

Abstract
When fluorescently tagged oligonucleotides are located near metal surfaces, their emission intensity is impacted by both electromagnetic effects (i.e., quenching and/or enhancement of emission) and the structure of the nucleic acids (e.g., random coil, hairpin, or duplex). We present experiments exploring the effect of label position and secondary structure in oligonucleotide probes as a function of hybridization buffer, which impacts the percentage of double-stranded probes on the surface after exposure to complementary DNA. Nanowires containing identifiable patterns of Au and Ag segments were used as the metal substrates in this work, which enabled us to directly compare different dye positions in a single multiplexed experiment and differences in emission for probes attached to the two metals. The observed metal–dye separation dependence for unstructured surface-bound oligonucleotides is highly sensitive to hybridization efficiency, due to substantial changes in DNA extension from the surface upon hybridization. In contrast, fluorophore labeled oligonucleotides designed to form hairpin secondary structures analogous to solution-phase molecular beacon probes are relatively insensitive to hybridization efficiency, since the folded form is quenched and therefore does not appreciably impact the observed distance-dependence of the response. Differences in fluorescence patterning on Au and Ag were noted as a function of not only chromophore identity but also metal–dye separation. For example, emission intensity for TAMRA-labeled oligonucleotides changed from brighter on Ag for 24-base probes to brighter on Au for 48-base probes. We also observed fluorescence enhancement at the ends of nanowires and at surface defects where heightened electromagnetic fields affect the fluorescence.
Introduction
When chromophores are placed close to a metal surface, their fluorescence emission intensity can be altered due to quenching or electromagnetic enhancement.1–5 Quenching occurs at distances very close to planar metal surfaces, usually ≤5–10 nm, while surface-enhanced fluorescence (SEF) generally occurs at tens of nanometers from the surface, and is sensitive to the nanoscale roughness of the metal.1–16 Although much smaller in magnitude than the related phenomenon of surface-enhanced Raman scattering (SERS), SEF has been shown to provide up to ~10-fold increases in emission, which could be analytically important in detection strategies.1–5,17–20 Predicting the optimal separation for a given experiment with precision can be difficult, as it varies with the identity and curvature of the metallic surface.1–20 The behavior of fluorescent dyes on or near metal surfaces is becoming increasingly important for biosensing applications, where fluorescence is used to indicate the occurrence of specific binding events.3,5
Bioassays can take advantage of fluorescence enhancement, quenching, or both. Incorporation of a metal film can provide additional discrimination between surface-bound and free chromophores by increasing emission under evanescent wave excitation.5,21 For example, Hong and Kang recently reported detection of cardiac markers via metal particle-enhanced fluorescence in a surface immunoassay on optical fibers.18 Fluorescence quenching was employed by Perez-Luna et al. in a displacement immunoassay, where target binding led to release of labeled receptors from a Au film and a concomitant ~10-fold increase in intensity.22 In some cases, probe conformational changes alter the metal–chromophore separation upon target binding. Maxwell et al. bound 3′ thiolated, 5′ dye-labeled oligonucleotides to 2.5 nm diameter Au particles as probes for complementary target DNAs.23 In the absence of target, the 5′ dye moiety was able to approach the metal surface and fluorescence was quenched; hybridization removed the fluorophore from the particle whereupon emission increased substantially. This approach is conceptually similar to that of solution-phase molecular beacon probes, which are oligonucleotides having regions of partial complementarity at their 3′ and 5′ ends, such that they form hairpin loop structures. Traditionally, a fluorescent dye is appended to one end of the DNA sequence, and a quencher molecule, to the opposite end. When in solution, they exist in “dark” hairpin loop structures with the dye and quencher in close proximity. Upon addition of a complementary target sequence, the hairpin unfolds to bind the target, allowing fluorescence to occur.24–29 Libchaber and co-workers have demonstrated that 1.4 nm gold particles can be used in place of molecular quenchers, providing quenching efficiencies >99%.30
