

Abstract
This review focuses on the emerging evidence that reactive oxygen species (ROS) derived from glucose metabolism, such as H2O2, act as metabolic signaling molecules for glucose-stimulated insulin secretion (GSIS) in pancreatic beta-cells. Particular emphasis is placed on the potential inhibitory role of endogenous antioxidants, which rise in response to oxidative stress, in glucose-triggered ROS and GSIS. We propose that cellular adaptive response to oxidative stress challenge, such as nuclear factor E2-related factor 2 (Nrf2)-mediated antioxidant induction, plays paradoxical roles in pancreatic beta-cell function. On the one hand, induction of antioxidant enzymes protects beta-cells from oxidative damage and possible cell death, thus minimizing oxidative damage-related impairment of insulin secretion. On the other hand, the induction of antioxidant enzymes by Nrf2 activation blunts glucose-triggered ROS signaling, thus resulting in reduced GSIS. These two premises are potentially relevant to impairment of beta-cells occurring in the late and early stage of Type 2 diabetes, respectively. In addition, we summarized our recent findings that persistent oxidative stress due to absence of uncoupling protein 2 activates cellular adaptive response which is associated with impaired pancreatic beta-cell function.
Keywords: ROS, oxidative stress, antioxidant, Nrf2, pancreatic beta-cells, insulin secretion, Ucp2
1. Introduction
Type 2 diabetes (T2D) has become a serious public health problem in the world. Approximately 150 million people worldwide had T2D in the year 2000, with the prediction that this number could double by 2025 (Zimmet et al., 2001). The medical and socioeconomic burdens of the disease caused by its associated complications, impose enormous strains on health-care systems and economic wealth (Stumvoll et al., 2005). Although the primary causes of T2D are unknown, insulin resistance plays an early role in its pathogenesis and defective glucose-stimulated insulin secretion (GSIS) from pancreatic beta-cells is instrumental in the progression to hyperglycemia (Fridlyand and Philipson, 2004; Krauss et al., 2005). GSIS is regulated by the rate of glucose metabolism within beta-cells. Following glucose uptake and phosphorylation, glucose oxidation involves both cytosolic and mitochondrial processes that generate signals leading to insulin secretion (Newgard and McGarry, 1995; Henquin, 2004; Jensen et al., 2008). Nevertheless, the precise spectrum of signals that couples glucose catabolism to insulin secretion are still incompletely understood. Previous studies (Bindokas et al., 2003; Armann et al., 2007; Leloup et al., 2009; Morgan et al., 2009) including our own (Pi et al., 2007) suggest that reactive oxygen species (ROS), such as H2O2, derived from glucose metabolism serve as one of the metabolic signals for GSIS. However, endogenous antioxidant enzymes that can be robustly induced in response to exposure to oxidative stressor have the potential to blunt such a glucose-triggered ROS signal and thus inhibit GSIS (Pi et al., 2007). This review focuses on the emerging evidence that ROS derived from glucose metabolism may function as metabolic signaling molecules for GSIS. Particular emphasis is placed on the potential inhibitory role of endogenous antioxidants, which rise in response to oxidative challenges, in glucose-triggered ROS and GSIS.
2. Metabolic signaling in GSIS
GSIS is regulated by the rate of glucose metabolism within beta-cells. Following its initial uptake and phosphorylation, glucose metabolism involves both cytosolic and mitochondrial processes and generates signals leading to insulin secretion (Newgard and McGarry, 1995; Jensen et al., 2008). It has become established in the field that glycolytic and oxidative processes leading to an increased ATP/ADP ratio are key transduction events in beta-cell signaling (Krauss et al., 2005). However, the precise signals that couple glucose catabolism to insulin secretion are still incompletely understood. The consensus model explaining how glucose generates a triggering signal in beta-cells involves the following sequence of events: entry of glucose by facilitated diffusion, metabolism by oxidative glycolysis, rise in ATP/ADP ratio, closure of KATP channels, depolarization of the plasma membrane potential, opening of voltage-operated Ca2+ channels, influx of Ca2+, rise in cytosolic Ca2+ and activation of exocytotic machinery (Henquin, 2000; Wollheim and Maechler, 2002). Over the past few years, many novel players have been implicated in the control of insulin secretion. This extensive collection includes glutamate (Maechler and Wollheim, 1999; Lehtihet et al., 2005), protein phosphatases (Ostenson et al., 2002; Sjoholm et al., 2002; Lehtihet et al., 2005; Pagliarini et al., 2005), GTP (Sjoholm et al., 2002), GTP-binding proteins (Kowluru, 2003), cGMP (Soria et al., 2004), cAMP-guanosine nucleotide exchange factor II (Ozaki et al., 2000), diadenosine polyphosphates (Soria et al., 2004), NADH shuttle (Eto et al., 1999), transcription factor NF-κB (Norlin et al., 2005), protein kinases including cyclin-dependent kinase 5 (Wei et al., 2005), cAMP-dependent protein kinase (Wollheim and Maechler, 2002), PKC (Jones and Persaud, 1998) and cyclic pathways of pyruvate metabolism (Jensen et al., 2008). Thus, there is no shortage of potential players, although the precise nature by which glucose controls insulin secretion remains elusive.
3. ROS: overlooked signaling molecules in pancreatic beta-cells?
ROS such as superoxide anion (O2·−) and H2O2 are produced in aerobic cells either during mitochondrial electron transport or by several oxidoreductases and metal-catalyzed oxidation of metabolites (Forman and Torres, 2002). For many years ROS have been exclusively thought of as the unfortunate byproducts of respiratory energy production in mitochondria and believed to be deleterious to biological systems (Finkel, 1998). However, ROS generation is not always a useless or harmful process but, rather, may be an essential element required for many biological responses (Finkel, 2003; Buetler et al., 2004). Small variations of intracellular ROS have been shown to modulate many physiological processes including redox-dependent transcriptional regulation (Arrigo, 1999; Liu et al., 2005), ion transport (Kourie, 1998), as well as protein phosphorylation (Finkel, 1998; Rhee et al., 2005).
