

Abstract
Homeodomain proteins are transcription factors that play a critical role in early development in eukaryotes. These proteins previously have been classified into numerous subgroups whose phylogenetic relationships are unclear. Our phylogenetic analysis of representative eukaryotic sequences suggests that there are two major groups of homeodomain proteins, each containing sequences from angiosperms, metazoa, and fungi. This result, based on parsimony and neighbor-joining analyses of primary amino acid sequences, was supported by two additional features of the proteins. The two protein groups are distinguished by an insertion/deletion in the homeodomain, between helices I and II. In addition, an amphipathic alpha-helical secondary structure in the region N terminal of the homeodomain is shared by angiosperm and metazoan sequences in one group. These results support the hypothesis that there was at least one duplication of homeobox genes before the origin of angiosperms, fungi, and metazoa. This duplication, in turn, suggests that these proteins had diverse functions early in the evolution of eukaryotes. The shared secondary structure in angiosperm and metazoan sequences points to an ancient conserved functional domain.
Homeodomain proteins originally were defined as gene products from the family of homeotic genes critical in development of Drosophila (1, 2). The term since has been applied to transcription factors that meet two criteria: four highly conserved residues including the absolutely conserved Trp-49 and a conserved secondary structure consisting of a helix–loop–helix–turn–helix motif (3–5). Approximately 1,850 homeodomain proteins currently meet these criteria and occur in angiosperms, fungi, and metazoa. Understanding the evolutionary relationships of these transcription factors is of tremendous interest, because they play a vital role in a wide range of biological phenomena, including mating-type recognition, pathogenesis response, and early morphological development.
Parsimony and neighbor-joining analyses of primary amino acid sequences of representative eukaryotic homeodomain proteins suggested a close relationship between certain angiosperm [e.g., knotted-like homeobox (KNOX)], metazoan [e.g., extradenticle (EXD)], and fungal [e.g., Cu homeostasis (CUP 9)] sequences. This result was further supported by two shared characters: an insertion/deletion in the homeodomain, and an alpha-helical structure in the region N terminal of the homeodomain in the angiosperm and metazoan proteins. These characters differentiate this group of homeodomain proteins from metazoan antennapedia-like (ANTP) and other angiosperm, fungal, and metazoan sequences. This observation leads to the hypothesis that, in addition to recent duplications within each kingdom, there was at least one ancient duplication of homeodomain-encoding genes before the origin of angiosperms, fungi, and metazoa. The secondary structure shared by some angiosperm and metazoan proteins suggests an ancient shared function. On the other hand, the phylogenetic pattern of distribution of known protein–protein interactions in metazoa suggests that other interactions arose after the origin of metazoa.
METHODS
Protein Sequences.
Sampling of sequences from protein and DNA databases was done by using blast (http://www.ncbi.nlm.nih.gov/BLAST/) (6). Angiosperm sequences were used as query sequences, with the purpose of obtaining the widest possible sample. Two to three sequences that were most like, and others that were least like, the query sequence were selected from each search. Additional searches were conducted by using these sequences that included metazoan and fungal proteins. The search was stopped when no new sequences were identified. A total of 152 sequences was downloaded and aligned. The list of 152 sequences with accession numbers is available on the web site http://www-plb.ucdavis.edu/sinha/homeo.html. An alignment of 60 of these sequences (see below) is available on the same site. Alignment followed previous studies (4), with the exception of an insertion/deletion of three amino acids in positions 24–26 between helix I and II (7–9). The insertion/deletion was not included in the phylogenetic analyses.
Phylogenetic Analyses.
