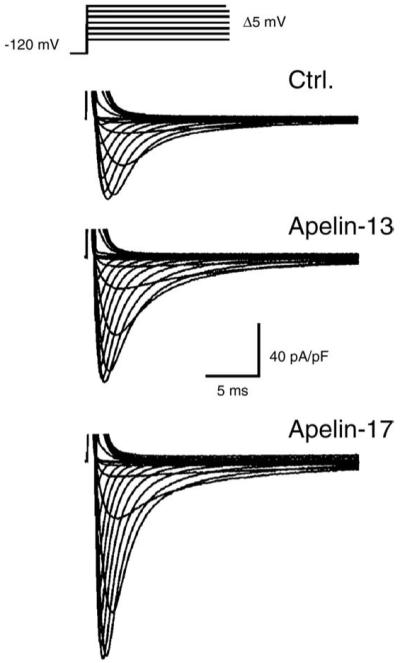
Apelin increases INa amplitude in cardiomyocytes isolated from the canine left ventricle. Protocol used for characterization of the current-voltage relationship (I/V) of INa. Representative recordings of INa in control (Ctrl.), and following perfusion (20 min) with apelin-13 or apelin-17 (100 nM).
Fig. 3.
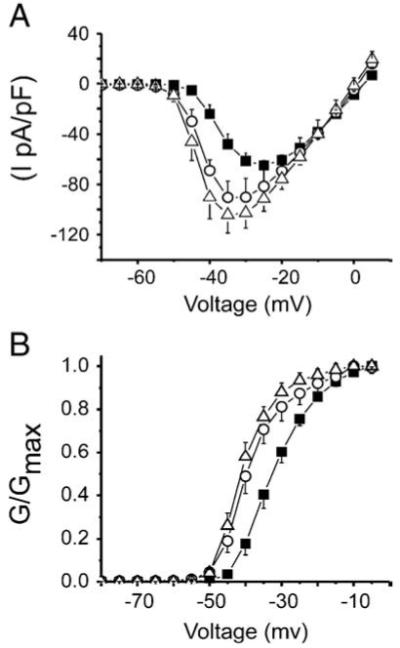
Effect of apelin on the I/V and activation curves of INa in isolated myocytes. (A) Peak INa was normalized to the capacitance of their respective cells. Current densities (pA/pF) were plotted against their respective test potential in control (■ n = 14) and following application of 100 nM apelin-13 (○ n = 7) or apelin-17 (△ n = 7). Peak currents were significantly increased by both active fragments compared to control (p < 0.01). (B) Conductance (G) at each potential was calculated as the ratio INa/(Vm − ENa) where Vm represents the membrane test potential and ENa the reversal potential for INa. Each G values were normalized to the maximum conductance (GMax). Data were plotted against the test potentials to obtain INa activation curve for each condition presented in A. A Boltzmann equation fit to data yielded mid-activation potential (V1/2) values of −31.9 ± 0.3 mV, −38.7 ± 0.5 mV and −40.5 ± 0.3 mV for control, Apelin-13 and Apelin-17, respectively. There was no significant difference between the two active fragments of apelin. Slope factors (k) were similar for control (5.6 ± 0.3 mV), apelin-13 (5.1 ± 0.4 mV) and apelin-17 = (4.3 ± 0.3 mV). Both apelin-13 and apelin-17 activation curves are significantly shifted to more negative potentials compared to control (p < 0,05, F-test). Data are presented as Mean ± SEM.
Analysis of the I/V relationship revealed that A-13 and A-17 negatively shifted mid-activation potential of INa (V1/2) by −6.8 ± 0.6 mV and −17 ± 1 mV respectively (Fig. 3B). Therefore apelin increased INa amplitude and the opening probability of the sodium channels at more negative voltage.
3.3. Effects of apelin stimulation on steady state inactivation, sodium window current and INa recovery
We next tested for changes in h∞ (Fig. 4) but did not observe differences in mid-inactivation voltage. Thus, apelin specifically modulates the activation of INa.
Fig. 4.