In most cells, ROS can be produced in the presence of high concentrations of glucose via multiple mechanisms. These include oxidative phosphorylation, glucose auto-oxidation, the Shiff reaction during glycation, PKC activation, methylglyoxal formation, and hexosamine metabolism (Brownlee, 2001; Kowluru, 2003). Mitochondria, under non-stress conditions, are one of the major sources of ROS in most cells (Turrens, 2003; Brand et al., 2004). The main sites of ·O2− generation are in the inner mitochondrial membrane: NADH dehydrogenase at complex I, and the interface between ubiquinone and complex III (Nishikawa et al., 2000; Brand et al., 2004). During the process of cellular respiration, there are at least three events that can lead to dramatically increased ·O2− generation in beta-cells (Fridlyand and Philipson, 2004) - increased glycolytic flux, decreased ADP concentration, and increased intracellular Ca2+ concentration.
·O2−, the parental form of intracellular ROS, is a very reactive molecule but it can be converted to H2O2 by superoxide dismutase (SOD) isoenzymes, and then to oxygen and water by several enzymes including catalase (CAT), glutathione peroxidase (GPx) and peroxiredoxin (Prx). Pancreatic beta-cells are equipped with ·O2−-inactivating SODs in the cytosol and mitochondria at levels of about 50% of those in the liver (Lenzen et al., 1996; Tiedge et al., 1997). However, the expression levels of the H2O2-inactivating enzymes GPx and CAT are extremely low in islets, at levels of only about 1% of those in the liver (Lenzen et al., 1996; Tiedge et al., 1997). This reduced antioxidant capacity potentially makes pancreatic beta-cells sensitive to ROS–mediated signal transduction and cellular response.
H2O2 is a small, uncharged, freely diffusible molecule that can be synthesized and destroyed rapidly in response to external stimuli (Rhee et al., 2003; Rhee, 2006). These features meet all of the important criteria for an intracellular messenger. Indeed, a growing body of evidence supports the notion that H2O2 is a ubiquitous intracellular messenger (Rhee, 2006). To date, many important signal transduction molecules or processes have been recognized as downstream targets of ROS, and H2O2 in particular. These molecules and processes include Ca2+-dependent protein phosphatases (Kamsler and Segal, 2004), protein tyrosine phosphatases (Denu and Tanner, 1998; Pagliarini et al., 2005), voltage-gated K+ channels (Archer et al., 2004), Ca2+ influx and release (Krippeit-Drews et al., 1995; Todt et al., 2001; Kraft et al., 2004; Tabet et al., 2004), tumor suppressor phosphatase PTEN (Rhee et al., 2005), c-Jun N-terminal kinase (Nemoto et al., 2000), extracellular signal-regulated kinases (Aikawa et al., 1997), NF-κB (Schmidt et al., 1996), and SIRT1 deacetylase (Brunet et al., 2004; Moynihan et al.,2005). Many of these molecules may potentially regulate insulin secretion. It should be noted that the gradient of H2O2 in cells is very steep, as evidenced by microscopic measurements in human keratinocytes (Pi et al., 2003) and mouse islets (Pi et al., 2007; Pi, 2009), so that all signaling by this molecule would require the target to be in very close proximity to the source.
ROS generation occurs in glucose metabolism and correlates with insulin secretion. Nevertheless, relatively little attention has been paid to the potential role of ROS in glucose signaling and insulin secretion. In 1999, Wollheim’s group found, that H2O2 and alloxan, which acutely increased intracellular H2O2, caused a rapid elevation of intracellular Ca2+ and briefly increased insulin release in rat islets at basal, nonstimulatory glucose concentrations (Janjic et al., 1999; Maechler et al., 1999). Consistent with these observations, short-term exposure of isolated rat islets to alloxan and xanthine oxidase/hypoxanthine, a well known ·O2− generation system, led to a temporarily elevated insulin release (Ebelt et al., 2000). In addition, attenuation of expression of glutamate-cysteine ligase (GCL), which reduced GSH synthesis and thus potentially raised intracellular ROS, enhanced insulin secretion in MIN6 cells (Kondo et al., 2000). In our recent studies (Pi et al., 2007), exogenous H2O2 as well as diethyl maleate, which enhanced intracellular H2O2 levels, increased insulin secretion in INS-1 (832/13) cells and isolated mouse islets, whereas high concentration of exogenous antioxidants inhibited the GSIS in these in vitro models. Support for the notion that ROS mediate glucose action also comes from Leloup et al. who reported that mitochondrial ROS are required for hypothalamic glucose sensing (Leloup et al., 2006) and GSIS (Leloup et al., 2009). More recently, we determined the acute effects (30-min challenge) of various chemicals, including oxidants and antioxidants, on insulin secretion using INS-1 (832/13) cells (Table 1). Low concentrations (without significant cytoxicity by 24-hr exposure) of oxidants, which increase intracellular H2O2 level, such as H2O2 solution and diethyl maleate (Pi et al., 2007), hypochlorous acid (Pi et al., 2008) and alloxan (Janjic et al., 1999; Maechler et al., 1999), facilitated insulin secretion, whereas antioxidants, including N-acetyl-cysteine, catalase, cell permeable glutathione, DL-dithiothreitol and reduced α-lipoic acid, blunted insulin secretion (Table 1). Interestingly, inhibiting the mitochondrial electron transport chain by various agents such as rotenone, antimycin A, oligomycin, which increase mitochondria-derived ROS but inhibit ATP generation, led to decreased GSIS. These results show that in beta-cells the glucose-induced rise in ROS alone cannot promote insulin release, and are consistent with the wealth of literature showing that glucose-induced elevations in ATP are necessary for GSIS (Henquin, 2000; Wollheim and Maechler, 2002). Since influx of Ca2+ is a major event to activate the insulin exocytotic machinery in the early phase of GSIS, the Ca2+-dependence of H2O2- stimulated insulin secretion was investigated in our previous studies (Pi et al., 2007). Our results indicate that H2O2-stimulated insulin secretion is an extracellular Ca2+-dependent process, suggesting H2O2 may be involved in Ca2+ influx. In contrast, the evidence that exogenous antioxidants marginally inhibit KCl-induced insulin release could represent either a small nonspecific effect on secretion, or indicate that H2O2 is also involved in some aspect of secretion after Ca2+ influx (Pi et al., 2007). Taken together, these studies suggest that ROS derived from glucose metabolism are potential metabolic signals that facilitate insulin secretion.