An important advantage of molecular beacon-style assays is that no washing or target labeling is required. A disadvantage is the limited degree to which solution-phase beacons can be multiplexed, due to the requirement for spectrally distinguishable dyes and efficient quenching. One solution would be to immobilize the beacon probes on a surface, where they could be used in an array format. Toward this end, beacon probes have been immobilized on glass,31,32 polystyrene,33 agarose,34 and metal35–37 surfaces. On metals, the surface itself can serve as the quencher. Krauss and co-workers introduced beacon-style probes on macroscopic, planar Au surfaces for target DNA sequence detection and demonstrated excellent discrimination between fully complementary and single nucleotide mismatched targets.35,36 We recently reported a surface-bound beacon assay where Au/Ag striped nanowires (marketed as Nanobarcodes or NBCs) were used for simultaneous detection of five human pathogenic target sequences.37 These particles are cylindrical, several microns in length, and ~300 nm in diameter and contain multiple segments of Au and Ag in patterns readily differentiated by reflectance optical microscopy.38–40
Here we report the effect of dye–metal separation on fluorescence emission from cylindrical nanowires composed of Ag and Au segments. Emission from dye-labeled oligonucleotides at metal surfaces has been investigated previously. Lakowicz and co-workers have reported a number of surface effects on fluorescence, including enhanced intrinsic fluorescence from unlabeled DNA,41 improved photostability,42 and increased resonant energy transfer between DNA-bound molecular donors and acceptors.43 This group has also reported increased apparent quantum yields and decreased lifetimes for Cy3- and Cy5-labeled dsDNA adsorbed nonspecifically onto Ag particles.44 The kinetics of labeled complementary strands to surface-bound probe DNAs have been followed in real time by both surface plasmon field enhanced21 and epi-illumination45,46 fluorescence spectroscopy. Despite this prior work, guidance on optimum length or base position for fluorescently tagging surface-bound probe strands is lacking. The effect of dye–Ag island film separation has been investigated by preparation of protein multilayers to which cyanine dye labeled oligonucleotides were attached.47 The maximum enhancement was about 12-fold, at ~9 nm from the metal surface (i.e., one layer of streptavidin and biotinylated BSA between the metal and the DNA).47 For labeled DNA bound to the surface without intervening protein layers, the conformation of the DNA can dramatically alter the dye–surface separation, and with it the emission intensity. Rant, Tornow, and co-workers used this effect to probe conformational changes in 5′ thiolated, 3′ dye labeled 12- and 24-base single-stranded oligonucleotides.48 Fluorescence increased with increasing DNA surface density, due to steric interactions forcing the strands to “stand up” to a greater extent on the surface.
We present data which build on these results to elucidate the effect of chromophores incorporated after the 8th, 12th, 24th, 36th, and 48th position along a 52-base 5′ thiolated probe strand extended from the surface by hybridization to a nonfluorescent complementary oligonucleotide. By performing the fluorescence measurements for each length simultaneously in a multiplexed format, we were able to directly and quantitatively compare the effect of dye incorporation at each position as a function of probe surface coverage and hybridization buffer. We found that the observed metal–dye separation dependence was highly sensitive to hybridization efficiency, due to increased DNA extension from the surface upon hybridization. This effect was substantial, with 10-fold lower dye-labeled probe coverages leading to higher emission intensities due to improved hybridization.
To explore the effect of probe secondary structure, we also tested molecular beacon-style probe sequences having 5′ thiols and 3′ chromophores. The beacon probes were less sensitive to hybridization efficiency than their unstructured counterparts: among 24-, 34-, and 48-base beacon probes, the 34-mer gave the highest fluorescence intensities in the presence of DNA target regardless of the hybridization buffer used. We interpret this as a result of differences in conformational flexibility for unquenched states of unstructured single-stranded probes (which are quite flexible at these lengths) and the beacon probes, which should unfold into unquenched states only upon binding to complementary target strands. Thus, while fluorescence from traditional single-stranded DNA probes lacking secondary structure occurs from molecules sampling a range of metal–dye separations, any emission observed from beacon probes should be from the double-stranded state (i.e., maximum metal–dye separation).
Because the striped nanowires contained segments of both Au and Ag, the effects of each metal could be directly compared within each experiment. Differences in fluorescence patterning on Au and Ag were observed as a function of chromophore identity and metal–dye separation. For example, emission intensity for TAMRA-labeled oligonucleotides changed from brighter on Ag for 24-base probes to brighter on Au for 48-base probes. We also observed regions of brighter fluorescence at the ends of nanowires and at surface defects where heightened electromagnetic fields affect the fluorescence. It is hoped that our results on fluorescence emission for different metal–dye separations, probe conformations, dye identities, and metal substrates will prove useful in the design of bioassays in which these effects are important.