Two different methods, maximum parsimony and neighbor-joining (10), were used to analyze amino acid sequences. Fitch parsimony was implemented by using heuristic searches on the program paup*4.0 (11). Neighbor-joining analyses were done by using p- and poisson distances on the program mega (12). Preliminary neighbor-joining analyses of all of the sequences showed certain well supported groups, e.g., angiosperm KNOX proteins. Smaller datasets were constructed that contained randomly chosen representatives from these well supported groups or from well recognized groups such as the HOM/Hox cluster. Parsimony analyses were not conducted on the large data sets because of computational constraints. Constrained parsimony analyses were conducted in which sequences from angiosperms, fungi, and metazoa were forced to remain together (constraint option in paup*4.0). The significance of difference in total length of trees in constrained and unconstrained analyses was assessed by using the Templeton test (13) as implemented on paup*4.0. Robustness of results was assessed by bootstrapping the data (14) in neighbor-joining analyses. Because homeodomain sequences have been found only in angiosperms, fungi, and metazoa, no outgroup sequence was available, so results are presented as unrooted trees.
Tree Mapping.
The minimum number of duplication events in the history of homeodomain proteins was estimated by obtaining reconciled trees by using the program component (15). This method compares the organismal phylogeny with the gene tree and assumes that the only processes acting on genes are duplication and loss (i.e., no horizontal transfer). Branches are added to the gene tree to indicate duplications and genes that have been lost or are yet to be identified (16).
Secondary Structure Prediction.
Secondary structure was predicted for the N terminal regions by using Predict-protein server (http://www.embl-heidelberg.de/predictprotein/predictprotein.html) (17–19). This analysis was done for 50 sequences, 40 of which are in the 60-sequence dataset.
RESULTS AND DISCUSSION
Our analyses indicate that the homeodomain proteins form several statistically well supported subgroups such as the angiosperm KNOX, BELL, HAT, ZM-HOX, and GL2, and the metazoan SIX2, EXD, ANTP, POU, and PAX (Fig. 1 A and B). The metazoan subgroups were also identified by Agosti et al. (20). Clusters of subgroups, such as the metazoan HOM/Hox cluster, are not strongly linked by primary sequence data. However, these proteins are believed to be closely related based on shared similarity of N terminal regions, genome structure, and expression patterns (21–24). The internal nodes in the trees do not have strong bootstrap support (e.g., Fig. 1B). This result is not surprising, given the short length of the homeodomain sequence and the length of time since divergence. Nonetheless, we find some robust conclusions and tantalizing results that emerge from this analysis.
Figure 1.
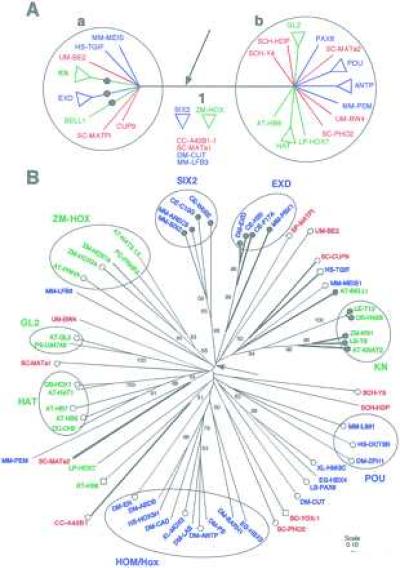
(A) Phylogenetic relationships between eukaryotic homeodomain protein sequences indicate an ancient duplication that occurred before the origin of angiosperms, metazoa, and fungi. Homeodomain proteins are divided into two groups, a and b, each containing well supported subgroups from all three kingdoms: angiospermae (green), fungi (red), and metazoan (blue). This tree is a consensus of results from different phylogenetic analyses of a dataset of 60 sequences from which a 3-aa insertion/deletion site was removed. The strict consensus of 59 trees was obtained after removing 14 sequences including subgroups ZM-HOX and SIX2. These 14 sequences occupy variable positions on the tree in all analyses. Results are presented as unrooted trees, because no outgroup sequence is known. Similar results were obtained from neighbor-joining analyses of larger datasets. All sequences in group a have a 3-aa insertion (arrow) in the homeodomain. Several sequences in group a share an amphipathic helical secondary structure in the region N terminal to the homeodomain (•). (B) The distributions of two protein characteristics are consistent with the phylogenetic tree based on primary sequence data. This tree was obtained from neighbor-joining analyses of pairwise p-distances. Strongly supported angiosperm protein subgroups (green) are associated with fungal (red) and metazoan (blue) subgroups. Sequence names are indicated as follows: the first two letters represent the Latin name and are followed by the name of the gene. Angiospermae: AT, Arabidopsis thaliana; DC, Daucus carota; LE, Lycopersicon esculentum; LP, Lycopersicon peruvianum; OS, Oryza sativa; PS, Phalaenopsis sp.; PC, Petroselinium crispum; ZM, Zea mays. Metazoa: CE, Caenorhabditis elegans; DM, Drosophila melanogaster; EG, Echinococcus granulosus; HS, Homo sapiens; LS, Lineus sanguineus; MM, Mus