Table 1.
Acute effects of oxidants, antioxidants and other agents on insulin secretion in INS-1 (832/13) cells *
| Agents | Function | Concentrations | Insulin secretion |
|
|---|---|---|---|---|
| 3 mM glucose | 20 mM glucose | |||
| ROS or oxidants: | ||||
| H2O2 | 1–100 μM | ↑ | ↑ | |
| Hypochlorous acid | 10–100 μM | ↑ | ↑ | |
| SIN-1 | NO/ONOO− donor | 500 μM | – | ↑ |
| S-nitrosoglutathione | NO donor | 100–500 μM | – | ↑ |
| 4-hydroxy-2-nonenal | lipid peroxide | 10 μM | – | ND |
| Diethyl maleate (DEM) | GSH depletor | 0.1–5 mM | ↑ | ↑ |
| Antioxidants: | ||||
| N-acetyl-cysteine | thiol | 100–400 μM | ↓ | ↓ |
| Catalase (CAT) | metabolizes H2O2 | 5K–10K U/ml | ↓ | ↓ |
| PEG-catalase | Cell permeable CAT | 1K–5K U/ml | – | ↓ |
| PEG-SOD | Cell permeable SOD | 10–30 U/ml | – | – |
| GSH-EE | Cell permeable GSH | 1 mM | ↓ | ↓ |
| DL-dithiothreitol | bithiols | 100–500 μM | ↓ | ↓ |
| α-Lipoic acid (reduced) | bithiols | 1 mM | ↓ | ↓ |
| α-Lipoic acid (oxidized) | 1 mM | – | – | |
| Others: | ||||
| Alloxan | 10–100 μM | ↑ | – | |
| tert-Butylhydroquinone | Nrf2 activator | 10 μM | – | – |
| Rotenone | Complex I inhibitor | 0.1–1 μM | – | ↓ |
| Antimycin A | Complex III inhibitor | 2 μM | – | ↓ |
| Sodium azide | heme-containing peroxidases | 0.6–1 mM | ↓ | ↓ |
| Mercaptosuccinic acid | GPx inhibitor | 100 μM | – | – |
| Carboxymethoxylamine | 100 μM | – | ↓ | |
| Doxorubincin | ·O−2 producer | 2 μM | – | ↓ |
| CCCP | Mitochondria uncoupler | 200 μM | – | ↓ |
| α-CHCAa | 250 μM | – | – | |
| Oligomycinb | 2 μg/ml | – | ↓ | |
| Phenylarsine oxidec | 1 μM | – | ↓ | |
| Tolbutamided | 0.5 mM | ↑ | – | |
| KCl | Open voltage-gated Ca2+ channels | 30 mM | ↑ | ↑ |
Insulin secretion was determined as described previously (Pi et al., 2007). ↑, increased; ↓, decreased; ND, not detected.
SIN-1, 3-morpholinosydnonimine hydrochloride; PEG-Catalase, catalase-polyethylene glycol; PEG-SOD, superoxide dismutase-polyethylene glycol; GSH-EE, glutathione ethyl ester; CCCP, Carbonyl cyanide m-chlorophenylhydrazone;
α-cyano-4-hydroxycinnamic acid (α-CHCA), inhibits glycolysis-derived pyruvate transport into mitochondria;
inhibits mitochondrial ATPase and phosphoryl group transfer;
complexes vicinal thiols and inhibits phosphotyrosine phosphatase;
closes KATP channels and elicits electrical activity.
4. Oxidative stress, Nrf2-mediated antioxidant response and ROS signaling
ROS clearly possess the capacity to behave in a sporadic and destructive fashion (Finkel, 2003). Persistent elevation of ROS resulted from an imbalance between ROS production and scavenging by endogenous antioxidants can directly or indirectly disturb physiological functions of many cellular macromolecules such as DNA, protein, and lipids, and activate cellular stress-sensitive signaling pathways (Droge, 2002; Evans et al., 2003). Having evolved in an oxygen environment, most cells, including beta-cells, have acquired intricate mechanisms to defend against ROS toxicity. Among them, induction of a family of antioxidant/detoxification enzymes that enhance cellular ROS-scavenging capacity is a key element in the maintenance of cellular redox homeostasis and in reducing oxidative damage (Wasserman and Fahl, 1997; Nguyen et al., 2003; Itoh et al., 2004). These enzymes include NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO1), GCL, glutathione reductase (GR), glutathione synthetase (GS), GPx, CAT and many others (Wasserman and Fahl, 1997; Nguyen et al., 2003; Itoh et al., 2004). These antioxidant genes are coordinately regulated through consensus cis-elements called antioxidant response elements (AREs) in their 5′-flanking promoter regions (Nguyen et al., 2003). Transcription factor Nrf2, a member of the Cap ’n’ Collar family of bZIP proteins, is a central regulator in both constitutive and inducible ARE-related gene expression (Itoh et al., 2004) and innate immune response (Thimmulappa et al., 2006). Nrf2 knockout mice show a deficiency in this coordinated gene regulatory program and have a higher susceptibility to oxidative damage and chemical carcinogenesis (Chan and Kan, 1999; Chan et al., 2001; Ramos-Gomez et al., 2001).