Materials and Methods
Materials
Nanobarcodes (NBCs) patterned 000010, 001100, 010100, 011110, 100001, 001010, 010010, and 011000, where 0 represents a 0.75 μm segment of Au and 1 represents a 0.75 μm segment of Ag, were purchased from Nanoplex, Inc. (now Oxonica). Each batch of nanowires consisted of ~1 × 109 wires per 1 mL of ethanol. Nanowires were rinsed 3 times in water (by centrifugation) to remove the ethanol prior to use. Hybridization buffer (0.3 M PBS) was made in-house and was prepared using 0.3 M NaCl and 10 mM sodium phosphate at pH 7.0, and 0.01 M PBS buffer (0.01 M phosphate buffered saline; 0.138 M NaCl; 0.0027 M KCl; pH 7.4) was purchased from Sigma. Commercial hybridization buffer (HS114) was obtained from Molecular Research Centers, Inc. CAC buffer was prepared according to Krauss and co-workers, using 0.5 M NaCl, 20 mM cacodylic acid, and 0.5 mM EDTA, pH = 7.35 All water used in these experiments and in the preparation of the buffers was purified through a Barnstead Nanopure System to an 18 MΩ resistivity.
Surface–Dye Separation Experiments with Unstructured DNA Probes
Probes for dye–surface separation experiments were designed by inserting a nonhybridizing, internal rhodamine red-X NHS ester labeled thymine base after the 8th, 12th, 24th, 36th, or 48th base position of a 51-base oligonucleotide. We anticipate that this labeled thymine forms a single base bulge loop,49 minimizing interference of the dye molecule with hybridization. This insertion does not appreciably decrease the stability of duplex formation at any of the positions used here (predicted melting temperatures for these sequences in 0.3 M salt are all ~82 °C). Because the bulge does not increase the overall length of the duplex formed, we refer throughout this manuscript to the separation in terms of the base just prior to the labeled thymine. All sequences are listed in Table 1 and were purchased from Integrated DNA Technologies, Inc. DNA sequences were received as disulfides, which were cleaved before use, resulting in a single thiol moiety terminating the sequence. To cleave the disulfide, the DNA was first dissolved in a 100 mM solution of DTT (dithiothreitol) in 1 mL of 0.1 M Na3PO4 buffer (pH 8.3) for a 0.5 h, and then the small thiol fragments were removed using Centri-Spin separation columns (Princeton Separations). The resulting DNA sequences (terminated with a single –SH group) were diluted in water to a concentration of 10 μM and were stored in the freezer at −80 °C.
Table 1.
Dna Sequences Used in This Worka
| sequence name | sequence (5′–3′) | description |
|---|---|---|
| 8D | thiol C6-TTT CAT GG*T TAG CGT ATG CTA GAT CGC GTA AAT GAA TGC CTA GAT CAG CGA T |
5′ thiol, internal dye after the 8th of 52 bases |
| 12D | thiol C6-TTT CAT GGT AGC *TGT ATG CTA GAT CGC GTA AAT GAA TGC CTA GAT CAG CGA T |
5′ thiol, internal dye after the 12th of 52 bases |
| 24D | thiol C6-TTT CAT GGT AGC GTA TGC TAG ATC *TGC GTA AAT GAA TGC CTA GAT CAG CGA T |
5′ thiol, internal dye after the 24th of 52 bases |
| 36D | thiol C6-TTT CAT GGT AGC GTA TGC TAG ATC GCG TAA ATG AAT *TGC CTA GAT CAG CGA T |
5′ thiol, internal dye after the 36th of 52 bases |
| 48D | thiol C6-TTT CAT GGT AGC GTA TGC TAG ATC GCG TAA ATG AAT GCC TAG ATC AGC G*TA T |
5′ thiol, internal dye after the 48th of 52 bases |
| NFD | thiol C6-TTT CAT GGT AGC GTA TGC TAG ATC GCG TAA ATG AAT GCC TAG ATC AGC GAT |
5′ thiol, 51 bases (no dye) |
| CD | ATC GCT GAT CTA GGC ATT CAT TTA CGC GAT CTA GCA TAC GCT ACC ATG |
48 base complement to 8D, 12D, 24D, 36D, 48D, and NFD |
| 48B | thiol C6-GCG AGT AAA AGA GAC CAT CAA TGA GGA AGC TGC AGA ATG GGA TAC TCG-TAMRA |
5′ thiol, 3′ TAMRA 48-base beacon probe with 6-base “stem” |
| 34B | thiol C6-GCG AGG AGA CCA TCA ATG AGG AAG CTG CAC TCG C-TAMRA | 5′ thiol, 3′ TAMRA 34-base beacon probe with 5-base “stem” |
| 24B | thiol C6-GCG AGA TCA ATG AGG AAG CCT CGC-TAMRA | 5′ thiol, 3′ TAMRA 24-base beacon probe with 5-base “stem” |
| 24S | thiol C6-GAG ACC ATC AAT GAG GAA GCT GCA -TAMRA | 5′ thiol, 3′ TAMRA 34-base unstructured probe (no “stem”) |
| C24B | GGC TTC CTC ATT GAT | 15-base complement to 24B |
| C34B | TGC AGC TTC CTC ATT GAT GGT CTC | 24-base complement to 34B and 25S |
| C48B | GTA TCC CAT TCT GCA GCT TCC TCA TTG ATG GTC TCT TTT A | 40-base complement to 48B |
The * marks the location of an internal rhodamine red-X NHS ester fluorophore. Italicized portions of sequences show stem regions of beacon sequences.
