

Abstract
The replication-defective herpes simplex virus 2 (HSV-2) dl5-29 mutant virus strain with deletions in the UL5 and UL29 genes has been shown to protect mice and guinea pigs against challenge with wild type (wt) HSV-2 and to protect against ocular disease caused by HSV-1 infection. The dl5-29 strain is currently being prepared for clinical trials as a herpes vaccine candidate. As a possible approach to improve the efficacy of dl5-29 as a genital herpes vaccine, we replaced the UL41 gene encoding the virion host shutoff function (vhs) with the UL41 gene from HSV-1. While the HSV-2 UL41 and HSV-1 UL41 gene products have analogous functions, vhs-1 is 40-fold less active than vhs-2. Previously, it was shown that disruption of the UL41 gene can increase the efficacy of dl5-29 as a vaccine against HSV-2. These properties led us to hypothesize that replacement of vhs-2 by vhs-1 would decrease cytopathic effects in infected host cells, allowing longer survival of antigen-presenting cells and induction of stronger immune responses. The new recombinant dl5-29-41.1 virus shows nearly the same immunogenicity and protection against HSV-2 challenge as the parental dl5-29 virus or a triply deleted mutant virus, dl5-29-41, in the murine model of infection, and grows to higher titers than the parental strain in complementing cells, which is important for GMP production. The results have implications for the design of future HSV-2 vaccine candidates and mechanisms of induction of protective immunity against genital herpes.
Introduction
Herpes simplex virus 2 (HSV-2) causes a sexually transmitted infection that is a common cause of genital lesions. The worldwide HSV-2 seroprevalence has been reported to be approximately 16% in the adult population [1]. The highest prevalence of antibodies to HSV-2 among developed counties has been reported in the USA with an overall age adjusted rate of approximately 17% [2, 3], although in certain groups the seroprevalence is reported to be nearly 40% [3]. The incidence of HSV-2 infection is particularly high in developing counties and reaches above 90% in groups of high risk [4].
Primary HSV-2 infection is particularly dangerous in pregnant woman due to a high risk of transmission of the virus to the fetus and newborn. This can lead to significant morbidity and mortality [5]. Recent studies have shown that HSV-2 infection is associated with increased risk of acquisition of human immunodeficiency virus type 1 (HIV-1) [6, 7] and that the use of antiviral drugs targeting HSV in co-infected individuals reduces the HIV-1 viral load in both genital lesions and plasma [8, 9]. These findings have direct clinical implications, suggesting that HIV-1 transmission and/or disease in the population may be reduced by therapy directed against HSV-2.
Infection with HSV results in a life-long latent infection which may lead to intermittent reactivation, shedding, and recurrences of disease [4]. While antiviral chemotherapy can reduce HSV disease, it does not eliminate the latent viral reservoir and even the most stringent regimen is not 100% effective at preventing transmission [10]. The most practical measure to prevent HSV acquisition and transmission would be an effective prophylactic and/or therapeutic vaccine.
Numerous attempts to develop an HSV-2 vaccine have been undertaken [11]. The most successful candidate to date, a recombinant gD subunit vaccine with MPL adjuvant, gave promising results in animal models [12, 13]; however, when tested in the general population, it gave protection only in women seronegative for both HSV-1 and HSV-2 [14]. A vaccine candidate based on gB and gD glycoproteins also failed to prevent HSV-2 infection when tested in the general population [15, 16]. Another attempt to develop an HSV-2 vaccine was undertaken using a single-cycle HSV-2 mutant containing a deletion of glycoprotein H. This candidate was ineffective as a therapeutic in previously infected patients [17].
Replication-defective mutant viruses are also good potential candidates as HSV vaccines because they activate innate immune responses through Toll-like receptors, can stimulate humoral and cellular immune responses and have a high biosafety profile because of their inability to replicate and spread and to establish latency. Studies have shown that replication-defective mutants of HSV are able to stimulate immune responses similar in magnitude and effectiveness to replication-competent viruses [18]. Because of these properties, replication-defective HSV-2 mutant strains are being considered as vaccine candidates. An HSV-2 mutant virus containing a deletion of the UL29 gene, which encodes infected cell protein 8 (ICP8), has been shown to protect mice and guinea pigs against challenge with wild-type HSV-2 [19, 20]. To reduce the potential for the generation of replication-competent virus due to recombination with a wild-type HSV-2, a second mutation in the UL5 gene, which encodes a component of viral helicase-primase complex, was introduced into the virus containing the UL29 deletion [21]. This double-deletion mutant virus, dl5-29, was as effective as the UL29 mutant virus, dl29 when used as a vaccine against HSV-2 in an animal model [22]. The dl5-29 HSV-2 mutant has also been shown to protect against ocular infection with HSV-1 [23].
All alphaherpesviruses contain a homolog of the HSV UL41 gene, which encodes an abundant tegument protein essential for shutoff of host protein synthesis and therefore is also known as the virion shut-off (vhs) protein. Vhs has RNase activity [24] and is involved in degrading both viral and host mRNA [25]. At early times post-infection, the high levels of viral gene transcription allow the continued expression of viral genes, but the vhs activity prevents over-expression of immediately early (IE) and early (E) genes, allowing for efficient transition between gene classes [26]. At later times post-infection the vhs activity is neutralized by interaction with another virion tegumet protein, VP16 [27, 28]. Importantly, vhs RNase activity contributes to HSV pathogenesis and neurovirulence [29, 30] and plays a role in HSV evasion of host immunity. There is evidence that 1) vhs degrades mRNA of the genes involved in the antiviral immune response [31], 2) is required for inhibition of dendritic cell (DC) activation [32, 33], and 3) inhibits dendritic cell apoptosis [34]. Studies with replication-defective [35] and replication-competent [36] HSV-1 viruses have shown that deletion of UL41 increases the immunogenicity of these viruses.
To improve the efficacy of dl5-29 as a vaccine against HSV-2, we disrupted the UL41 gene in dl5-29 [37]. Experiments in mice showed that the additional deletion in the UL41 gene significantly increases the efficacy of double-deletion mutant, when tested against challenge with HSV-2 G strain. However, dl5-29 and the dl5-29-41L triple-deletion mutant were equally efficient in reducing the genital disease and viral shedding after challenge with the wild-type HSV-2 333 strain in guinea pigs and mice [38].
In this study we tested the effect on the immunogenicity of dl5-29 of replacement of the HSV-2 UL41 gene with the UL41 gene of the HSV-1 KOS strain. While vhs-1 and vhs-2 have analogous functions in HSV-1 and HSV-2, vhs-1 is 40-fold less active than vhs-2 [39]. Therefore, we hypothesized that replacement of vhs-2 with vhs-1 in the dl5-29 virus would result in slower degradation of viral and host mRNA in infected cells and longer survival of infected cells and greater immunogenicity.