Thus, the Nrf2-mediated antioxidant response represents a critically important cellular defense mechanism that serves to maintain intracellular redox homeostasis and limit oxidative damage (Nguyen et al., 2003; Cullinan and Diehl, 2004; Itoh et al., 2004). In spite of this protective role, antioxidant enzymes may function as an off switch for the signal transduction pathways mediated by ROS (Forman and Torres, 2002). Thus the Nrf2-mediated antioxidant response has a potential to be “too much of a good thing”, producing undesirable effects. It could dampen normal ROS/redox signaling that could be triggered, for instance, by glucose oxidation. Therefore, we propose that Nrf2-mediated antioxidant response plays a paradoxical role in insulin secretion (Fig. 1). On the one hand, it protects beta-cells from oxidative damage and possible cell death, thus minimizing oxidative damage-related impairment of insulin secretion. On the other hand, situations leading to chronic induction of endogenous antioxidants due to oxidative stress may blunt endogenous ROS signaling, resulting in reduced GSIS.
FIG. 1. Proposed mechanisms for oxidative stress-induced impairment of pancreatic beta-cells.
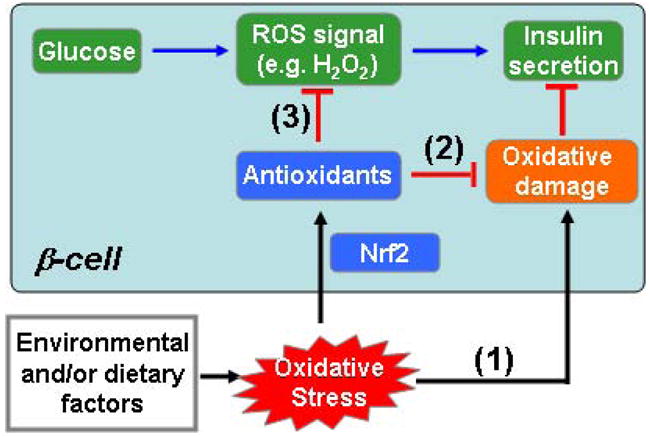
(1) Environmental and/or dietary factors-induced oxidative stress might result in oxidative damage and directly diminish beta-cell function; (2) Oxidative stress activates the Nrf2-mediated antioxidant response and thus protects cells from oxidative damage; (3) Enhanced antioxidant capacity by Nrf2 activation could negatively affect glucose-derived ROS signaling that contributes to GSIS.
To understand the quantitative nature of ROS signaling, we resorted to an ordinary differential equation-based model to describe the Nrf2-mediated adaptive response to oxidative stress (Zhang and Andersen, 2007; Zhang et al., 2008). We investigated in silico whether pre-existing conditions of oxidative stressor might interfere with subsequent ROS signaling, as triggered, for instance, by glucose. Our simulation indicates that in the absence of oxidative challenge, a stimulus mimicking glucose treatment causes a large spike in intracellular H2O2 concentration. However, when the cell is under continuous oxidative challenge, the same glucose-mimicking stimulus triggers a much smaller increase in H2O2 (Pi et al., 2007). These in silico results demonstrate that by adaptively upregulating overall antioxidant capacity chronic oxidative stress is expected to blunt acute ROS signaling.
Thus, we envisage the following scenarios for chronic oxidative stress-induced impairment of beta-cell function. Under low-level oxidative stress (without cytoxicity), beta-cells will adapt to the condition adequately, by activating cellular adaptive response, such as the Nrf2-ARE system, thereby keeping oxidative damage/cell death-related impairment of GSIS at a minimum. However, persistent induction of endogenous antioxidants would attenuate glucose-dependent ROS signaling leading to reduced GSIS. In contrast, under high-level oxidative challenge, the Nrf2 system may be overwhelmed and unable to protect the cells from oxidative damage and/or death. In this situation, excessive oxidative damage and possible cell death become the primary cause for the impaired GSIS. These two premises are potentially relevant to impairment of beta-cells occurring in the early and late stage of Type 2 diabetes, respectively.
5. New findings on Ucp2 and endogenous ROS production in beta-cell function
Mitochondria are one of the primary sources of ROS under basal conditions (Turrens, 2003; Brand et al., 2004). The ·O2− production from the mitochondrial matrix is very sensitive to the proton motive force (Jezek et al., 2004), so mild uncoupling can substantially decrease mitochondria-derived ROS and is believed to aid in preventing oxidative damage (Miwa and Brand, 2003; Brand et al., 2004; Jezek et al., 2004). Ucp2 is a widely expressed mitochondrial inner membrane carrier protein that was discovered through its homology to the brown fat Ucp1, which dissipates caloric energy in the form of heat by uncoupling mitochondrial respiration from ATP production (Ricquier and Bouillaud, 2000). Ucp2 is not a physiologically relevant “uncoupling protein” as is Ucp1 and does not contribute to adaptive thermogenesis (Krauss et al., 2002; Rousset et al., 2004; Brand and Esteves, 2005). However, there is evidence that UCP2 increases the proton conductance of the mitochondrial inner membrane when it is activated by ·O2− (Echtay et al., 2002a; Echtay et al., 2002b) and/or free radical-derived alkenals such as 4-hydroxy-2-nonenal (HNE) (Echtay et al., 2003; Brand and Esteves, 2005).