musculus; XL, Xenopus laevis. Fungi: SC, Saccharomyces cerevisiae; SCH, Schizophyllum commune; UM, Ustilago maydis. Branches are drawn proportional to p-distance. The scale represents p-distance. Numbers along each branch indicate bootstrap values over 50%. Most internal branches have low statistical support. Branch 1 derives support from evidence external to primary sequence data. Presence of three amino acids in the insertion/deletion (thick branches) marks most of the sequences in group a. The SIX2 subgroup is assumed to have lost three amino acids on this tree, but not in other trees where its phylogenetic position is outside of group a. The phylogenetic distribution of the amphipathic helix in the N terminal region (•), its absence (○), and a short N terminal region (□) indicates that the N terminal structure characterizes sequences in group a.
We have identified a clear division among homeodomain proteins in a consensus of results from several different analyses (Fig. 1A). Proteins on either side of the node labeled 1 (groups a and b) are distinguished not only by their sequences but also by presence or absence of a three amino acid insertion/deletion between helices I and II of the homeodomain. A striking feature is that groups a and b each contain representatives from all three kingdoms (Fig. 1). Thus, the angiosperm KNOX subgroup (group a) is more closely related to fungal and metazoan sequences in group a than to angiosperm proteins in group b. Similarly, the ANTP cluster (group b) is more closely related to angiosperm and fungal sequences in group b than to metazoan sequences in group a. If the root of the tree is between the two protein groups, then both may be orthologous (25); if it is within one of these groups, then the other protein group would be orthologous. Thus, groups a and b are candidates for ancient orthologies, i.e., duplicated genes present in the common ancestor and passed on to the three lineages during their divergence. More recent duplications have resulted in multiple copies within each kingdom.
This result also is supported by several constrained parsimony analyses in which sequences within each kingdom are forced to remain together. For instance, for the 60-sequence dataset the total tree length in constrained analysis is 1,414, whereas in unconstrained analysis it is 1,379. This difference is statistically significant (P < 0.05) by using the Templeton test (13). In parsimony analyses the shortest tree is taken to be the best hypothesis of relationships. Therefore we conclude that the data support a close relationship of some angiosperm sequences to fungal and metazoan sequences rather than to other angiosperm sequences, and similarly for sequences from the other two kingdoms.
We found that all sequences in group a contain an additional three amino acids, in positions 24–26, between helices I and II of the homeodomain (7, 8) that are absent from most other homeodomains, even though the protein tree is based on alignments from which the insertion/deletion was excluded. The exceptions are fungal sequences, CC-A42B1 and SC-MATα2, which contain the additional amino acids, yet are placed outside of group a. This discrepancy may be the result of either independent insertion/deletion events or incorrect placement of the fungal sequences outside of group a, perhaps owing to their highly divergent primary sequence (3) (suggested by Val-50 in SC-MATα2 instead of the highly conserved Phe-50).
In several sequences we also identified two alpha helical regions N terminal to the homeodomain (Fig. 2). The first region, the ELK domain (26), is immediately adjacent to the homeodomain and contains two short helices. The second region lies further N terminal (16–54 amino acid residues away) and consists of one amphipathic helix of 9–13 turns with conserved residues on one face of the helix. This structure is seen in the angiosperm KNOX and metazoan EXD subgroups. None of the sequences examined outside of group a has this secondary structure. The distribution of features of the region N terminal to the homeodomain on the protein tree (Fig. 1B) suggests one of three possibilities: (i) the structure arose once, was spliced to the homeodomain in a single event, but was lost several times, (ii) it arose once and was spliced to the homeodomain in independent events, or (iii) it arose independently in angiosperms and metazoa. It is less likely that such a complex structure evolved several times, so we infer a single origin. Assuming a single origin, it is remarkable that this protein secondary structure has been maintained across the metazoa and angiosperms. Conserved secondary structure and amino acid motifs are taken to indicate common function, so the amphipathic helix of the KNOX and EXD subgroups suggests a protein–protein interaction that has been conserved in evolution.