Materials and Methods
Cells and viruses
Vero (ATCC #CRL-1586), Hep2 (ATCC #CCL-23), and V529 [21] cells which contain the HSV-2 UL5 and UL29 (ICP8) genes, were grown and maintained as described [22]. The HSV-2 dl5-29 replication-defective mutant virus contains deletions in the UL5 and UL29 genes [21, 22], and the HSV-2 dl5-29-41L mutant virus also contains lacZ coding sequences within the UL41 locus of dl5-29 virus, [37]. The dl5-29-41 triple deletion mutant virus was constructed by co-transfection of dl5-29-41 viral DNA with linearized pUC41.1 plasmid DNA into V529 cells and selection of white plaques (T. Dudek and D. Knipe, unpublished results). The replication-defective mutants dl5-29, dl5-29-41, dl5-29-41L and dl5-29-41.1 were propagated on V529 cells as described [21]. The wild-type HSV-2 G strain [40] and 186syn+-1 [41] viruses were grown in Vero cells. Supernatant virus and cell lysate stocks were prepared and stored as described [19].
Plasmids
The pUC41.1 plasmid was constructed as follows. A 3585 bp HindIII-HpaI DNA fragment containing the HSV-1 UL41 gene and flanking sequences was isolated from the pSG124 plasmid [42] and cloned into the Hind III-Sma I sites of plasmid pUC19 (Promega) to generate the pUC41.1 plasmid. The UL41 gene and flanking sequences in pUC41.1 were determined at the Dana Farber/Harvard Cancer center DNA Resource Core using standard primers specific for the multiple cloning site (MCS) of pUC19. Viral genome nucleotide numbers correspond to the published sequences of HSV-1 strain 17 [43] or HSV-2 strain HG52 [44].
Antibodies
Mouse monoclonal antibodies specific for gB (ab6506), gD (ab6507), β−actin (ab 8226), and GAPDH (ab8245) antibodies were purchased from Abcam, Inc. Mouse monoclonal antibodies specific for HSV ICP5, and ICP0 were purchased from East Coast Bio, Inc. The 3-83 rabbit polyclonal anti-ICP8 serum has been described [45].
Viral growth assays
For viral growth curve experiments, dl5-29, dl5-29-41 and dl5-29-41.1 were used to infect V529 cells at multiplicity of infection (MOI) of 3 PFU/cell as described [21] .
Analysis of protein expression by western blotting
Viral protein expression was examined by western blotting as described [46]. Briefly, cultures of Hep2 cells were infected at an MOI=10 with dl5-29, dl5-29-41 or dl5-29-41.1 mutant viruses and harvested at the indicated times. Cells were lysed with Laemmli gel sample buffer with complete Mini protease inhibitors (Roche Applied Science, Indianapolis, IN), and the lysates were boiled for 10 min. Equal amounts of cell lysate from each infection were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent Western blotting.
Analysis of protein expression by radioisotopic labeling
Cultures of Hep2 cells were infected at MOI=10 with dl5-29, dl5-29-41L, dl5-29-41.1, HSV-2 186syn+-1, HSV-1 KOS, or mock-infected. Cells were labeled with radioactive amino acids according to [47, 48] with the following modifications. At 30 min before the indicated times (7-8 hpi) the cells were washed twice with methionine/cysteine-free DME medium (Invitrogen, Carlsbad, CA) and then incubated in methionine/cysteine-free DMEM supplemented with 50 μCi/ml of NEG-772 Easy Tag EXPRESS 35-S Protein Labeling Mix containing 35S L-methionine and 35S L-cysteine (Perkin Elmer Life and Analytical Sciences, Waltham, MA) for 30 min. Cells were washed twice with cold PBS and lysed with gel sample buffer containing complete Mini protease inhibitors (Roche Applied Science, Indianapolis, IN). Proteins were resolved in diallyltartardiamide cross-linked 9.25% polyacrylamide gels by SDS-PAGE. The gel was dried under vacuum, and the dried gel was exposed to Kodak BioMAX film for autoradiography.
Animal studies
All procedures were according to a protocol approved by the Institutional Animal Use Committee of Harvard Medical School.
Immunization and challenge
Five week old female C57Bl/6 mice (Taconic Farms) were immunized twice, subcutaneously in the rear flank [49], 4 weeks apart, with 1×106 plaque forming units (pfu) of extracellular supernatant virus in a 50μl volume. At 4 weeks after the second immunization, mice were challenged with HSV-2 G stain by intravaginal infection as described [19].
Measurement of antibody titers
At 7 days after the second immunization, blood was taken via cardiac puncture, and sera were prepared using Microtrainer tubes (BD Biosciences, Franklin Lakes, NJ), pooled and stored at −20° C. Total HSV-2-specific IgG titers were determined by ELISA assays [21]. Neutralizing antibody titers were determined using a standard plaque-reduction assay [18].
Measurement of T cell responses
For MHC-I pentamer staining of HSV gB specific CD8+ T-cells, mice were bled via the tail vein at days 6, 7 and 8 post-immunization. The quantification of circulating HSV gB-specific CD8+ T-cells was conducted by MHC-I pentamer staining as described [37].
For intracellular cytokine staining, splenocytes from C57Bl/6 mice were harvested at day 7 after the second immunization. Analysis of the HSV gB-specific CD8+ T-cells by intracellular cytokine staining was performed as described [37].
Measurement of viral shedding
On day 1 to 5 post challenge, the vaginal cavities of mice were swabbed with pre-wetted dacron swabs (PurFybr, Munster, IN) twice. For each mouse, the collected sample was placed in 1.0 ml of sterile phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 10 mM MgCl2, 1 mM CaCl2, pH 7.4)-0.1% glucose −1% bovine calf serum (BCS) and stored at −80°C. Viral titers were determined by standard plaque assay on Vero cells [48]
Clinical observations
Between days 1 and 10 post challenge mice were monitored for signs of genital lesions. The severity of disease was scored as follows: 0, no sign; 1, slight genital erythema and edema; 2, moderate genital inflammation; 3, purulent genital lesions; 4, hind-limb paralysis. [19].
Measurement of GAPDH mRNA levels
Hep-2 cells were either mock-infected or infected with the indicated viruses at an MOI of 15 PFU/cell. Cells were harvested at 5 hpi in Trizol reagent (Invitrogen), and the sample was extracted with 0.5 volume of chloroform. The samples were mixed with isopropanol and kept at room temperature for 10 minutes, and the precipitate was collected by centrifugation and washed twice with 70% ethanol. The RNAs were dissolved in RNase-free water and stored at −80°C. To obtain cDNAs, three hundred nanograms of each RNA sample was reverse transcribed using oligo-(dT) primers and 200 units of SuperScriptTM II RT (Invitrogen) in a 20 μl volume according to the manufacturer’s instructions. Real-time PCR were performed using 0.5 μl of cDNA, 12.5 μl of Power SYBR green PCR Master Mix (Applied Biosystem) and 250nM of each primer set. The sequence of GAPDH primers (5′-GGACGGGAAGCTTGTCATCAATGG-3′ and 5′-TGTGGTCATGAGTCCTTCCACGAT-3′) and 18S rRNA primers (5′-GTAACCCGTTGAACCCCATT-3′ and 5′CCATCCAATCGGTAGTAGCG’-3) were designed as described [50]. Each reaction was performed in triplicate in 20 μl volumes. The reaction conditions were as follows: 50°C for 2 min, 95°C for 10 min initially, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The GAPDH RNA level was normalized to the 18S rRNA level using the PFAFFL method [51]. The GAPDH mRNA level in mock infected cells was set as 100%, and the levels of GAPDH mRNA in infected cells were calculated as the percentage of mock-infected cells.