Preparation of Distance Dependence Assays
Aliquots of 25 μL of each type of NBC were rinsed 3 times in water and resuspended in 100 μL of 0.01 M PBS buffer. Each internally labeled DNA sequence was added at a final concentration ranging between 1 and 2 μM to different patterned NBCs (concentrations were held constant within a single experiment but varied slightly from experiment to experiment). Nanowire patterns allowed for identification of the corresponding attached DNA sequence. Thiolated DNA (8D, 12D, 24D, 36D, or 48D) was allowed to self-assemble to the wires for at least 4 h while tumbling at room temperature, and then the wires were rinsed 3 times in 0.01 M PBS buffer. The samples were resuspended in 100 μL of either HS114 hybridization buffer or 0.3 M PBS for hybridization. Complementary DNA (CD) was added at a final concentration of 5 μM and was allowed to hybridize overnight at room temperature while tumbling. Samples were then rinsed 3 times (by centrifugation) in 0.3 M PBS and resuspended in 250 μL of the same buffer. The samples were then imaged using reflectance microscopy (see below).
Hybridization Efficiency
Two separate aliquots of NBCs patterned 000010, one containing 150 μL of wires, and the other containing 300 μL, were rinsed in water and resuspended in 0.01 M PBS buffer to their original volumes. To the 150 μL sample, 48D DNA was added, and to the 300 μL sample, NFD DNA was added for surface attachment, at a final concentration of 5 μM. The DNA was allowed to attach for 4 h at room temperature while tumbling. The samples were then rinsed 4 times in 0.01 M PBS buffer and were resuspended in 0.01 M PBS buffer to the original volumes. The 150 μL sample was then divided into a total of three tubes each containing 50 μL of wires, such that three samples were available to collect surface coverage data from (see below). The 300 μL sample was divided into 2 tubes (150 μL each). The buffer from one of the tubes was removed and replaced with HS114 hybridization buffer. A total of 10 μM 5′ TAMRA-labeled CD DNA was added to each tube and was allowed to hybridize overnight at room temperature. Both samples were then rinsed in 0.3 M PBS buffer and were further divided into three separate samples each containing 50 μL of nanowires. Mercaptoethanol was then added to these six samples for determination of surface coverage (see below).
Beacon Performance as a Function of Buffer and Beacon Length
Beacon probes were each attached to a different patterned NBC by first washing 50 L of each type of NBC 2 times in 0.01 M PBS and resuspending them in 50 μL of 0.01 M PBS. Beacon probes (either 24B, 34B, or 48B) were then added to the wires (500 μL of 5 μM probe in water) and were allowed to attach overnight at room temperature while rotating. Next, 300 μL of 0.3 M PBS were added for 2 h at room temperature while rotating, and then the samples were washed 3 times in 0.3 M PBS buffer. From these beacon coated wire samples, 3 μL aliquots were each added to separate 42 μL volumes of 0.3 M PBS, 0.5 M NaCl CAC, or HS114 buffer, and the DNA targets were hybridized at a final concentration of 5 μM while rotating at 37 °C for 1 h. The samples were then rinsed two times in 0.3 M PBS buffer before imaging.
Sensitivity of Nanowire Beacon Assay
Beacon probe (48B) was attached to NBCs patterned 00001, in the same manner as that described in the previous section. A range of target DNA concentrations was prepared such that the final concentrations of target when added to 50 μL 0.3 M PBS buffer ranged from 0 M to 1 × 10−6 M. To the separate 50 μL aliquots of target in buffer, 3 μL of beacon-coated wires were added. Beacon targets were allowed to hybridize at 50 °C for 3 h while tumbling. Samples were rinsed 3 times in 0.3 M PBS before being imaged.