Figure 2.

Secondary structure in the region immediately N terminal to the homeodomain is conserved across some angiosperm and metazoan proteins. This alignment of N terminal regions for some group a proteins shows N terminal helical regions (shaded amino acids), nonhelical linker region, and the homeodomain. Helical regions were predicted for the N terminal regions by using Phomeodomain Sec (http://www.embl-heidelberg.de/predictprotein/predictprotein.html/). We identified two alpha helical regions in the angiosperm KN and metazoan EXD subgroups. The first is immediately adjacent to the homeodomain and contains two short helices (ELK domain) and was not detected in any other sequences. The second region lies further N terminal and consists of a long amphipathic helix. This helix, if found in other protein subgroups, was either short, or not amphipathic and not alignable. By using a helical wheel representation it was possible to align the sequence such that conserved amino acids (boxed and numbered) were positioned on one (hydrophobic) face of the helix. Gaps correspond to one or two turns of the helix and thus maintain the conserved face of the helix.
Biased sampling of genes and organisms, as well as uncertainty regarding relationships between protein subgroups in this study, do not allow us to infer the precise number of ancient homeodomain proteins, although it appears that more than two copies must have existed in the common ancestor. Mapping of a reduced set of sequences on the organismal tree by using reconciled trees produced on the program component reveals that at least seven gene duplication events must be postulated to occur in the history of homeodomain sequences to reconcile the gene and organism trees (Fig. 3). Multiple tree mapping by using several alternative topologies gave similar result (7–11 duplications). Mapping also points to “missing” sequences, for instance a metazoan representative for the protein group that contains the angiosperm AT-HAT1 and fungal SC-MATA1 sequences. The homeobox–leucine zipper genes, the HAT group, are believed to have evolved after divergence of angiosperms and metazoa (27). Our results, although lacking statistical support, suggest that the homeodomain of the HAT group may be most closely related to homeodomain sequences in fungi and, possibly, metazoa. A search for representatives from the “missing” kingdom (in this case, metazoa) may be fruitful. It is likely that such genes will have new and intriguing functions.
Figure 3.
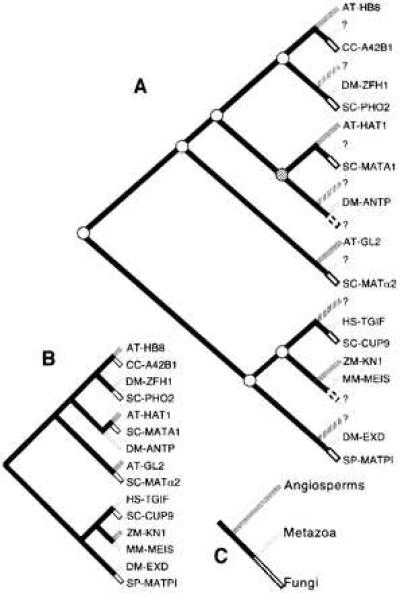
Reconciled tree (A) showing seven gene duplication events (circles) postulated to have occurred in the common ancestor of angiosperms, metazoa, and fungi before their diversification. The reconciled tree (A) is obtained from the gene tree (B) and organism tree (C) by adding leaves (hatched) such that the gene tree and the organismal association of the sequences can be explained by shared common history alone. The reconciled tree suggests that nine sequences are missing (?), because of either lack of sampling or gene loss, or because they arose in the common ancestor of fungi and animals and are therefore absent from angiosperms. Reconciled trees based on alternative gene trees gave estimates of 7–11 duplications. These numbers are merely illustrative, and a precise estimate can be made only with the acquisition of a wider sample of sequences.