Statistical analysis
Statistical analysis was performed using the two-sided Student’s t-test.
Results
Construction of an HSV-2 dl5-29 recombinant virus encoding HSV-1 vhs
A replication-defective triple mutant strain of HSV-2 with mutations in the UL5, UL29 and UL 41 genes showed increased immunogenicity and protective capacity relative to the dl5-29 virus double mutant [21, 22, 52] in some [37] but not all situations [38]. It therefore appeared that reduction in vhs activity might increase the immunogenicity of dl5-29. HSV-2 has a 40-fold more active virion host shutoff activity than HSV-1 [39]; therefore we asked if replacement of the HSV-2 UL41 gene encoding vhs with the HSV-1 UL41 gene could also increase immunogenicity of dl5-29.
We isolated a new dl5-29 recombinant encoding HSV-1 UL41 by co-transfection of dl5-29-41L viral DNA containing a lacZ expression cassette in the UL41 gene locus (Figure 1C) with linearized pUC41.1 plasmid containing the HSV-1 UL41 gene (Figure 1D) into V529 cells. Progeny viruses were harvested, and white plaques were isolated in medium containing X-gal, as described previously [20]. The virus was plaque-purified three times, and the new virus was named dl5-29-41.1.
Figure 1. Schematic diagrams of the HSV and recombinant viral genomes.

(A) Genome of wild-type HSV. Shaded boxes represent repeated sequences, and lines represent unique sequences. (B) Genome of the dl5-29-41L triple mutant virus. The UL5 and UL29 genes contain deletions, and the UL41 gene contains an insertion of a lacZ expression cassette. (C) An expanded view of the UL41 gene disrupted by the lacZ expression cassette in dl5-29-41L. (D) The HSV-1 KOS DNA fragment containing the UL41 gene and flanking regions introduced into plasmid pUC19 to generate the pUC41.1 plasmid. Dashed lines define the boundaries of the homologous sequences in which recombination may have occurred to generate dl5-29-41.1. Nucleotide numbers in (C) correspond to the HSV-2 HG-52 viral genome and in (D) to the HSV-1 strain 17 viral genome.
Expression of HSV-1 vhs protein, by the dl5-29-41.1 virus, was verified by Western blotting analysis of infected V529 cell lysates. HSV-1 and HSV-2 vhs proteins (ICP25) show different electrophoretic mobilities [53], and we observed that in our gel system, the HSV-2 strain 186syn+-1 vhs protein had a reduced mobility as compared to that of HSV-1 strain KOS vhs (Figure 2, Panel A). The HSV-2 dl5-29 vhs protein co-migrated with the wild-type HSV-2 vhs, while dl5-29-41L did not express any vhs protein. In contrast, the dl5-29-41.1 virus expressed a vhs protein that co-migrated with HSV-1 KOS vhs. The dl5-29-41.1 recombinant did not express β-galactosidase, as dl5-29-41L did (Figure 2, panel B), indicating that the HSV-1 vhs gene had replaced the lacZ expression cassette in the dl5-29-41.1 virus. Western blot analysis with HSV-1 specific ICP4 anti-serum showed that only HSV-1 KOS expressed HSV-1 ICP4 (Figure 2, panel C), confirming that dl5-29-41.1 was an HSV-2 virus expressing an HSV-1 vhs protein, not an HSV-1 contaminant. Western blot analysis with anti-serum specific for both HSV-1 and HSV-2 ICP8 proteins provided a control for expression of viral proteins in all infected cultures (Figure 2, panel D). These blots confirmed that dl5-29-41.1 was a recombinant of HSV-2 dl5-29 expressing the HSV-1 vhs protein.
Figure 2. Expression of viral and transgene proteins by the recombinant viruses.

V529 cells were infected with the indicated viruses, and lysates were prepared at 10 hours post-infection. Protein expression was analyzed by SDS-PAGE followed by Western blotting with following antibodies: vhs (A), β-gal (B), ICP4 (C) and ICP8 (D).
Growth properties of dl5-29-41.1
We further examined the properties of dl5-29-41.1 by studying its plaque size and replication kinetics in the V529 complementing cell line. We observed that plaques formed by dl5-29-41.1 were larger than the double deletion mutant virus, dl5-29, or the triple deletion mutant virus, dl5-29-41 (Figure 3A). Similarly, in single cycle growth analysis, dl5-29-41.1 replicated to titers 8-fold higher than dl5-29 or dl5-29-41 (Figure 3B). Therefore, replacement of the HSV-2 vhs with HSV-1 vhs enhanced the replication of dl5-29.
Figure 3. Growth properties of the recombinant viruses.
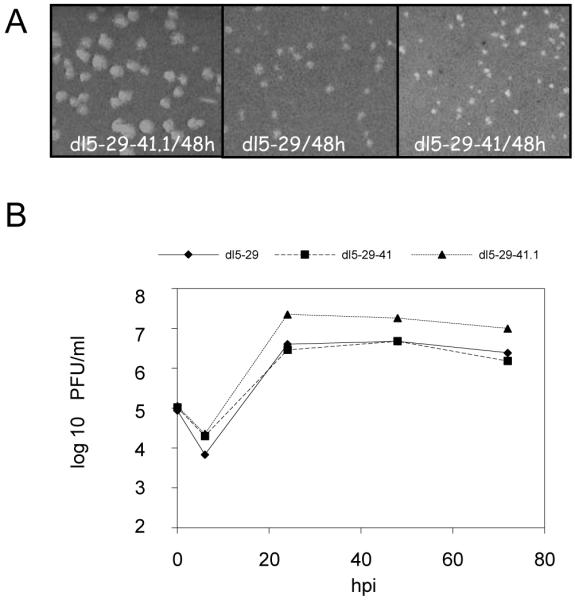
(A) Plaque morphologies of recombinant viruses on the V5-29 complementing cell line. V529 cells were infected with dl5-29-41.1, dl5-29 or dl2-29-41 viruses, overlaid with medium supplemented with anti-HSV IgG, and fixed and stained at 48 hours post-infection. (B) Replication kinetics of the viruses in V529 cells. V529 cells were infected with dl5-29-41.1, dl5-29 or dl5-29-41 at an MOI = 3 pfu and harvested at the indicated times. The yields of progeny viruses were measured on V529 cells by standard plaque assay.