Fluorescence Patterning
For each set of experiments, 50 μL of NBCs patterned 000111 were rinsed 3 times in water by centrifugation and were resuspended in 5 μM DNA beacon in 0.3 M PBS buffer at a final volume of 150 μL. Beacons used for the different dye study were sequenced the same as 34B except terminated with different fluorophores (Cy3, Cy5, or 6-FAM). For the study using different length DNA, 24B, 34B, or 48B DNA was used. Beacons were added to each tube at a final concentration of 5 μM in 150 μL of 0.3 M PBS buffer and were allowed to hybridize with gentle agitation at room temperature for 4 h. The samples were then rinsed 3 times in 0.3 M PBS and were resuspended in 50 μL of 0.3 M PBS buffer for imaging.
Imaging and Emission Intensity Quantification
Brightfield reflectance images were acquired using a Nikon TE-300 inverted microscope equipped with a 12 bit high-resolution Coolsnap HQ camera (Photometrics). A CFI plan fluor 60× oil immersion lens (N.A. = 1.4) was used in conjunction with Image-Pro Plus software (version 4.5) to image the samples. The light source was a 175 W ozone-free Xe lamp, and a Sutter Instruments filter wheel (Lambda 10–2) allowed for wavelength selection. Sample preparation for analysis by optical microscopy involved dropping a 10 μL aliquot of sample onto a glass coverslip (Fisher 12–542-C), allowing the wires to settle to the surface of the cover slip, then sandwiching the sample between the coverslip and a glass slide. All reflectance images were taken at 430 nm, due to high reflectance contrast between Au and Ag at this wavelength.38,39
Fluorescence intensity values for samples were obtained quantitatively using NBSee Software designed by Nanoplex Technologies, Inc., for identification of patterned wires and quantification of corresponding fluorescence.50 Over 30 (and typically several hundred) of each nanowire striping pattern were analyzed and averaged to generate the log normal mean fluorescence intensities (Cox’s Method of normalization).51 The error bars reported are the 95% confidence interval.
DNA Surface Coverage
Surface coverage determination methods used were similar to those described by Demers et al.52 To obtain coverages of fluorescent DNA bound to nanowires, 5 μL of mercaptoethanol were added to the nanowire solutions that remained after imaging, and the samples were allowed to tumble on a rotator at room temperature overnight. The DNA was displaced into solution, which was collected above the wires, and the fluorescence intensity was determined using a fluorolog-3 fluorimeter. This fluorimeter was equipped with a 450 W Xe lamp, a double grating excitation spectrometer, and a single grating emission spectrometer. Calibration standards were used to determine the concentration of DNA in each sample.
Results and Discussion
Characterization of fluorescence behavior on nanowires of different metals can provide insight for future design and optimization of fluorophore-based bioassays on metal substrates and guidance for other metal–fluorophore investigations. Using barcoded metal nanowires for this work enabled us to directly compare results from the two adjacent metals on each wire (Au and Ag in this case), as well as from multiple experiments performed simultaneously on particles having different patterns. This avoids irregularities from variability in lamp intensity, focus, etc. and makes direct, quantitative comparison of fluorescence on different wires and/or adjacent metals possible.
Dye–Surface Separation Dependence for Unstructured Probe DNAs: Effect of Hybridization Efficiency
True distance dependence data for quenching and SEF on metal nanowires could be acquired by coating the wires with inorganic or organic films of known thickness and attaching the chromophore to the film.8,11,14–16 However, the relevant variable for optimization of surface-based nucleic acid assays is not the absolute distance dependence of the response but rather the optimal number of nucleobases between the metal and the fluorophore. Thus, variables such as surface coverage and whether the DNA is single- or double-stranded may be as important as oligonucleotide length. To avoid potentially substantial differences in surface coverage or hybridization efficiency between different DNA probe lengths, we used a single length of DNA for all distance dependence experiments and varied the position of dye-labeled thymine bases along this length. Scheme 1 illustrates the experimental design. Dye-labeled DNAs were attached to the barcoded nanowires via 5′ thiol moieties and hybridized to unlabeled complementary strands, to increase the rigidity and extension of the DNA and to approximate DNA hybridization assay conditions. We chose to incorporate the labeled thymine bases in the thiolated strand that would be attached to the surface rather than the complementary strand, as this more closely mimics surface-bound molecular beacon probes. Rhodamine red dye was chosen based on availability for internally labeling DNA sequences and for spectral similarity to TAMRA, which we have used in molecular beacon assays.