Apelin does not modulate sodium channel availability (steady state inactivation) in isolated dog cardiomyocytes from the left ventricle. (A) Standard voltage clamp protocol to measure steady state inactivation consisting of a series of conditioning pulses in increments of 5 mV from a holding potential of −120 mV followed by a test pulse to −15 mV. (B) INa inactivation curves were obtained by plotting the ratio of INa to its maximum value against the conditioning pulse voltage. Data represent values in control (■ n = 12) and with 100 nM apelin-13 (○ n = 5) or apelin-17 (△ n = 7). Mid-inactivation potentials (V1/2) and slopes (k) from a Boltzmann equation fit to data are not statistically significant (p > 0,05). (V1/2; control: −70.9 ± 0.1 mV, apelin-13: −70.4 ± 0.2 mV, apelin-17: −72.1 ± 0.1 mV; k; control: 5.7 ± 0.1, apelin-13: 5.7 ± 0.2, apelin-17: 13 = 5.2 ± 0.1).
The overlap between h∞ and m∞ curves generates a sustained window current (IW) as the channel population dynamically equilibrates between activated and inactivated states (Fig. 5). IW is an important component of intracellular Na+ homeostasis. By negatively shifting activation, A-13 increased the area of overlap between activation and inactivation (Fig. 5B). Interestingly, A-17 only slightly increased the slope of the inactivation curve and yielded area under the intersection of the inactivation and activation curves similar to control values. However, both A-13 and A-17 shifted the intersection point between inactivation and activation yielding larger IW at voltages near the normal resting membrane potential of cardiomyocytes.
Fig. 5.
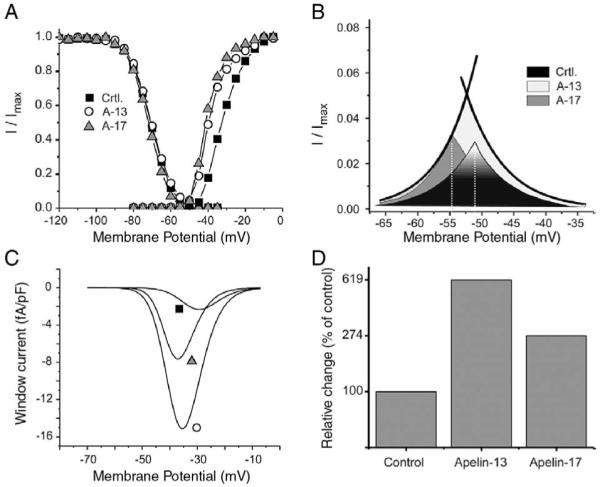
Overlap of steady state activation (m∞) and steady state inactivation (h∞) curves reveals an increase in sodium window current (IW) by apelin. (A) m∞ and h∞ curves in control (■) and following perfusion with 100 nM apelin-13 (○) or apelin-17 (▲). (B) Expanded view of the overlap between the h∞ and m∞ curves from the Boltzmann equation fit to data in A. Intersection points (dotted line) were at −51 mV, −53 mV, −55 mV for control, apelin-13 and apelin-17, respectively. (C) Theoretical IW current voltage relationship calculated from the Hodgkin-Huxley model (IW = GMaxm3h (Vm − ENa), see Materials and methods) in control (■) and following perfusion with 100 nM of apelin-13 (■) or apelin-17 (▲). Maximum IW amplitude was negatively shifted from −29 mV in control to −35 mV and −37 mV with apelin-13 and apelin-17, respectively. (D) Total electric charge carried by IW as obtained from the integral of its I/V relationship presented in C and expressed as percent of the control values.