Since mitochondria are one of the major sources of ROS in cells and the production of ·O2− is sensitive to the uncoupling of respiration, the Ucp2-mediated feedback regulation on ROS generation may be considered as a compensatory mechanism alleviating oxidative stress (Ruzicka et al., 2005). Consistent with this idea, Ucp2-ablated mice exhibit increased ROS production in macrophages and have a higher susceptibility to ROS-induced damage (Arsenijevic et al., 2000; Blanc et al., 2003; Krauss et al., 2003; Horimoto et al., 2004; Bai et al., 2005). In contrast, overexpression of Ucp2 protects cells from oxidative damage (Li et al., 2001; Kizaki et al., 2002; Diano et al., 2003; Ryu et al., 2004). Furthermore, as with most antioxidant responses, the transcription of the Ucp2 gene is highly inducible under oxidative stress conditions. For instance, the treatments by H2O2 (Li et al., 2001), lipopolysaccharide (Pecqueur et al., 2001; Ruzicka et al., 2005), TNFa (Lee et al., 1999), free fatty acids (Chevillotte et al., 2001; Medvedev et al., 2001; Medvedev et al., 2002), and irradiation (Voehringer et al., 2000) increase Ucp2 expression in vitro and in vivo. Thus, there is good evidence that a major function of Ucp2 is to attenuate mitochondrial production of ROS and activation/induction of Ucp2 may function as an alternate adaptive response to mitochondria-derived ROS generation and oxidative damage.
The first report of Ucp2−/− mice from Collins and colleagues described a phenotype of exaggerated ROS and phagocytosis in macrophages (Arsenijevic et al., 2000). Another report of Ucp2−/− mice showed dramatically enhanced GSIS in Ucp2−/− mice (Zhang et al., 2001). This finding in beta-cells of Ucp2−/− mice was replicated by Collins and Corkey (Pi, 2009). Of note, all studies on Ucp2−/− mice during that time were conducted with mice that were a C57BL/6J and 129 mixed background. However, after the mice were backcrossed onto one of three strains of inbred mice (C57BL/6J, A/J, or 129/SvImJ) for 8–20 generations to eliminate the potential influence of genetic background on their phenotypes, a significantly decreased GSIS in islets was observed in the Ucp2−/− mice (Pi, 2009). The consistent observation of diminished GSIS from three highly congenic lines are highly suggestive that decreased GSIS of islets is the phenotype that results from absence of Ucp2.
Extensive studies have indicated that Ucp2 negatively regulates mitochondria-derived ROS generation (Arsenijevic et al., 2000; Bai et al., 2005). Thus, elimination of Ucp2 in vivo by targeted gene “knockout” has a potential to promote oxidative stress and activate adaptive antioxidant response. Consistent with this hypothesis, the expression of a broad array of antioxidant enzymes including SOD2, GPx2, GCLC, GS, CAT and HO-1 was significantly elevated in the islets of Ucp2−/− mice (Pi, 2009). Importantly, despite enhanced basal H2O2 levels in Ucp2−/− islets, glucose-stimulated ROS production was markedly lower than in Ucp2+/+ mice. Together with our previous results (Arsenijevic et al., 2000; Bai et al., 2005), these findings provide evidence to suggest that chronic absence of Ucp2 results in persistent oxidative stress and thereby activation of the antioxidant defense system, which likely blunt GSIS in islets.
6. Antioxidants as therapeutics in treating T2D: beneficial or harmful?
In contrast to the prevailing detrimental view of ROS in many cellular responses and disease states, including T2D, elevated levels of antioxidant molecules have been observed in ob/ob mice (Nakao et al., 2000), subjects at risk of T2D (Costa et al., 2002) and diabetic patients (Chen et al., 2003; Chen et al., 2004). Thus, induction of endogenous antioxidant enzymes may have a role during the development of T2D. More importantly, overexpression of H2O2-scavenging enzymes, such as CAT and Gpx1, actually increased the likelihood of developing diabetes (McClung et al., 2004; Li et al., 2006). In humans, the clinical outcomes of antioxidant therapy in the treatment of T2D remain very inconclusive (Wiernsperger, 2003; Scott and King, 2004). A recent report even showed that use of antioxidants actually increased the incidence of all-cause death (Bjelakovic et al., 2007). These results have cast doubt on the usefulness of antioxidant supplements, and possibly on the fundamental concept that enhancing antioxidant capacity to counteract ROS benefits T2D patients.
In the general population, many people take antioxidant supplements, such as vitamin C and E, and beta-carotene, believing these agents will be beneficial to their general health and prevent diseases. In fact, there are a number of clinical studies that have failed to find any benefit of such antioxidant vitamin supplement (Bjelakovic et al., 2007). Our hypotheses raise a serious concern about antioxidant supplements, at least in the context of T2D. Given that ROS signaling is attenuated by antioxidants, we cannot rule out the possibility that the increasing incidence of T2D over the decades could be due, at least in part, to our self-prescribed preventive measures. For T2D patients, taking antioxidant supplements may even exacerbate their diseased conditions because of the further dampening of ROS signaling by exogenous antioxidants. For cancer prevention, phase II enzyme inducers, which activate Nrf2, are being considered as new candidates for chemoprevention. Though likely to be effective in controlling cancers, such treatments may predispose patients to T2D due to antioxidant-hampered beta-cell function.
Although antioxidants may function as negative regulators for ROS signaling, ROS- mediated signal transduction is relatively resistant to ROS scavenging. This assumption is supported by the evidence that ROS-mediated signal transduction occurs in normal cells which usually contain considerable amount of antioxidants. Thus, unless we could identify and eliminate specific oxidative stressors from our environmental and dietary exposures, antioxidants are still needed to cope with oxidative stress and its associated diseases. Given the complex role of antioxidants in T2D, the question should focus on how exogenous antioxidants can alleviate the stressed condition without interfering with physiological ROS signaling. This principle implies a new type of antioxidant supplementation and treatment paradigm. If the source of oxidative stress from environmental or dietary exposure can not be avoided, successful antioxidant supplements should be those that do not easily penetrate the cell membrane. In this way, the exogenous oxidative agents will be neutralized extracellularly before reaching the inside of cells. More importantly, since these new antioxidant supplements can not penetrate the cell membrane, they would not interfere with the intracellular ROS signal in beta-cells. Membrane-penetrating antioxidant supplements would be still useful in situations where the oxidative stress is internally originated, for instance, as a result of increased cellular metabolism or dysfunction of an intracellular antioxidant enzyme. To avoid interference with intracellular ROS signaling for insulin secretion, which is mostly required postprandially, the antioxidant supplements may be taken sometime after the meal. In addition, antioxidants with relatively short life and can be cleared at the time of the next meal would be more efficient. With this multi-stage uptake paradigm, the exogenous antioxidant will be bioavailable only between meals. In this way, oxidative stress would be combated for most of the day, keeping the adaptive induction of endogenous antioxidant at a low basal level. Thus, the physiological postprandial ROS signals would be spared from attenuation by endogenous and exogenous antioxidants.