Scheme 1.

Illustration of Distance Dependent Fluorescence Study
In this experiment, five different sequences with fluorophores positioned after the 8th, 12th, 24th, 36th, and 48th base of separate 52-base long sequences were investigated. Each of these five sequences was bound to nanowires having different barcode patterns, as illustrated in Scheme 1. Nanowire patterns were composed in each case of six ~750 nm long segments, where two of the segments were Ag (denoted in the scheme as 1) and the other four were Au (denoted in the scheme as 0). The position of the Ag segments was changed to alter the nanowire barcode pattern; for example, pattern 100001 had a Ag segment at either end, while 001100 had a two-segment Ag stripe in the center. The relative number of Au and Ag segments was held constant in these experiments in order to minimize any differences in fluorescence intensity due to possible differences in DNA surface coverage or fluorescence emission intensity between Au and Ag segments. DNA 52 bases long was used to ensure extension of a few bases beyond the furthest positioned internal fluorophore (49th base) such that similar DNA–fluorophore interactions were experienced by all of the fluorophores. Estimated fluorophore–metal separations (for the base immediately 5′ to the labeled thymine, calculated as 3.4 Å/base plus the C6 spacer)29 for the fully extended nanowire bound DNA sequences are as follows: 8th base ~3.6 nm, 12th base ~4.8 nm, 24th base ~8.6 nm, 36th base ~12.3 nm, and 48th base ~16.0 nm. The five DNA/nanowire conjugates were mixed together prior to imaging, enabling simultaneous data collection for all five labeled positions, and eliminating factors such as variations in lamp intensity between samples with time and the need for internal reference standards.53
Figure 1 shows a representative reflectance and corresponding fluorescence image from this experiment. The reflectance image enables identification of the nanowire patterns (Ag segments are brighter than Au with blue illumination), while the corresponding fluorescence images provide emission intensity.39 These data, hybridized in 0.3 M PBS buffer, suggest that base positions 8 and 12 are very efficiently quenched, while positions 24, 36, and 48 give much higher fluorescence intensities. Some patterning in the fluorescence image is also visible. We note that the degree of patterning is not constant for the different dye–metal separations (i.e., it is more noticeable when the dye is 5′ to the 24th than 48th base) and will return to this point in a later section of the manuscript.
Figure 1.

Reflectance and corresponding fluorescence images of internal rhodamine labels at five different positions within thiolated 51-base oligonucleotides attached to Au/Ag encoded metal nanowires. Unlabeled complementary strands were hybridized in 0.3 M PBS buffer. The numbers in the images indicate which DNA base is labeled for each nanowire pattern (e.g., wires with bright Ag ends and a dark Au middle in the reflectance image are coated with DNA labeled at the 24th base). Five separate samples were mixed after DNA attachment to obtain this image.
Quantification of fluorescence response was obtained from multiple reflectance and fluorescence images using NBSee data analysis software.50 This software first identified individual nanowires (i.e., clumped wires are omitted from analysis) and identified their barcode patterns based on linescans through each wire in the reflectance images. It then referred to the corresponding fluorescence images to determine the fluorescence intensity for each wire. The fluorescence values were averaged across each wire and then compiled to give an overall mean fluorescence intensity for each wire pattern (i.e., each labeled base position). Because the relative total lengths of Au (four ~750 nm segments) and Ag (two ~750 nm segments) were held constant in these experiments, averaging across each wire should provide directly comparable results uninfluenced by the nanowire striping pattern. Figure 2 gives the relative averaged fluorescence intensities of samples hybridized in two different buffers (0.3 M PBS and commercially available HS114) and with two different surface densities of thiolated DNA. We draw the reader’s attention first to the upper two curves, for which surface coverage was (7–8) × 1011 DNA/cm2. Two distinct trends were observed, depending on the hybridization buffer used. In the HS114 samples, as the position of the fluorophore was moved further from the surface of the wire, the fluorescence intensity increased (Figure 2, dashed lines). The HS114 hybridization buffer, although its composition is not revealed by the manufacturer, presumably contains surfactant (noted by the foamy nature of the buffer) and other reagents to reduce nonspecific binding. The same experiment hybridized in 0.3 M PBS buffer resulted in a different fluorescence trend (Figure 2, solid lines). The fluorescence intensities in these samples were highest with a metal–fluorophore separation of 24 bases (~8.6 nm if fully extended) from the metal surface, then remained relatively constant, and/or decreased slightly beyond that distance.