Because sustained sodium current amplitude is strongly reduced by inactivating membrane potentials [25], the rate of depolarization used in traditional voltage ramp protocols strongly influence the amplitude of IW and impairs accurate measurement of its amplitude. To calculate the I/V relationship of IW (Fig. 5C), we used the classical Hodgkin &Huxley model for INa. Since activation of INa follows m∞3 kinetics, the calculated peak of IW is more positive than the actual crossover of the h∞ and m∞ curves and, as predicted, A-13 and A-17 negatively shifted peak IW by 6 and 8 mV respectively. The total electric charge (QW) carried by IW over its active voltage range was calculated from the area under the I/V relationship (Fig. 5D). A-13 and A-17 respectively increased QW by 619% and 274% compared to controls. Although the crossover point for IW at −50 mV is above the normal resting membrane potential, during phase 2 and 3 repolarization of each action potential, ventricular cells spend between 100 to 200 ms between −70 mV and −30 mV, a range of voltages where IW is active. Such increase in IW amplitude and the shift in activation voltage have the ability to elevate resting intracellular sodium concentration.
An important determinant of INa amplitude in-vivo is recovery from inactivation, which determines the refractory period of the myocardium. To evaluate the effect of apelin on this parameter, we used a standard two pulse protocol with incremental time intervals between them. A-17 and A-13 both significantly slowed INa recovery from inactivation (Fig. 6B, Table 1).
Fig. 6.
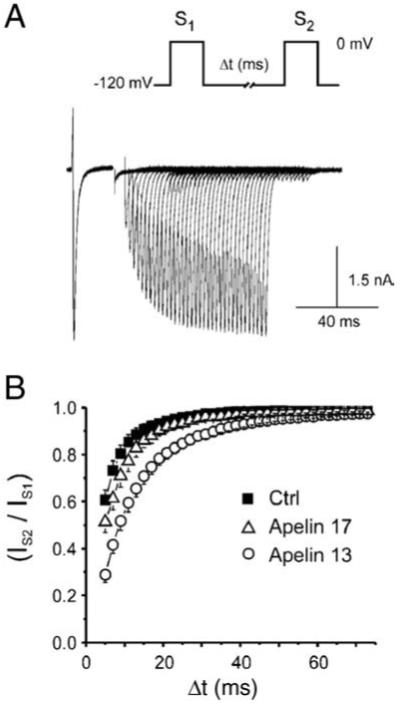
Apelin slows INa recovery from inactivation in isolated dog LV myocytes. (A) Standard electrophysiological double pulse protocol (S1-S2) to −20 mV for 20 ms used for measurements of recovery from inactivation. (B) INa recovery from inactivation expressed as the fraction of the initial current elicited during the second pulse and plotted against the inter-pulse interval duration (△t) in control (■ n = 11) and following application of 100 nM apelin-13 (○ n = 5) or apelin-17 (△ n = 7). Data were fitted to a sum of two exponentials. Statistical comparison between the fit using an F-test shows that both apelin isoforms significantly slow recovery from inactivation
Table 1.
Reactivation parameters from the sum of two exponentials fit to data presented in Fig. 6 in control and following exposure to 100 nM apelin. As and Af respectively represent the contribution of the fast and slow exponential components to recovery and τs and τf: The time constants obtained from a two exponential fit to the data, as described in Fig. 6
| Parameters for recovery from inactivation (2 exp. fit) |
||||
|---|---|---|---|---|
| As (%) | τs (ms) | Af (%) | τf (ms) | |
| Control | 13 ± 2 | 18 ± 2 | 86 ± 2 | 4.4 ± 0.1 |
| Apelin-13 | 100 | 11.5 ± 0.5 | ||
| Apelin-17 | 10 ± 3 | 25 ± 8 | 89 ± 3 | 6.3 ± 0.2 |
3.4. Role of PKA and PKC on sodium channel activation by apelin
Previous studies showed involvement of protein kinase A (PKA) and C (PKC) in the effect of apelin [7–9]. Phosphorylation of sodium channels by these kinases also modulates the amplitude and gating of INa [26–29]. We then evaluated whether the effects of A-13 on INa involved activation of PKA or PKC (Fig. 7). In myocytes pre-treated with IBMX (100 mM) and 8-Bromo-cAMP (50 μM) to fully activate PKA, addition of A-13 increased INa by 55 ± 9% (Fig. 7A), as observed for apelin alone without prior activation of PKA (Fig. 2). Peak INa was negatively shifted alike (Fig. 7B). These results suggest that the effects of Apelin on INa do not involve the PKA signalling cascade.