7. Conclusions and Perspectives
If valid, the proposed mechanism for impaired GSIS by oxidative stress will generate a major paradigm shift in our understanding of the roles of ROS and antioxidants in T2D and perhaps other diseases. Support for the hypotheses would lead to radically different strategies for the treatment of T2D and could suggest possible strategies for early intervention for metabolic syndrome and T2D. The potential paradoxical roles of ROS in beta-cell function suggest that site- and function-specific antioxidants or ROS donors may need to be considered in future therapeutic approaches.
Acknowledgments
This research was supported in part by the NIH grant DK76788 (JP), DK54024 (SC), ES016005 (JP) and the Long-Range Research Initiative of the American Chemistry Council. The content is solely the responsibility of the authors, and they have no conflicts of interest to disclose. The authors wish to thank Dr. Christopher Newgard for providing INS-1 (832/13) cells.
Abbreviations
- CAT
catalase
- GCLC
γ-glutamate cysteine ligase catalytic subunit
- GPx
glutathione peroxidase
- GSIS
glucose-stimulated insulin secretion
- ·O2−
superoxide
- GSH
reduced glutathione
- GS
glutathione synthetase
- GR
glutathione reductase
- GSSG
oxidized glutathione
- HO-1
heme oxygenase 1
- NQO1, NAD(P)H
quinone oxidoreductase 1
- Nrf2
nuclear factor E2-related factor 2
- Prx
peroxiredoxin
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SOD
superoxide dismutase
- Trx
thioredoxin
- Ucp2
uncoupling protein 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Wu XC, Thebaud B, Moudgil R, Hashimoto K, Michelakis ED. O2 sensing in the human ductus arteriosus: redox-sensitive K+ channels are regulated by mitochondria-derived hydrogen peroxide. Biol Chem. 2004;385:205–216. doi: 10.1515/BC.2004.014. [DOI] [PubMed] [Google Scholar]
- Armann B, Hanson MS, Hatch E, Steffen A, Fernandez LA. Quantification of basal and stimulated ROS levels as predictors of islet potency and function. Am J Transplant. 2007;7:38–47. doi: 10.1111/j.1600-6143.2006.01577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- Bai Y, Onuma H, Bai X, Medvedev AV, Misukonis M, Weinberg JB, Cao W, Robidoux J, Floering LM, Daniel KW, Collins S. Persistent nuclear factor-kappa B activation in Ucp2−/− mice leads to enhanced nitric oxide and inflammatory cytokine production. J Biol Chem. 2005;280:19062–19069. doi: 10.1074/jbc.M500566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW, Philipson LH. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J Biol Chem. 2003;278:9796–9801. doi: 10.1074/jbc.M206913200. [DOI] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390. doi: 10.1161/01.cir.0000051722.66074.60. [DOI] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol Sci. 2004;19:120–123. doi: 10.1152/nips.01514.2003. [DOI] [PubMed] [Google Scholar]
- Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wildman RP, Hamm LL, Muntner P, Reynolds K, Whelton PK, He J. Association between inflammation and insulin resistance in U.S. nondiabetic adults: results from the Third National Health and Nutrition Examination Survey. Diabetes Care. 2004;27:2960–2965. doi: 10.2337/diacare.27.12.2960. [DOI] [PubMed] [Google Scholar]
- Chen X, Scholl TO, Leskiw MJ, Donaldson MR, Stein TP. Association of glutathione peroxidase activity with insulin resistance and dietary fat intake during normal pregnancy. J Clin Endocrinol Metab. 2003;88:5963–5968. doi: 10.1210/jc.2003-030544. [DOI] [PubMed] [Google Scholar]
- Chevillotte E, Rieusset J, Roques M, Desage M, Vidal H. The regulation of uncoupling protein-2 gene expression by omega-6 polyunsaturated fatty acids in human skeletal muscle cells involves multiple pathways, including the nuclear receptor peroxisome proliferator-activated receptor beta. J Biol Chem. 2001;276:10853–10860. doi: 10.1074/jbc.M008010200. [DOI] [PubMed] [Google Scholar]
- Costa A, Iguala I, Bedini J, Quinto L, Conget I. Uric acid concentration in subjects at risk of type 2 diabetes mellitus: relationship to components of the metabolic syndrome. Metabolism. 2002;51:372–375. doi: 10.1053/meta.2002.30523. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, Horvath TL. Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology. 2003;144:5014–5021. doi: 10.1210/en.2003-0667. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Ebelt H, Peschke D, Bromme HJ, Morke W, Blume R, Peschke E. Influence of melatonin on free radical-induced changes in rat pancreatic beta-cells in vitro. J Pineal Res. 2000;28:65–72. doi: 10.1034/j.1600-079x.2001.280201.x. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. Embo J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002a;277:47129–47135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002b;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- Eto K, Tsubamoto Y, Terauchi Y, Sugiyama T, Kishimoto T, Takahashi N, Yamauchi N, Kubota N, Murayama S, Aizawa T, Akanuma Y, Aizawa S, Kasai H, Yazaki Y, Kadowaki T. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283:981–985. doi: 10.1126/science.283.5404.981. [DOI] [PubMed] [Google Scholar]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. 2003;52:1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- Fridlyand LE, Philipson LH. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic beta-cells? Diabetes. 2004;53:1942–1948. doi: 10.2337/diabetes.53.8.1942. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes. 2004;53(Suppl 3):S48–58. doi: 10.2337/diabetes.53.suppl_3.s48. [DOI] [PubMed] [Google Scholar]