Virion host shutoff activity
We investigated the ability of the dl5-29-41.1 virus to shut off host protein synthesis by measuring both protein synthesis and mRNA levels in infected Hep-2 cells. In cells labeled with 35S-L-methionine and L-cysteine, HSV-1 or HSV-2 infection caused a reduction in the host protein synthesis, as shown by a decrease in specific proteins, e.g., bands H1 and H2, and by a decrease in the smear of host proteins through the lane (Figure 4). In contrast, dl5-29-41 showed no decrease in host protein synthesis, and the new recombinant dl5-29-41.1 showed a moderate decrease in host protein synthesis, similar to wild-type HSV-1.
Figure 4. Analysis of infected cell protein synthesis in Hep2 cells infected with recombinant viruses.

Hep2 cells were mock-infected, or infected with wild-type HSV-1, wild-type HSV-2, dl5-29-41.1, dl5-29, or dl5-29-41 at an MOI = 10. Cells were labeled with 35S-methionine and 35S-cysteine at 6.5 and 11.5 hours post-infection and harvested 30 min. later. Proteins in the resulting lysates were resolved by SDS-PAGE, and an autoradiogram of the dried gel is shown.
A more direct test of vhs activity is measurement of host cell mRNA levels; therefore, we determined the levels of cellular GAPDH mRNA by reverse transcriptase-polymerase chain reaction (RT-PCR) assay of RNA from Hep-2 cells at 5 hours post-infection. Cells infected with wild-type HSV-2 strain 186syn+-1 or dl5-29 virus showed an 80% reduction in GAPDH mRNA, while cells infected with wild-type HSV-1 strain KOS virus showed a 50% reduction (Figure 5) while dl5-29-41 showed no decrease in GAPDH mRNA levels. In contrast, the dl5-29-41.1 virus-infected cells showed approximately 65% reduction in GAPDH mRNA levels under these conditions; therefore, the reduction in mRNA was substantially greater in cells infected with dl5-29-41.1 than in cells infected with dl5-29-41. The ability of dl5-29-41.1 to reduce GAPDH mRNA levels seemed to be less than wild-type HSV-2, but greater than wild-type HSV-1, suggesting that HSV-2 specific factors other than vhs may affect the ability of HSV-2 to reduce GAPDH mRNA levels.
Figure 5. Levels of GAPDH gene transcript levels in cells infected with different viral strains.
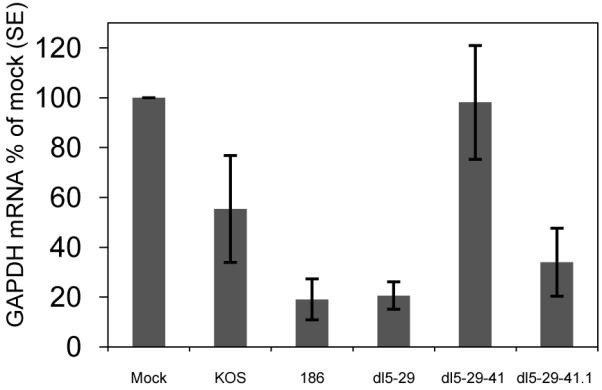
Hep2 cells were mock-infected or infected with dl5-29, dl5-29-41 or dl5-29-41.1 at an MOI of 10. The amounts of GAPDH mRNA in the samples were determined by reverse transcription and real-time PCR (RT-PCR) and normalized to the amounts of 18S rRNA. Average values obtained from 3 experiments are shown.
Gene expression by dl5-29-41.1 virus in non-complementing cells
Comparison of viral cell protein synthesis in 35S-L-methionine- and cysteine-labeled cells infected with the replication-defective dl5-29, dl5-29-41 or dl5-29-41.1 mutant viruses showed similar levels of viral proteins such as ICP5, ICP6 and ICP25 (Figure 4). The viral glycoproteins B (gB) and D (gD) were difficult to measure in the total infected cell profile, in particular gD, so we measured expression of these viral proteins by western blotting (Figure 6).
Figure 6. Levels of accumulation of gB and gD glycoproteins in cells infected with different recombinants.
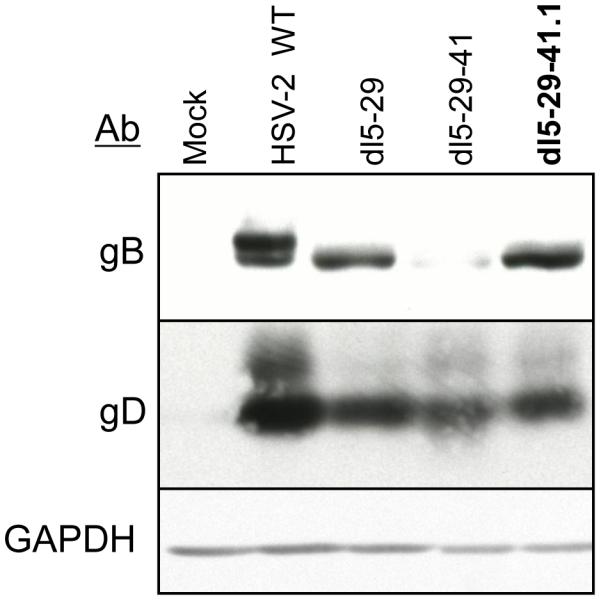
Hep-2 cells were mock infected or infected with dl5-29, dl5-29-41 or dl5-29-41.1 at an MOI = 10 and then harvested at 6 hours post-infection. Protein expression was analyzed by Western blotting with antibodies specific for HSV gB, HSV gD, or cell GAPDH.
Viral surface glycoproteins are key targets for neutralizing antibodies and T-cell responses. Among the 11 known herpes glycoproteins, gB and gD appear to be the most immunodominant [54, 55] and have been tested in subunit vaccines against HSV-2 [12, 15, 16, 56]. Because a T-cell epitope found within gB is highly dominant in C57Bl/6 mice and can be used to assess the CD8+ T-cell responses elicited by HSV infection, we wished to determine the levels of gB and gD expressed by dl5-29-41.1 as compared to the levels expressed by dl5-29 and dl5-29-41. We performed Western blot analysis of Hep-2 cells infected with wild-type HSV-2 or the dl5-29, dl5-29-41 or dl5-29-41.1 mutant viruses. The dl5-29 virus expressed gB at lower levels than wild-type HSV-2, dl5-29-41.1 expressed less gB than dl5-29, and dl5-29-41 expressed substantially less gB than dl5-29-41.1 (Figure 6). All of the mutant viruses expressed gD at similar levels, approximately 50% less than that of wild-type HSV-2. Reduced gB expression was previously observed for dl5-29 when compared to wild-type and single dl5 and dl29 deletion mutants [21]. Therefore, additional mutations seemed to reduce gB expression but did not affect gD expression.