Fig. 7.

The effect of apelin on INa amplitude is mediated by PKC. (A) Left: Representative current recordings on cardiomyocytes pre-treated with IBMX (100 mM) + 8Br-cAMP (50 μM) for 20 min (control) to inhibit PKA activation followed by concurrent perfusion with Apelin-13 for 15 min. Right: Representative recordings from same protocol as left panel but this time myocytes were pre-treated with chelerythrine (3 μM) for 30 min to inhibit PKC. (B) Left: PKA inhibition did not prevent the increase in INa by apelin-13, as shown by the I/V relationship under perfusion with PKA inhibitor (■ n = 6) and apelin-13 (100 nM) (○ n = 6). Apelin-13 significantly increased the maximum amplitude of INa from −17,8 ± 2,0 pA/pF to −27,6 ± 2,4 pA/pF as seen in Fig. 2 without pre-treatment. Right: I/V curves following concurrent but sequential application of the PKC inhibitor (■ n = 5) and apelin-13 (○ n = 5). Inhibition of PKC activity by chelerethrine reduced the amplitude of INa compared to untreated cells. Apelin-13 increase in INa amplitude was lost following inhibition of PKC (maximum INa chelerythrine: −11,1 ± 2,4 pA/pF, chelerythrine + Apelin-13: −12.1 ± 1.9 pA/pF).
Activation of PKC is known to increase INa. Consequently, chelerythrine, a potent and selective inhibitor of PKC [30] reduced the amplitude of peak INa in control myocytes. As opposed to PKA activation, pre-treatment with chelerythrine (3 μM) prevented the Apelin-induced increase in INa and changes in steady-state activation (Fig. 7B). Since the action of Apelin on INa was prevented by chelerythrine, these results suggest the APJ-R signalling pathway involves PKC activation.
3.5. Action of Apelin on cardiac excitability
Since changes in INa amplitude are likely to modulate cardiac excitability, we directly tested the effect of Apelin on canine ventricular action potentials (Fig. 8). Apelin at a concentration of 100 nM significantly increased the maximum rate of depolarization from 137 ± 23 V/s in control to 166 ± 20 V/s during pacing at a cycle length of 800 ms (Fig. 8A-B). Effects on Vmax could be observed for concentrations as low as 30 nM (Fig. 8C). Apelin therefore increases cardiac excitability as predicted by its effects on INa.
Fig. 8.
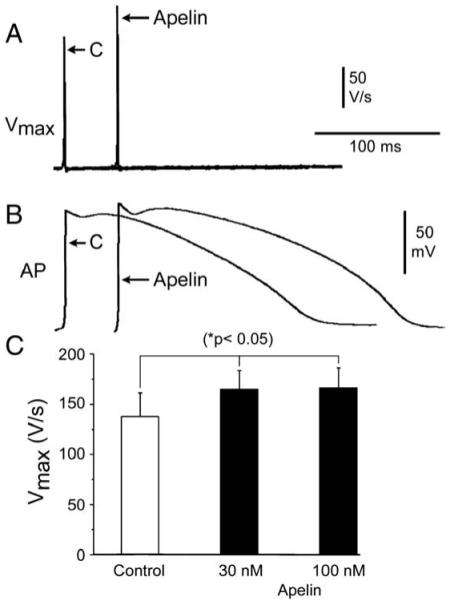
Apelin increase excitability in canine ventricular preparations. Representative action potential recordings and corresponding maximum rate of depolarization (Vmax) values (A–B) following stimulations at a cycle length (CL) of 800 ms under control and in the presence of Apelin (100 nM) in canine superfused ventricular preparations. Bar graph summarizes the effect of Apelin on Vmax at concentrations of 30 nM and 100 nM (C). *p < 0.05 versus respective control (C); n = 4.