- Horimoto M, Fulop P, Derdak Z, Wands JR, Baffy G. Uncoupling protein-2 deficiency promotes oxidant stress and delays liver regeneration in mice. Hepatology. 2004;39:386–392. doi: 10.1002/hep.20047. [DOI] [PubMed] [Google Scholar]
- Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- Janjic D, Maechler P, Sekine N, Bartley C, Annen AS, Wolheim CB. Free radical modulation of insulin release in INS-1 cells exposed to alloxan. Biochem Pharmacol. 1999;57:639–648. doi: 10.1016/s0006-2952(98)00346-3. [DOI] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2008;295:E1287–1297. doi: 10.1152/ajpendo.90604.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek P, Zackova M, Ruzicka M, Skobisova E, Jaburek M. Mitochondrial uncoupling proteins--facts and fantasies. Physiol Res. 2004;53(Suppl 1):S199–211. [PubMed] [Google Scholar]
- Jones PM, Persaud SJ. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr Rev. 1998;19:429–461. doi: 10.1210/edrv.19.4.0339. [DOI] [PubMed] [Google Scholar]
- Kamsler A, Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol. 2004;29:167–178. doi: 10.1385/MN:29:2:167. [DOI] [PubMed] [Google Scholar]
- Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoe K, Day NK, Good RA, Ohno H. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci U S A. 2002;99:9392–9397. doi: 10.1073/pnas.142206299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Mori S, Takino H, Kijima H, Yamasaki H, Ozaki M, Tetsuya I, Urata Y, Abe T, Sera Y, Yamakawa K, Kawasaki E, Yamaguchi Y, Kondo T, Eguchi K. Attenuation of expression of gamma-glutamylcysteine synthetase by ribozyme transfection enhance insulin secretion by pancreatic beta cell line, MIN6. Biochem Biophys Res Commun. 2000;278:236–240. doi: 10.1006/bbrc.2000.3776. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol. 1998;275:C1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Kowluru A. Regulatory roles for small G proteins in the pancreatic beta-cell: lessons from models of impaired insulin secretion. Am J Physiol Endocrinol Metab. 2003;285:E669–684. doi: 10.1152/ajpendo.00196.2003. [DOI] [PubMed] [Google Scholar]
- Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci U S A. 2002;99:118–122. doi: 10.1073/pnas.012410699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, Lowell BB. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest. 2003;112:1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krippeit-Drews P, Haberland C, Fingerle J, Drews G, Lang F. Effects of H2O2 on membrane potential and [Ca2+]i of cultured rat arterial smooth muscle cells. Biochem Biophys Res Commun. 1995;209:139–145. doi: 10.1006/bbrc.1995.1481. [DOI] [PubMed] [Google Scholar]
- Lee FY, Li Y, Zhu H, Yang S, Lin HZ, Trush M, Diehl AM. Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating mice [correction of rat] liver. Hepatology. 1999;29:677–687. doi: 10.1002/hep.510290320. [DOI] [PubMed] [Google Scholar]
- Lehtihet M, Honkanen RE, Sjoholm A. Glutamate inhibits protein phosphatases and promotes insulin exocytosis in pancreatic beta-cells. Biochem Biophys Res Commun. 2005;328:601–607. doi: 10.1016/j.bbrc.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Leloup C, Magnan C, Benani A, Bonnet E, Alquier T, Offer G, Carriere A, Periquet A, Fernandez Y, Ktorza A, Casteilla L, Penicaud L. Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes. 2006;55:2084–2090. doi: 10.2337/db06-0086. [DOI] [PubMed] [Google Scholar]
- Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani AL, Ktorza A, Casteilla L, Penicaud L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58:673–681. doi: 10.2337/db07-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- Li LX, Skorpen F, Egeberg K, Jorgensen IH, Grill V. Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal beta-cells. Biochem Biophys Res Commun. 2001;282:273–277. doi: 10.1006/bbrc.2001.4577. [DOI] [PubMed] [Google Scholar]
- Li X, Chen H, Epstein PN. Metallothionein and Catalase Sensitize to Diabetes in Nonobese Diabetic Mice: Reactive Oxygen Species May Have a Protective Role in Pancreatic {beta}-Cells. Diabetes. 2006;55:1592–1604. doi: 10.2337/db05-1357. [DOI] [PubMed] [Google Scholar]
- Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–689. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, Liu F, Lei XG. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci U S A. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev AV, Robidoux J, Bai X, Cao W, Floering LM, Daniel KW, Collins S. Regulation of the uncoupling protein-2 gene in INS-1 beta-cells by oleic acid. J Biol Chem. 2002;277:42639–42644. doi: 10.1074/jbc.M208645200. [DOI] [PubMed] [Google Scholar]
- Medvedev AV, Snedden SK, Raimbault S, Ricquier D, Collins S. Transcriptional regulation of the mouse uncoupling protein-2 gene. Double E-box motif is required for peroxisome proliferator-activated receptor-gamma-dependent activation. J Biol Chem. 2001;276:10817–10823. doi: 10.1074/jbc.M010587200. [DOI] [PubMed] [Google Scholar]
- Miwa S, Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans. 2003;31:1300–1301. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- Morgan D, Rebelato E, Abdulkader F, Graciano MF, Oliveira-Emilio HR, Hirata AE, Rocha MS, Bordin S, Curi R, Carpinelli AR. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology. 2009;150:2197–2201. doi: 10.1210/en.2008-1149. [DOI] [PubMed] [Google Scholar]