Vhs-1 protein is packaged into the dl5-29-41.1 virion
Vhs is an abundant tegument protein [25], and HSV-1 X HSV-2 recombinants isolated previously suggest that the HSV-1 and HSV-2 vhs proteins are interchangeable [53]. To confirm that vhs-1 protein was incorporated into dl5-29-41.1 virions, we performed western blot analysis of virions from extracellular preparations of virus. We compared the virion proteins of dl5-29-41.-1 virus with equivalent numbers of pfu of dl5-29 and dl5-29-41 viruses. Vhs protein was observed at high levels in dl5-29-41.1 virions (Figure 7). The darker band seen for vhs in dl5-29-41.1 virions could be due to increased amounts of vhs protein in virions or due to preferential recognition by the antibody with the HSV-1 protein. To confirm that we had compared the equal amounts of virions, the samples were also probed for gB, gD and ICP5. Equal amounts of these proteins were observed (Figure 7), indicating that equal amounts of virions had been analyzed. Therefore, vhs-1 protein was assembled in dl5-29-41.1 virions.
Figure 7. Analysis of virion proteins of different recombinant viruses.

Equal amounts of infectious virus (5×105 pfu) from extracellular preparations of dl5-29, dl5-29-41 and dl5-29-41.1 viruses were analyzed by Western blotting for vhs, gB, gD or ICP5.
Immunogenicity Studies
HSV-2-specific antibody responses elicited by immunization with dl5-29-41.1 were similar to those of dl5-29 and dl5-29-41
To measure humoral immune responses, we determined antibody titers at one week after the second immunization using the pooled sera collected from all eight mice of each group. Total IgG responses to HSV-2 were assayed by ELISA, using HSV-2 virions as the antigen. Mice immunized with dl5-29 showed titers of 800 while dl5-29-41 and dl5-29-41.1 gave similar titers of 400 (Table 1). When neutralizing antibody titers were measured, mice immunized with dl5-29-41 showed titers of 256 while dl5-29 and dl5-29-41.1 showed titers of 128 (Table 1). Thus, the viruses elicited similar antibody responses in mice
Table 1. Antibody responses in mice immunized with recombinant viruses.
| Immunogen | Antibody titer |
|
|---|---|---|
| Total IgG | Neutralization | |
| Mock | <100 | <4 |
| dl5-29 | 800 | 128 |
| dl5-29-41 | 400 | 256 |
| dl5-29-41.1 | 400 | 128 |
C57Bl/6 mice were mock-immunized or immunized with dl5-29, dl5-29-41, or dl5-29-41.1 viruses and boosted at 4 weeks. Blood was collected one week after the boost. The sera were pooled from mice of each group. The total HSV-2-specific antibody titers was measured by ELISA, and neutralizing antibody titers were measured by plaque reduction assay against the HSV-2 G strain.
The dl5-29-41.1 virus induces CD8+ T-cell responses to HSV gB-specific epitopes at levels similar to those induced by dl5-29 and dl5-29-41
In HSV-infected C57Bl/6 mice, approximately 70-90% of all HSV-specific CD8+ T-cells recognize a gB epitope, SSIEFARL [54], providing an easily quantifiable assay for the CD8+ T-cell response against HSV. Therefore, we used this epitope to measure and compare the HSV-specific CD8+ T-cell responses elicited in mice immunized with dl5-29-41.1 virus, the dl5-29 double mutant virus, or the dl5-29-41 triple mutant virus. The primary gB-SSIEFARL specific CD8+ T-cell responses were quantified using MHC-I pentamers on days 6 to 8 post-immunization. The peak responses were observed at day 7 in all groups of immunized animals, and similar levels of circulating gB-specific CD8+ T-cells were observed in mice immunized with the three viruses (Figure 8A).
Figure 8. CD8+ T-cell responses in mice immunized with recombinant viruses.

C57Bl/6 mice were immunized with dl5-29, dl5-29-41 or dl5-29-41.1 viruses. (A) Blood samples were collected at days 6 to 8 post-immunization, and CD8+ T-cells were stained with MHC-I pentamers specific for the HSV-2 gB epitope SSIEFARL. The results are shown as the percentage of CD8+ positive cells that are also pentamer-positive (± SEM). (B) Whole splenocytes were collected one week after the boost. The splenocytes were stimulated with the gB-SSEFARL peptide. The results are shown as the percentage of CD8+ positive cells that express IFN-γ after stimulation with gB-SSEFARL peptide (±SEM).
Secondary HSV gB-specific CD8+ T-cell responses were also assayed in splenocytes at one week after the second immunization. The collected splenocytes were stimulated with the gB-specific SSIEFARL peptide, and the percentages of CD8+ T-cells that produced IFN-γ were measured by an intracellular cytokine staining (ICS) assay. Similar to the primary responses, we observed comparable responses among the animals immunized with the three viruses (Figure 8B). Thus, both MHC-I pentamer and ICS assays showed that the three recombinant viruses induced similar HSV gB-specific CD8+ T-cell responses.
dl5-29-41.1 immunization protects mice from vaginal challenge with HSV-2
We investigated the ability of the dl5-29-41.1 virus to elicit protective immunity against HSV-2 infection in a murine model of genital infection. To compare the protection induced by the three viruses, we mock-immunized or immunized Balb/c mice twice, one month apart, by subcutaneous injection in the rear flank with dl5-29, dl5-29-41 or dl5-29-41.1 virus. All groups were challenged with HSV-2 strain G at one month after the second immunization. Mice were then scored for the severity of the induced genital lesions and assayed for levels of viral shedding within the genital tract.
Immunization with dl5-29-41.1 virus reduces viral shedding from the vaginal tract after challenge with HSV-2
Virus shedding was measured in the genital tract from 1 to 5 days after genital challenge. The mock-immunized (control) group showed high levels of virus shedding from days 1 to 5 post-challenge with maximal shedding at day 2 (Figure 9A). In contrast, all three groups of immunized mice showed a significant reduction in virus shedding as compared to the control group from days 1 to 5 (p < 0.05). Similar to the control group, maximal viral shedding for all immunized mice was observed on day 2. Of the groups of immunized mice, the greatest reduction of viral shedding was in animals immunized with dl5-29-41. However, this shedding was not statistically different from the reduction in viral shedding in mice from the groups immunized with dl5-29 (p = 0.36) or dl5-2941.1 (p = 0.18). Thus, immunization with all of the replication-defective viruses tested efficiently reduced viral shedding after challenge with wild-type HSV-2. In particular, dl5-29-41.1 reduced viral shedding as effectively as either dl5-29 or dl5-29-41.
Figure 9. Protection against genital challenge with HSV-2.

Balb/c mice were immunized at weeks 0 and 4 with 105 pfu of dl5-29, dl5-29-41 or dl5-29-41.1 supernatant virus. At 4 weeks after the boost, mice were challenged intravaginally with 50 times the lethal dose50 (50 LD50) of the wild-type HSV-2 G strain virus. (A) Vaginal swabs were collected from day 1 to day 5 after the challenge, and virus titers from the samples were measured by standard plaque assay on Vero cells. (B) Disease progression and survival. Severity of genital disease and progression of encephalitis were monitored from day 4 to 10 post challenge.