4. Discussion
Messenger RNA for APJ-R was previously detected in rat, mouse and human [12,15,31]. Our immunoblots demonstrate for the first time that APJ-Rs are expressed in dog ventricle, a cardiac model electrophysiologically similar to humans. We also observed two extra bands at 80 kDa and 160 kDa specific to APJ-R in LV. We attribute these bands to homodimerics and oligomerics forms of the APJ receptor since members of the GPCR family are prone to form homomultimers in cardiac tissue [32]. We localized APJ-R at the Z-lines in isolated cardiomyocytes in agreement with previous findings in rat and human cardiomyocytes [6,15]. Since the T-Tubules and sodium channels are also localized at Z-lines [5,33], our results suggest a close interaction between them and APJ-R. Such colocalization with the APJ-R may facilitate the apelin inotropic effect.
We found that apelin increases INa and shifts steady state activation towards more negative voltages but the maximum conductance of the I/V curve remains unchanged. Therefore, the increase in INa is due to increased driving force from the change in activation voltage and changes in the kinetics of resident membrane channels rather than recruitment or synthesis of new channels.
The increase in INa by Apelin accelerated the initial depolarization (phase 0) of the ventricular action potential as shown by the increase in Vmax. Modulation of INa by Apelin therefore facilitates firing of the cardiac action potential and increases excitability. The lack of a hyperpolarizing shift in Na+ channels availability will further potentiate this effect. These results are in agreement with the increased frequency of spontaneous activation, speed of conduction and excitability previously reported [6,34].
We show that A-13 and A-17 increase the total electric charge carried by IW by 619% and 274% respectively. This will increase the entry of sodium during the plateau and phase 3 repolarization of the AP and may lead to a reduction in the Na+ gradient (▽Na+) between the intra- and extracellular milieu. ▽Na+ generates the driving force for NCX and its reduction will slow extrusion of calcium and favour its accumulation in the sarcoplasmic reticulum. We hypothesize that these changes in INa may lead to alterations of Na+ homeostasis and contribute to the positive inotropic effect of apelin, in agreement with Szokodi et al. [9]. Thus, the increase in INa and IW amplitude and the negative shift in steady state activation and IW current-voltage relationship will concur to increase cardiac excitability and may contribute to the inotropic effects of Apelin.
Interestingly, apelin also slows recovery from inactivation, an effect that is likely to prolong the duration of the refractory period. On the one hand, the increased excitability could potentially create an arrhythmogenic substrate, particularly in the setting of heart failure, but re-entry and tachycardia may be minimized by the increase in refractory period. This dual effect on INa may confer interesting antiarrhythmic properties to apelin.
4.1. Increase in sodium current by PKC
Our results show that the APJ-R signalling pathway involves activation of PKC. These results are in agreement with Sokodi et al. [9] who reported only a 50% inhibition of the inotropic effects of apelin by staurosporine and GF-109203X on cardiomyocytes. In contrast we found 90% inhibition of the effects of apelin on INa with chelerythrine.
Farkasfalvi et al. [6] suggested that apelin increases NHE activity, leading to intracellular alkalinisation and an increased calcium sensitivity of the myofilaments. The effects of apelin on INa go in opposite direction. NHE extrude protons in exchange of sodium entry. Because of the augmentation of intracellular Na+ concentration the turnover of NHE will be slowed down thus leading to a decrease in intracellular pH and desensitization of the myofilaments. In our experiments, apelin increased INa within 5 min of perfusion and reached steady state at 20 min. This may explain the transient effect of apelin on NHE (1 to 2 min) observed by Farkasfalvi et al. [6] Apelin through APJ-R could initially enhance NHE activity and later on potentiate INa, both mechanisms leading to a positive inotropic effect.
Interestingly, apelin has a vasodilating effect in hypertensive arteries. However, in denuded arteries where endothelium does not produce NO, apelin constricts vascular smooth muscle by a PKC mechanism [8]. Thus, apelin seems to enhance both cardiac and smooth muscle contraction through similar mechanisms.
4.2. Clinical relevance
Our results show that Apelin has the beneficial effects of increasing INa amplitude and cellular excitability. These characteristics give apelin a strong potential not only as a positive inotropic drug but also as a new antiarrythmic agent. Compounds that enhance peak INa and prolong the recovery from inactivation with little effects on availability of the channels could also be beneficial in inherited channelopathies such as Brugada syndrome where the amplitude of INa is significantly reduced.