These insights into the evolution of homeodomain proteins can help to explain and predict patterns of genomic distribution. For instance, the metazoan HOM/Hox group is clustered in the genome (28), whereas the Knox family (knotted-like homeobox genes) are dispersed throughout the genome (29). This result is not surprising, because the HOM/Hox cluster is believed to have diversified within the metazoa, and our results suggest that the HOM/Hox and Knox gene lineages diverged early in evolution. Similarly, there can be no a priori expectation that other homeodomain gene families will be clustered.
Interestingly, early metazoan development involves interactions between proteins of the two groups identified here. Members of the ANTP cluster (group b) have a conserved hexapeptide sequence N terminal to the homeodomain that is recognized by EXD (group a). This protein–protein interaction leads to cooperative binding to promoter sequences of genes that play a role in development (30, 31). Three lines of evidence suggest that the interaction arose after divergence from angiosperms and, possibly, the origin of metazoa. First, the conserved hexapeptide is absent in the Abd-B class (32), which is phylogenetically distinct from other Antp genes (24), so only a subset of ANTP proteins show the interaction. Second, the hexapeptide is absent in the early metazoan group Cnidaria (32), so not all metazoan ANTP proteins show this interaction. Third, homeodomain proteins that regulate development in angiosperms belong to two protein subgroups, KNOX and BELL (group a), which so far are not known to interact with other homeodomain proteins. Therefore, interactions between ANTP and EXD classes may have evolved within metazoa and may characterize a set of metazoa excluding Cnidaria.
Our study also revealed evidence of modularity in homeodomain proteins. Our survey showed that the domain N terminal to the homeodomain is significant in most sequences, but is very short in a few subgroups (Fig. 1B). The phylogenetic distribution of this trait indicates that, regardless of where the tree might be rooted, the N terminal domain must have been lost independently at least twice (in HS-TGIF, AT-HB8, or SC-YOX1). Modularity of homeodomain proteins raises the possibility that the shared structure of the N terminal region noted by us for angiosperm and metazoan sequences may represent independent exon shuffling events involving the same domain. However, we were not able to detect a similar region in any other proteins. We have found suggestive evidence for modularity of the homeodomain itself. We discovered a fragment of sequence in Toxoplasma (Alveolata, Eukaryota; Toxoplasma EST project, accession no. W00079) that is similar to helix I of the homeodomain. Flanking sequences reveal no primary or secondary structure similarity to either helix II or III. If we include this sequence in a phylogenetic analysis, it attaches within group a near the fungal sequence SC-MATPI. Because Toxoplasma may represent an outgroup in our study (33), this finding raises the intriguing possibility that the homeodomain (currently known only from angiosperms, fungi, and metazoa) arose by the evolutionary assembly of an helix I-like sequence (detected in Toxoplasma) with one containing the helix–turn–helix–motif present in helix II and III. Future sampling of a wide range of protists would provide data to test this hypothesis of stepwise assembly.
Our results suggest that there were at least two, and quite possibly multiple, genes coding for homeodomain proteins in the last common ancestor of angiosperms, fungi, and metazoa. These phylogenetic results in combination with conserved secondary protein structure, putative modularity of homeodomain proteins, and known patterns of expression contribute to greater understanding of the origin and evolution of these proteins and their role in the diversification of eukaryote lineages.
Acknowledgments
We thank David Swofford for permission to use a beta test version of paup*4.0, and Mike Sanderson for helpful discussions. This work was supported by a Katherine Esau Fellowship to G.B., National Science Foundation Grant DEB-9419748 to E.A.K., and National Science Foundation Grant IBN-96-32013 to N.S.