Effect of immunization with dl5-29-41.1 on disease progression
To determine if dl5-29-41.1 could protect against disease induce by vaginal challenge with HSV-2, we monitored the progression of clinical disease in mice immunized with dl5-29, dl5-29-41 or dl5-29-41.1 compared to the control group. As observed previously, mock-immunized mice developed genital lesions by day 4, and by day 10 all mice from this group developed severe lesions in the genital tract, followed by hind limb paralysis, as reflected in disease scores (Figure 9B). In contrast, immunization with all of the replication-defective mutant viruses protected mice from genital disease. In immunized mice we observed mild inflammation and erythema of the genital area by day 5 reaching a maximum at day 7; however, all signs of disease were eventually cleared in all mice from the immunized groups and all immunized mice survived the challenge. Most importantly, the dl5-29-41.1 mutant virus protected against disease caused by genital challenge as effectively as either dl5-29 or dl5-29-41.
Discussion
In this study we constructed a modified replication-defective HSV-2 vaccine candidate virus by replacing the HSV-2 virion host shut-off (vhs) gene in the HSV-2 mutant virus dl5-29 with an HSV-1 vhs gene. The new dl5-29-41.1 recombinant virus grows to higher titers than the parental strain in complementing cells and shows nearly the same immunogenicity and protection against HSV-2 challenge as the parental virus or a triple mutant virus, dl5-29-41, containing a deletion within the HSV-2 vhs gene. Although dl5-29-41.1 does not show increased immunogenicity, these results have important implications for the design of future genital herpes vaccine candidates.
Enhanced growth of the dl5-29-41.1 virus
The new recombinant virus, dl5-29-41.1, showed up to 8-fold higher viral yields in the V529 complementing cell line relative to the dl5-29 or the dl5-29-41 viruses. The increased yield of this virus would be highly advantageous for growth of stocks of an HSV-2 vaccine candidate, because the growth kinetics and yields of HSV-2 can be limiting in GMP production [57, 58]. The increased growth of the recombinant with an HSV-1 vhs gene, as compared with the parental strain containing the HSV-2 vhs, argues that the strong HSV-2 vhs activity may actually limit viral yields, while the increased yields relative to dl5-29-41, which expresses no vhs protein, argues that either some vhs activity is helpful to viral growth or that the UL41 protein enhances the formation or stability of viral particles. Geiss et al. [35] did not observe any increased replication of an HSV-1 vhs mutant virus; therefore, the effect that we have observed may be HSV-2 specific.
Gene expression by the new recombinant
Viral gene expression by the dl5-29-41.1 virus was reduced slightly compared to the dl5-29 parental virus, but not as much as the reduction in gene expression observed with the dl5-29-41 virus containing a deletion of the UL41 gene. This argues that either the vhs activity promotes viral gene expression or that the vhs protein possibly enhances early steps in the infectious process such as viral entry or uncoating [4]. Alternatively, vhs has been shown to promote the transition from early to late viral gene expression [26], and this could contribute to the reduced gene expression with the vhs-negative viruses.
In addition, it appeared that introducing sequential mutations in the replication-defective mutant virus had a cumulative negative effect on the level of gB expression, while they had no effect on gD expression. It is unclear at this time why introducing mutations into the UL5, UL29 and UL41 genes affects expression of these two γ1 genes differently. There appear to be subtleties of HSV gene control yet to be elucidated.
Immunogenicity of the new recombinant
The new dl5-29-41.1 recombinant induced HSV gB-specific CD8+ T-cell responses, as measured by pentamer staining and intracellular cytokine expression assays, which were equivalent to those induced by either the dl5-29 virus or the dl5-29-41 virus. This was surprising because other groups have shown that disruption of the UL41 gene increases the immunogenicity of both replication-competent and replication-defective HSV-1 viruses [35, 36] and we have shown previously that disrupting the UL41 gene in the dl5-29 virus by introduction of a β-galactosidase expression cassette increases its immunogenicity [37]. Due to this previous body of work, it was unexpected that the dl5-29 virus and dl5-29-41 viruses would induce CD8+ T-cell responses of similar magnitude, and it was expected that addition of HSV-1 vhs, with significantly less host-shut off activity [24], would most likely give the dl5-29-41.1 virus an intermediate phenotype. The discrepancy between these results and the expected results may be explained by a combination of the differences in glycoprotein B (gB) expression levels, as the CD8+ T-cell responses were measured against a CTL epitope in this protein, and the effects that HSV-1 vhs and HSV-2 vhs have on immunogenicity. A higher level of antigen expression should result in increased CD8+ T-cell responses elicited against an antigen. dl5-29 and dl5-29-41.1 express similar levels of gB (Figure 6), which is comparable to that expressed by dl5-29-41L [37], while dl5-29-41 expresses gB at substantially lower levels (Figure 6). Regardless of the reduced expression levels of gB, dl5-29-41, containing a deletion of HSV-2 vhs, still elicited similar responses to those of dl5-29, with an intact HSV-2 vhs, and dl5-29-41.1, containing the less effective HSV-1 vhs. This suggests that the abrogation of HSV-2 vhs activity can compensate for the reduced viral gene expression levels. Additionally, replacement of HSV-2 vhs with HSV-1 vhs does not substantially increase the ability of the virus to induce a cellular immune response. The increased immunogenicity of strains with reduced vhs activity may be due to 1) increased innate responses in the infected cells; 2) increased survival of infected antigen-presenting cells; and/or 3) increased apoptosis of infected cells, engulfment by dendritic cells and cross-presentation, as proposed previously (Dudek et al., 2008). We conclude that inactivation of the vhs RNase activity is likely needed for increased immunogenicity.
Protection against genital HSV-2 infection
The new dl5-29-41.1 recombinant virus protected mice against challenge with wild-type HSV-2 virus with similar efficacy to that induced by dl5-29 or dl5-29-41. Again, this was surprising, as the dl5-29-41L virus, containing a disruption of the UL41 gene, has previously elicited stronger protection against HSV-2 G strain viral challenge than dl5-29 [37]. The most likely explanation for this result would follow the argument given above to explain the lack of differences seen in mutant viral immunogenicity, i.e. disruption of HSV-2 vhs increases the immunogenicity and protective capacity of a replication-defective HSV-2 virus even in the face of reduced viral gene expression, and while replacement of HSV-2 vhs with HSV-1 vhs does not reduce viral gene expression, it also does not increase the immunogenicity or protective capacity of a virus.
Implications for herpes vaccine design
At this point it is not clear whether the reduction in viral gene expression seen in the dl5-29-41 virus is an implicit result of the abrogation of all HSV vhs activity or an artifact of the recombination event that led to the removal of the vhs gene, as the dl5-29-41L virus does not appear to have a significant reduction in viral gene expression and shows a significantly higher immunogenicity and protective capacity as compared to dl5-29 in mice challenged with the HSV-2 G strain [37], although not against the HSV-2 333 strain in mice or guinea pigs [38].