Figure 2.

Quantification of the effect of dye label position on fluorescence intensity as a function of surface coverage of DNA. Hybridization was done in two different buffers; 0.3 M PBS (solid lines) and HS114 (dotted lines). The surface coverages of fluorescent DNA attached to the nanowires are reported in the figure for each experiment. Error reported is the 95% confidence interval.
We suspected that the difference in the two buffers was due to a higher stringency for the HS114 buffer, leading to lower hybridization efficiency and consequently smaller dye–surface separations than those for the PBS buffer. Scheme 2 illustrates this concept. Sample A, which has a lower percentage of double-stranded DNA, has a smaller average dye–surface separation than that in sample B, which has higher hybridization efficiency. The 0.3 M NaCl buffer is used to overcome electrostatic repulsion due to the high density of oligonucleotides on surfaces54 and therefore can be expected to be more similar to sample B, while the commercial HS114 buffer was presumably designed for use in solution-phase hybridization reactions and consequently can be expected to more closely approximate sample A. We interpret the data in the upper two curves of Figure 2 as follows: as the dye position is moved from the 8th base to the 24th base in PBS, or the 8th to the 48th base in HS114, emission intensity increases due to decreased quenching as the metal–dye separation increases. When the experiment was performed in PBS buffer, the fluorescence intensity levels off and may decrease slightly (our experimental error makes it unclear whether the very slight decrease we see is real). Leveling off is expected for dye–metal separations too large to allow quenching. The HS114 data do not level off because, at lower hybridization efficiencies, the average position of the dyes is not as far from the metal surface as it is in the PBS buffer (Scheme 2). Although the distance dependence of quenching is the same for both samples, the relationship between dye position and actual metal–dye separation depends on hybridization efficiency. A decrease in intensity at larger separations would indicate some degree of surface enhancement of emission.
Scheme 2.

Hybridization Efficiency Impacts Average Dye–Metal Separation
The above explanation hinges on the assumption that more of the DNA on the wires in the PBS experiment is double-stranded than in the HS114 experiment. To verify this, we compared the hybridization efficiencies of the two buffers under otherwise identical conditions. Separate samples, one with fluorescently labeled DNA (48D) attached to nanowires and the other with nonfluorescent DNA of the same sequence (NFD), were prepared to determine the surface coverage of probe and amounts of hybridization, respectively. From the surface-bound fluorescent DNA sample, the surface coverage of probe was determined to be 7 × 1012 oligos/cm2. The hybridization efficiency in 0.3 M PBS was 43%, while that in HS114 buffer was just 19% (note that hybridization efficiencies near 100% for surface-bound oligonucleotides, particularly for the lengths used here, generally require special attention to steric factors55–57). These hybridization results explain the existence of the two trends identified in the distance dependence data: in PBS buffer, more than twice as much of the labeled DNA was double-stranded as compared to when HS114 buffer was used, causing more DNA to be rigid and extended further from the surface. Single-stranded DNA is flexible, allowing the fluorophore to reside closer to the surface. Thus, the average metal–dye separation is much greater for the 0.3 M PBS samples (which correspond to Scheme 2B) than for the HS114 samples (Scheme 2A). We expect that neither set of samples provides metal–dye separations as high as are calculated based on label position and assuming fully extended DNA (i.e., 16 nm for the 48th base). Bearing this in mind, our observations for optimal base position are not inconsistent with decreased quenching after approximately 5 nm from the surface. A small SEF effect cannot be ruled out; however, if SEF was substantial in this case, we would expect to see a maximum in the emission intensity at intermediate separations, followed by a decrease prior to leveling off.1–5 The fact that we see only a very slight decrease after 24 bases in the PBS buffer indicates that either the optimum separation has not yet been exceeded for this sample or SEF is not significant.