4.3. Conclusion
Our study is the first to evaluate the effects of apelin on the cardiac sodium current. Our results indicate that modulation of INa gating and amplitude by apelin contribute to the increased cardiac excitability and inotropic effects previously reported for this endogenous peptide. Considering apelin as a potential heart-supporting drug, the increase in excitability suggests it might additionally have beneficial antiarrhythmic actions in hypertension and heart failure. Levels of apelin are 3 to 5 times higher than normal during heart failure [17,18,35]. Such increase in plasma level is consistent with the inotropic action of apelin as it will tend to counteract the loss of contractility associated to heart failure. Our data thus suggest that rising apelin plasma concentration (or optimizing cardiac apelin to APJ-R interactions) might be beneficial in this context.
Acknowledgments
The authors wish to thanks Mr. N. Beaudet for his helpful review of this manuscript.
This work was supported by the Canadian Institutes of Health MSc student award (CC), Fonds de la Recherche en Santé du Québec Student (CC) and postdoctoral fellowship (VH), NIH grant HL47678 from NHLBI (CA) and by New York State and Florida Grand Lodges Free and Accepted Masons(CA), Canadian Institutes of Health Grant # 74615 (RD), Pfizer Cardiovascular Award (RD). Philip Morris External Research Program (RD).
Footnotes
5. Conflict of interest statement
The authors have no conflict of interest to declare.
References
- [1].Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- [2].O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, et al. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–60. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- [3].Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–6. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- [4].Haufe V, Zimmer T, Benndorf K, Cordeiro JM, Scicchitano S, Iodice A, et al. Distribution of neuronal sodium channels in dog heart. Biophys J. 2003:84. abstract 2003. [Google Scholar]
- [5].Zimmer T, Biskup C, Dugarmaa S, Vogel F, Steinbis M, Bohle T, et al. Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J Membr Biol. 2002;186:1–12. doi: 10.1007/s00232-001-0130-1. [DOI] [PubMed] [Google Scholar]
- [6].Farkasfalvi K, Stagg MA, Coppen SR, Siedlecka U, Lee J, Soppa GK, et al. Direct effects of apelin on cardiomyocyte contractility and electrophysiology. Biochem Biophys Res Commun. 2007;357:889–95. doi: 10.1016/j.bbrc.2007.04.017. [DOI] [PubMed] [Google Scholar]
- [7].Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP. [(125)I]-(Pyr(1)) Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br J Pharmacol. 2001;132:1255–60. doi: 10.1038/sj.bjp.0703939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004;118:119–25. doi: 10.1016/j.regpep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- [9].Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–40. doi: 10.1161/01.res.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- [10].Edinger AL, Hoffman TL, Sharron M, Lee B, Yi Y, Choe W, et al. An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol. 1998;72:7934–40. doi: 10.1128/jvi.72.10.7934-7940.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, et al. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- [12].O’Carroll AM, Selby TL, Palkovits M, Lolait SJ. Distribution of mRNA encoding B78/apj, the rat homologue of the human APJ receptor, and its endogenous ligand apelin in brain and peripheral tissues. Biochim Biophys Acta. 2000;1492:72–80. doi: 10.1016/s0167-4781(00)00072-5. [DOI] [PubMed] [Google Scholar]
- [13].Sorhede WM, Magnusson C, Ahren B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul Pept. 2005;131:12–7. doi: 10.1016/j.regpep.2005.05.004. [DOI] [PubMed] [Google Scholar]
- [14].Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem. 2003;84:1162–72. doi: 10.1046/j.1471-4159.2003.01587.x. [DOI] [PubMed] [Google Scholar]
- [15].Kleinz MJ, Skepper JN, Davenport AP. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept. 2005;126:233–40. doi: 10.1016/j.regpep.2004.10.019. [DOI] [PubMed] [Google Scholar]