Regardless, these results argue that the replacement of the HSV-2 vhs with HSV-1 vhs in a replication-defective HSV-2 mutant virus will enhance the growth of the mutant virus in culture, without reducing its immunogenicity or protective capacity. This is a highly desired property, as the growth of HSV-2 viruses in GMP conditions can be limiting [57, 58]. Therefore, a higher yield of virus per cell would allow greater flexibility in manufacturing and immunization protocols for any replication-defective HSV-2 vaccine candidate. Future studies to design new vaccine constructs should attempt to combine the increased immunogenicity observed by knocking out the vhs activity with the increased growth observed here with the HSV-1 vhs protein.
Acknowledgments
This research was supported by National Institutes of Health grant AI057552.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Looker KJ, Garnett GP, Schmid GP. An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. Bull World Health Organ. 2008;86:805–12. doi: 10.2471/BLT.07.046128. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis. 2002;186(Suppl 1):S3–28. doi: 10.1086/343739. [DOI] [PubMed] [Google Scholar]
- [3].Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. Jama. 2006;296:964–73. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- [4].Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Virus. In: Knipe DM, Howley PM, editors. Fields Virology. 5th ed. Lippincott, Williams and Wilkins; Philadelphia: 2007. pp. 2501–602. [Google Scholar]
- [5].Kimberlin DW. Neonatal herpes simplex infection. Clin Microbiol Rev. 2004;17:1–13. doi: 10.1128/CMR.17.1.1-13.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wald A, Link K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis. 2002;185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- [7].Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS. 2006;20:73–83. doi: 10.1097/01.aids.0000198081.09337.a7. [DOI] [PubMed] [Google Scholar]
- [8].Schacker T, Zeh J, Hu H, Shaughnessy M, Corey L. Changes in plasma human immunodeficiency virus type 1 RNA associated with herpes simplex virus reactivation and suppression. J Infectious Diseases. 2002;186:1718–25. doi: 10.1086/345771. [DOI] [PubMed] [Google Scholar]
- [9].Nagot N, Ouedraogo A, Defer MC, Vallo R, Mayaud P, Van de Perre P. Association between bacterial vaginosis and Herpes simplex virus type-2 infection: implications for HIV acquisition studies. Sex Transm Infect. 2007;83:365–8. doi: 10.1136/sti.2007.024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Corey L, Wald A, Patel R, Sacks SL, Tyring SK, Warren T, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med. 2004;350:11–20. doi: 10.1056/NEJMoa035144. [DOI] [PubMed] [Google Scholar]
- [11].Koelle DM, Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clinical Microbiology Reviews. 2003;16:96–113. doi: 10.1128/CMR.16.1.96-113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stanberry LR, Bernstein DI, Burke RL, Pachl C, Myers MG. Vaccination with recombinant herpes simplex virus glycoproteins: protection against initial and recurrent genital herpes. Journal of Infectious Diseases. 1987;155:914–20. doi: 10.1093/infdis/155.5.914. [DOI] [PubMed] [Google Scholar]
- [13].Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- [14].Stanberry LR, Spruance SL, Bernstein DI, Mindel A, Sacks S, Tyring S, et al. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med. 2002;347(21):1652–61. doi: 10.1056/NEJMoa011915. [DOI] [PubMed] [Google Scholar]
- [15].Corey L, Langenberg AG, Ashley R, Sekulovich RE, Izu AE, Douglas JM, Jr., et al. Chiron HSV Vaccine Study Group Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection: two randomized controlled trials. JAMA. 1999;28:331–40. doi: 10.1001/jama.282.4.331. [DOI] [PubMed] [Google Scholar]
- [16].Straus SE, Wald A, Kost RG, et al. Immunotherapy of recurrent genital herpes with recombinant herpes simplex virus type 2 glycoproteins D and B: results of a placebo-controlled vaccine trial. J Infect Dis. 1997;176:1129–34. doi: 10.1086/514103. [DOI] [PubMed] [Google Scholar]
- [17].Loudon PT, Blakeley DM, Boursnell ME, Day DA, Duncan IA, Lowden RC, et al. Preclinical safety testing of DISC-hGMCSF to support phase I clinical trials in cancer patients. J Gene Med. 2001;3:458–67. doi: 10.1002/jgm.206. [DOI] [PubMed] [Google Scholar]
- [18].Morrison LA, Knipe DM. Mechanisms of immunization with a replication-defective mutant of herpes simplex virus 1. Virology. 1996;220:402–13. doi: 10.1006/viro.1996.0328. [DOI] [PubMed] [Google Scholar]
- [19].Morrison LA, Da Costa XJ, Knipe DM. Influence of mucosal and parenteral immunization with a replication-defective mutant of HSV-2 on immune responses and protection from genital challenge. Virology. 1998;243:178–87. doi: 10.1006/viro.1998.9047. [DOI] [PubMed] [Google Scholar]
- [20].Da Costa XJ, Bourne N, Stanberry LR, Knipe DM. Construction and characterization of a replication-defective herpes simplex virus 2 ICP8 mutant strain and its use in immunization studies in a guinea pig model of genital herpes. Virology. 1997;232:1–12. doi: 10.1006/viro.1997.8564. [DOI] [PubMed] [Google Scholar]
- [21].Da Costa XJ, Kramer MF, Zhu J, Brockman MA, Knipe DM. Construction, phenotypic analysis, and immunogenicity of a UL5/UL29 double deletion mutant of herpes simplex virus 2. J Virol. 2000;74:7963–71. doi: 10.1128/jvi.74.17.7963-7971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Da Costa XJ, Jones CA, Knipe DM. Immunization against genital herpes with a vaccine virus that has defects in productive and latent infection. Proc Natl Acad Sci USA. 1999;96:6994–98. doi: 10.1073/pnas.96.12.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].van Lint AL, Torres-Lopez E, Knipe DM. Immunization with a replication-defective herpes simplex virus 2 mutant reduces herpes simplex virus 1 infection and prevents ocular disease. Virology. 2007;368:227–31. doi: 10.1016/j.virol.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Everly DN, Jr., Feng P, Mian IS, Read GS. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. Journal of Virology. 2002;76:8560–71. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Read GS, Karr BM, Knight K. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J Virol. 1993;67:7149–60. doi: 10.1128/jvi.67.12.7149-7160.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oroskar AA, Read GS. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J Virol. 1989;63:1897–906. doi: 10.1128/jvi.63.5.1897-1906.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, et al. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO Journal. 1996;15:2575–81. [PMC free article] [PubMed] [Google Scholar]