References
- 1.McGinnis W, Levine M S, Hafen E, Kuroiwa A, Gehring W J. Nature (London) 1984;308:428–433. doi: 10.1038/308428a0. [DOI] [PubMed] [Google Scholar]
- 2.Scott M P, Weiner A J. Proc Natl Acad Sci USA. 1984;81:4115–4119. doi: 10.1073/pnas.81.13.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott M P, Tamkun J W, Hartzell G W. Biochim Biophys Acta. 1989;989:25–48. doi: 10.1016/0304-419x(89)90033-4. [DOI] [PubMed] [Google Scholar]
- 4.Kappen C, Schughart K, Ruddle F H. Genomics. 1993;18:54–70. doi: 10.1006/geno.1993.1426. [DOI] [PubMed] [Google Scholar]
- 5.Bürglin T R. In: Guidebook to the Homeobox Genes. Duboule D, editor. New York: Oxford; 1994. pp. 27–71. [Google Scholar]
- 6.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 7.Laughon A. Biochemistry. 1991;30:11358–11367. doi: 10.1021/bi00112a001. [DOI] [PubMed] [Google Scholar]
- 8.Wollberger C, Vershon A, Liu B, Johnson A, Pabo C. Cell. 1991;67:517–528. doi: 10.1016/0092-8674(91)90526-5. [DOI] [PubMed] [Google Scholar]
- 9.Clarke N D. Protein Sci. 1995;4:2269–2278. doi: 10.1002/pro.5560041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 11.Swofford D L. paup*: Phylogenetic Analysis Using Parsimony, Beta Test Version 4.0d54. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 12.Kumar S, Tamura K, Nei M. mega: Molecular Evolutionary Genetics Analysis, Version 1.01. University Park, PA: Institute of Molecular Genetics; 1993. [Google Scholar]
- 13.Templeton A R. Evolution. 1983;37:221–244. doi: 10.1111/j.1558-5646.1983.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 14.Felsenstein J. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 15.Page R D M. component, Version 2.0. London: Natural History Museum; 1993. [Google Scholar]
- 16.Page R D M. Syst Biol. 1994;43:58–77. [Google Scholar]
- 17.Rost B. Methods Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- 18.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 19.Rost B, Sander C. Proteins. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 20.Agosti D, Jacobs D, DeSalle R. Cladistics. 1996;12:66–82. doi: 10.1111/j.1096-0031.1996.tb00193.x. [DOI] [PubMed] [Google Scholar]
- 21.Slack J, Holland P, Graham C. Nature (London) 1993;36:470–492. [Google Scholar]
- 22.Carroll S B. Nature (London) 1995;376:479–485. doi: 10.1038/376479a0. [DOI] [PubMed] [Google Scholar]
- 23.Popodi E, Kissinger J C, Andrews M E, Raff R A. Mol Biol Evol. 1996;13:1078–1086. doi: 10.1093/oxfordjournals.molbev.a025670. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Nei M. Genetics. 1996;142:295–303. doi: 10.1093/genetics/142.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitch W M. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 26.Vollbrecht E, Veit B, Sinha N, Hake S. Nature (London) 1991;350:241–243. doi: 10.1038/350241a0. [DOI] [PubMed] [Google Scholar]
- 27.Schena M, Davis R W. Proc Natl Acad Sci USA. 1994;91:8393–8397. doi: 10.1073/pnas.91.18.8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gehring W J. In: Guidebook to the Homeobox Genes. Duboule D, editor. New York: Oxford; 1994. pp. 3–10. [Google Scholar]
- 29.Kerstetter R, Vollbrecht E, Lowe B, Veit J, Hake S. Plant Cell. 1994;6:1877–1887. doi: 10.1105/tpc.6.12.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauskolb C, Weischaus E. EMBO J. 1994;13:3561–3569. doi: 10.1002/j.1460-2075.1994.tb06663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang C-P, Shen W-F, Rozenfeld S, Lawrence H F, Largman C, Cleary M L. Genes Dev. 1995;9:663–674. doi: 10.1101/gad.9.6.663. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn K, Streit B, Schierwater B. Mol Phyl Evol. 1996;6:30–38. doi: 10.1006/mpev.1996.0055. [DOI] [PubMed] [Google Scholar]
- 33.Patterson D J, Sogin M L. In: The Origin and Evolution of Prokaryotic and Eukaryotic Cells. Hartman H, Matsuno K, editors. Singapore: World Scientific; 1992. pp. 13–46. [Google Scholar]