- [16].Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, et al. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, et al. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation. 2003;108:1432–9. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- [18].Berry MF, Pirolli TJ, Jayasankar V, Burdick J, Morine KJ, Gardner TJ, et al. Apelin has in vivo inotropic effects on normal and failing hearts. Circulation. 2004;110:II187–93. doi: 10.1161/01.CIR.0000138382.57325.5c. [DOI] [PubMed] [Google Scholar]
- [19].Wang C, Du JF, Wu F, Wang HC. Apelin decreases the SR Ca2+ content but enhances the amplitude of [Ca2+]i transient and contractions during twitches in isolated rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2540–6. doi: 10.1152/ajpheart.00046.2008. [DOI] [PubMed] [Google Scholar]
- [20].Dai T, Ramirez-Correa G, Gao WD. Apelin increases contractility in failing cardiac muscle. Eur J Pharmacol. 2006;553:222–8. doi: 10.1016/j.ejphar.2006.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cordeiro JM, Greene L, Heilmann C, Antzelevitch D, Antzelevitch C. Transmural heterogeneity of calcium activity and mechanical function in the canine left ventricle. Am J Physiol Heart Circ Physiol. 2004;286:H1471–9. doi: 10.1152/ajpheart.00748.2003. [DOI] [PubMed] [Google Scholar]
- [22].Dumaine R, Cordeiro JM. Comparison of K+ currents in cardiac Purkinje cells isolated from rabbit and dog. J Mol Cell Cardiol. 2007;42:378–89. doi: 10.1016/j.yjmcc.2006.10.019. [DOI] [PubMed] [Google Scholar]
- [23].Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, et al. Contribution of neuronal sodium channels to the cardiac fast sodium current I(Na) is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–27. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- [24].Fuller W, Eaton P, Medina RA, Bell J, Shattock MJ. Differential centrifugation separates cardiac sarcolemmal and endosomal membranes from Langendorff-perfused rat hearts. Anal Biochem. 2001;293:216–23. doi: 10.1006/abio.2001.5127. [DOI] [PubMed] [Google Scholar]
- [25].Dumaine R, Kirsch GE. Mechanism of lidocaine block of late current in long-QT mutant Na+ channels. Am J Physiol. 1998;274(43):H477–87. doi: 10.1152/ajpheart.1998.274.2.H477. [DOI] [PubMed] [Google Scholar]
- [26].Zhou J, Yi J, Hu N, George AL, Jr, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000;87:33–8. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- [27].Frohnwieser B, Chen LQ, Schreibmayer W, Kallen RG. Modulation of the human cardiac sodium channel alpha-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J Physiol (Lond) 1997;498(Pt 2):309–18. doi: 10.1113/jphysiol.1997.sp021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ono K, Fozzard HA, Hanck DA. Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ Res. 1993;72:807–15. doi: 10.1161/01.res.72.4.807. [DOI] [PubMed] [Google Scholar]
- [29].Murray KT, Hu NN, Daw JR, Shin HG, Watson MT, Mashburn AB, et al. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res. 1997;80:370–6. doi: 10.1161/01.res.80.3.370. [DOI] [PubMed] [Google Scholar]
- [30].Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–9. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- [31].Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem. 2000;275:21061–7. doi: 10.1074/jbc.M908417199. [DOI] [PubMed] [Google Scholar]
- [32].Nekrasova E, Sosinskaya A, Natochin M, Lancet D, Gat U. Overexpression, solubilization and purification of rat and human olfactory receptors. Eur J Biochem. 1996;238:28–37. doi: 10.1111/j.1432-1033.1996.0028q.x. [DOI] [PubMed] [Google Scholar]
- [33].Haufe V, Chamberland C, Dumaine R. The promiscuous nature of the cardiac sodium current. J Mol Cell Cardiol. 2007;42:469–77. doi: 10.1016/j.yjmcc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- [34].Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, et al. Vascular Effects Of Apelin In Vivo In Man. J Am Coll Cardiol. 2008;52:908–13. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- [35].Atluri P, Morine KJ, Liao GP, Panlilio CM, Berry MF, Hsu VM, et al. Ischemic heart failure enhances endogenous myocardial apelin and APJ receptor expression. Cell Mol Biol Lett. 2007;12:127–38. doi: 10.2478/s11658-006-0058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]