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Nakao C, Ookawara T, Sato Y, Kizaki T, Imazeki N, Matsubara O, Haga S, Suzuki K, Taniguchi N, Ohno H. Extracellular superoxide dismutase in tissues from obese (ob/ob) mice. Free Radic Res. 2000;33:229–241. doi: 10.1080/10715760000301401. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol. 2000;20:7311–7318. doi: 10.1128/mcb.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Norlin S, Ahlgren U, Edlund H. Nuclear factor-{kappa}B activity in {beta}-cells is required for glucose-stimulated insulin secretion. Diabetes. 2005;54:125–132. doi: 10.2337/diabetes.54.1.125. [DOI] [PubMed] [Google Scholar]
- Ostenson CG, Sandberg-Nordqvist AC, Chen J, Hallbrink M, Rotin D, Langel U, Efendic S. Overexpression of protein-tyrosine phosphatase PTP sigma is linked to impaired glucose-induced insulin secretion in hereditary diabetic Goto-Kakizaki rats. Biochem Biophys Res Commun. 2002;291:945–950. doi: 10.1006/bbrc.2002.6536. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Wiley SE, Kimple ME, Dixon JR, Kelly P, Worby CA, Casey PJ, Dixon JE. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem. 2001;276:8705–8712. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- Pi J, Bai Y, Daniel K, Liu D, Lyght O, Edelstein D, Brownlee M, Corkey BE, Collins S. Persistent oxidative stress due to absence of uncoupling protein 2 is associated with impaired pancreatic beta-cell function. Endocrinology. 2009 doi: 10.1210/en.2008-1642. in-press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- Pi J, Zhang Q, Woods CG, Wong V, Collins S, Andersen ME. Activation of Nrf2-mediated oxidative stress response in macrophages by hypochlorous acid. Toxicol Appl Pharmacol. 2008;226:236–243. doi: 10.1016/j.taap.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000;345(Pt 2):161–179. [PMC free article] [PubMed] [Google Scholar]
- Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53(Suppl 1):S130–135. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- Ruzicka M, Skobisova E, Dlaskova A, Santorova J, Smolkova K, Spacek T, Zackova M, Modriansky M, Jezek P. Recruitment of mitochondrial uncoupling protein UCP2 after lipopolysaccharide induction. Int J Biochem Cell Biol. 2005;37:809–821. doi: 10.1016/j.biocel.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Ryu JW, Hong KH, Maeng JH, Kim JB, Ko J, Park JY, Lee KU, Hong MK, Park SW, Kim YH, Han KH. Overexpression of uncoupling protein 2 in THP1 monocytes inhibits beta2 integrin-mediated firm adhesion and transendothelial migration. Arterioscler Thromb Vasc Biol. 2004;24:864–870. doi: 10.1161/01.ATV.0000125705.28058.eb. [DOI] [PubMed] [Google Scholar]
- Schmidt KN, Amstad P, Cerutti P, Baeuerle PA. Identification of hydrogen peroxide as the relevant messenger in the activation pathway of transcription factor NF-kappaB. Adv Exp Med Biol. 1996;387:63–68. doi: 10.1007/978-1-4757-9480-9_9. [DOI] [PubMed] [Google Scholar]
- Scott JA, King GL. Oxidative stress and antioxidant treatment in diabetes. Ann N Y Acad Sci. 2004;1031:204–213. doi: 10.1196/annals.1331.020. [DOI] [PubMed] [Google Scholar]
- Sjoholm A, Lehtihet M, Efanov AM, Zaitsev SV, Berggren PO, Honkanen RE. Glucose metabolites inhibit protein phosphatases and directly promote insulin exocytosis in pancreatic beta-cells. Endocrinology. 2002;143:4592–4598. doi: 10.1210/en.2002-220672. [DOI] [PubMed] [Google Scholar]
- Soria B, Quesada I, Ropero AB, Pertusa JA, Martin F, Nadal A. Novel players in pancreatic islet signaling: from membrane receptors to nuclear channels. Diabetes. 2004;53(Suppl 1):S86–91. doi: 10.2337/diabetes.53.2007.s86. [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- Tabet F, Savoia C, Schiffrin EL, Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2004;44:200–208. doi: 10.1097/00005344-200408000-00009. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- Todt I, Ngezahayo A, Ernst A, Kolb HA. Hydrogen peroxide inhibits gap junctional coupling and modulates intracellular free calcium in cochlear Hensen cells. J Membr Biol. 2001;181:107–114. doi: 10.1007/s00232001-0014-4. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voehringer DW, Hirschberg DL, Xiao J, Lu Q, Roederer M, Lock CB, Herzenberg LA, Steinman L. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc Natl Acad Sci U S A. 2000;97:2680–2685. doi: 10.1073/pnas.97.6.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei FY, Nagashima K, Ohshima T, Saheki Y, Lu YF, Matsushita M, Yamada Y, Mikoshiba K, Seino Y, Matsui H, Tomizawa K. Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat Med. 2005;11:1104–1108. doi: 10.1038/nm1299. [DOI] [PubMed] [Google Scholar]
- Wiernsperger NF. Oxidative stress as a therapeutic target in diabetes: revisiting the controversy. Diabetes Metab. 2003;29:579–585. doi: 10.1016/s1262-3636(07)70072-1. [DOI] [PubMed] [Google Scholar]
- Wollheim CB, Maechler P. Beta-cell mitochondria and insulin secretion: messenger role of nucleotides and metabolites. Diabetes. 2002;51(Suppl 1):S37–42. doi: 10.2337/diabetes.51.2007.s37. [DOI] [PubMed] [Google Scholar]
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Andersen ME. Dose response relationship in anti-stress gene regulatory networks. PLoS Comput Biol. 2007;3:e24. doi: 10.1371/journal.pcbi.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pi J, Woods CG, Jarabek AM, Clewell HJ, Andersen ME. Hormesis and adaptive cellular control systems. Dose Response. 2008;6:196–208. doi: 10.2203/dose-response.07-028.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]