- [28].Taddeo B, Sciortino MT, Zhang W, Roizman B. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc Natl Acad Sci U S A. 2007;104:12163–8. doi: 10.1073/pnas.0705245104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Strelow LI, Leib DA. Analysis of conserved domains of UL41 of herpes simplex virus type 1 in virion host shutoff and pathogenesis. J Virol. 1996;70(8):5665–7. doi: 10.1128/jvi.70.8.5665-5667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Strelow LI, Leib DA. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol. 1995;69:6779–86. doi: 10.1128/jvi.69.11.6779-6786.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smiley JR. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol. 2004;78:1063–8. doi: 10.1128/JVI.78.3.1063-1068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jones CA, Fernandez M, Herc K, Bosnjak L, Miranda-Saksena M, Boadle RA, et al. Herpes simplex virus type 2 induces rapid cell death and functional impairment of murine dendritic cells in vitro. J Virol. 2003;77:11139–49. doi: 10.1128/JVI.77.20.11139-11149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Samady L, Costigliola E, MacCormac L, McGrath Y, Cleverley S, Lilley CE, et al. Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: Potential of vhs(−) HSV vectors for dendritic cell-mediated immunotherapy. J Virol. 2003;77:3768–76. doi: 10.1128/JVI.77.6.3768-3776.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Barzilai A, Zivony-Elbom I, Sarid R, Noah E, Frenkel N. The herpes simplex virus type 1 vhs-UL41 gene secures viral replication by temporarily evading apoptotic cellular response to infection: Vhs-UL41 activity might require interactions with elements of cellular mRNA degradation machinery. J Virol. 2006;80:505–13. doi: 10.1128/JVI.80.1.505-513.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Geiss BJ, Smith TJ, Leib DA, Morrison LA. Disruption of virion host shutoff activity improves the immunogenicity and protective capacity of a replication-incompetent herpes simplex virus type 1 vaccine strain. J Virol. 2000;74:11137–44. doi: 10.1128/jvi.74.23.11137-11144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Walker J, Leib DA. Protection from primary infection and establishment of latency by vaccination with a herpes simplex virus type 1 recombinant deficient in the virion host shutoff (vhs) function. Vaccine. 1998;16:1–5. doi: 10.1016/s0264-410x(97)00164-3. [DOI] [PubMed] [Google Scholar]
- [37].Dudek T, Mathews LC, Knipe DM. Disruption of the U(L)41 gene in the herpes simplex virus 2 dl5-29 mutant increases its immunogenicity and protective capacity in a murine model of genital herpes. Virology. 2008;372:165–75. doi: 10.1016/j.virol.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hoshino Y, Pesnicak L, Dowdell KC, Lacayo J, Dudek T, Knipe DM, et al. Comparison of immunogenicity and protective efficacy of genital herpes vaccine candidates herpes simplex virus 2 dl5-29 and dl5-29-41L in mice and guinea pigs. Vaccine. 2008;26:4034–40. doi: 10.1016/j.vaccine.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Everly DN, Jr., Read GS. Mutational analysis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): characterization of HSV type 1 (HSV-1)/HSV-2 chimeras. J Virol. 1997;71(10):7157–66. doi: 10.1128/jvi.71.10.7157-7166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ejercito PM, Kieff ED, Roizman B. Characterization of herpes simplex virus strains differing in their effect on social behavior of infected cells. Journal of General Virology. 1968;2:357–64. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- [41].Spang AE, Godowski PJ, Knipe DM. Characterization of herpes simplex virus 2 temperature-sensitive mutants whose lesions map in or near the coding sequences for the major DNA-binding protein. J Virol. 1983;45:332–42. doi: 10.1128/jvi.45.1.332-342.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goldin AL, Sandri-Goldin RM, Levine M, Glorioso JC. Cloning of herpes simplex virus type 1 sequences representing the whole genome. J Virol. 1981;38:50–58. doi: 10.1128/jvi.38.1.50-58.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, et al. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. Journal of General Virology. 1988;69:1531–74. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- [44].Dolan A, Jamieson FE, Cunnigham C, Barnett BC, MdGeogh DJ. The genome sequence of herpes simplex virus type 2. J Virol. 1998;72:2010–21. doi: 10.1128/jvi.72.3.2010-2021.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Knipe DM, Senechek D, Rice SA, Smith JL. Stages in the nuclear association of the herpes simplex virus transcriptional activator protein ICP4. J Virol. 1987;61:276–84. doi: 10.1128/jvi.61.2.276-284.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Simpson-Holley M, Baines J, Roller R, Knipe DM. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J Virol. 2004;78:5591–600. doi: 10.1128/JVI.78.11.5591-5600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Knipe DM, Quinlan MP, Spang AE. Characterization of two conformational forms of the major DNA-binding protein encoded by herpes simplex virus 1. J Virol. 1982;44:736–41. doi: 10.1128/jvi.44.2.736-741.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Knipe DM, Spang AE. Definition of a series of stages in the association of two herpesviral proteins with the cell nucleus. J Virol. 1982;43:314–24. doi: 10.1128/jvi.43.1.314-324.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Morrison LA, Knipe DM. Immunization with replication-defective mutants of herpes simplex virus type 1: Sites of immune intervention in pathogenesis of challenge virus infection. J Virol. 1994;68:689–96. doi: 10.1128/jvi.68.2.689-696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Korom M, Wylie KM, Morrison L,A. Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice. J Virol. 2008;82:3642–53. doi: 10.1128/JVI.02409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hoshino Y, Dalai SK, Wang K, Pesnicak L, Lau TY, Knipe DM, et al. Comparative efficacy and immunogenicity of replication-defective, recombinant glycoprotein, and DNA vaccines for herpes simplex virus 2 infections in mice and guinea pigs. J Virol. 2005;79:410–8. doi: 10.1128/JVI.79.1.410-418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Morse LS, Pereira L, Roizman B, Schaffer PA. Anatomy of herpes simplex virus (HSV) DNA. X. Mapping of viral genes by analysis of polypeptides and functions specified by HSV-1 x HSV-2 recombinants. J Virol. 1978;26:389–410. doi: 10.1128/jvi.26.2.389-410.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wallace ME, Keating R, Heath WR, Carbone FR. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J Virol. 1999;73:7619–26. doi: 10.1128/jvi.73.9.7619-7626.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cooper D, Mester JC, Guo M, Nasar F, Souza V, Dispoto S, et al. Epitope mapping of full-length glycoprotein D from HSV-2 reveals a novel CD4+ CTL epitope located at the transmembrane-cytoplasmic junction. Cell Immunol. 2006;239(2):113–20. doi: 10.1016/j.cellimm.2006.04.005. [DOI] [PubMed] [Google Scholar]
- [56].Bernstein D. Glycoprotein D adjuvant herpes simplex virus vaccine. Expert Rev Vaccines. 2005;4:615–27. doi: 10.1586/14760584.4.5.615. [DOI] [PubMed] [Google Scholar]
- [57].O’Keeffe R, Johnson MD, Slater NKH. The primary production of an infectious recombinant herpes simplex virus vaccine. Biotechnology & Bioengineering. 1998;57:262–71. doi: 10.1002/(sici)1097-0290(19980205)57:3<262::aid-bit2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- [58].O’Keeffe R, Johnson MD, Slater NKH. The affinity adsorptive recovery of an infectious herpes simplex virus vaccine. Biotechnology & Bioengineering. 1999;62:537–45. doi: 10.1002/(sici)1097-0290(19990305)62:5<537::aid-bit5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]