Because of the large difference in persistence length for single-stranded versus double-stranded DNA and the distance-dependence of quenching by metals, differences in fluorescence intensity due to the extent of hybridization can be quite large. Indeed, Nie and co-workers have used this to their advantage to develop a molecular beacon-style DNA detection assay in which fluorescent DNA probe strands that lacked secondary structure (i.e., no hairpin was designed into the sequence) were attached via thiols to Au nanoparticles.23 When no target was present, the DNA looped around the particles where the fluorophore was quenched, but in the presence of target, the double-stranded DNA was extended from the particle surface, greatly increasing the emission.23 To investigate the importance of having the DNA double-stranded in our experiments, we attached 5′ thiolated, 3′ TAMRA-labeled, 24-base single-stranded DNA (24S) to nanowires, and either the complementary strand was added or no target was added. The samples with complementary DNA were 2.5 times more fluorescent than those that were single-stranded, despite the facts that (i) the same number of dye molecules were present on the wires in each case and (ii) this probe sequence was designed to avoid secondary structure.
In our distance dependence experiments, hybridization efficiency of surface bound DNA was critically important in determining the overall emission intensity. Figure 2 shows data for each buffer not only for (7–8) × 1011/cm2 dye-labeled DNA (discussed above) but also for (6–8) × 1012/cm2 DNA coverage. Both sets of samples hybridized in HS114 buffer show an increase in mean fluorescence, while those in 0.3 M PBS buffer reached a maximum then leveled off as a function of labeled base position. The two experiments with lower dye-labeled DNA coverage had more than 5-fold higher average fluorescence than samples with an order of magnitude greater number of dye molecules on the surface. These data underscore the critical importance of steric hindrance in determining hybridization efficiency in surface-based nucleic acid experiments. Decreased hybridization efficiencies for the higher surface coverage samples result in more flexible, less fully extended strands and consequently greater quenching, as described above. Additionally, at higher coverages, the fluorophores may be experiencing collisional quenching with neighboring fluorophores, thus further decreasing the intensities.
Dye–Surface Separation Dependence for Molecular Beacon-Style Probe DNAs: Effect of Secondary Structure
The principal difference between the DNA sequences used in the experiments described above and those used in molecular beacon-style experiments is the presence of secondary structure in the beacon probe sequences. To determine how well our predictions from the fluorophore-distance study translated to the design of DNA beacon assays, we studied three different length hairpin probe sequences (24, 34, and 48 bases in length, each with an identical 5 base pair “stem” region, and different “loop” lengths) attached to barcoded nanowires. To determine the separation dependence of fluorescence intensity, each sequence was hybridized to a complementary target sequence— C24B, C34B, or C48B— so that the fluorescence emission would not be quenched by the metal surface (as it should be for the beacon probes in the absence of target). Hybridization was performed in both the 0.3 M PBS and HS114 buffers used for distance dependence experiments and also in a third, higher salt buffer (0.5 M NaCl CAC) introduced by Krauss and co-workers for beacon probes on planar Au surfaces.35 Figure 3 shows the relative fluorescence intensities after hybridization for each beacon probe length in each of the three buffers; fluorescence intensities have been normalized to a scale of 0 to 100 for each of the three different buffers, in order to highlight length-dependent differences between the probes. The 34-base beacon probe gave the highest fluorescence intensity, with both longer and shorter probes giving lower intensities. This trend was observed for all three buffers, including the lowest hybridization efficiency HS114 buffer, for which unstructured probes gave higher intensities at the 48th than 36th position (Figure 2). We interpret this difference between the data in Figures 2 and 3 as resulting from probe secondary structure. For molecular beacon probes, unlike traditional single-stranded DNA probes (i.e., without secondary structure), fluorescence emission is quenched in the absence of target DNA, because the hairpin structure holds the fluorophore very close to the metal surface (Scheme 3). Thus, decreased hybridization efficiencies should not substantially change the average dye–metal separation for the subset of dyes that are fluorescent. The magnitude of emission intensity should increase with increasing hybridization efficiency, but the separation dependence of emission should not.
Figure 3.

Effect of molecular beacon probe length and hybridization buffer on fluorescence intensity after exposure to complementary DNA in various buffers: 0.3 M PBS (black bars), 0.5 M NaCl CAC (white bars), or HS114 (gray bars). Error reported is the 95% confidence interval.
Scheme 3.

Hybridization of Surface-Bound Molecular Beacon Probes
The decrease in intensity between the 34- and 48-base beacon probes in all three buffers (Figure 3) was greater than that between the 36th and 48th base positions of the unstructured probes in the higher hybridization efficiency 0.3 M PBS buffer (Figure 2). This may indicate some surface enhancement of fluorescence, in that the average metal–dye separation for the 48-base beacon probes is greater than that for the 48th labeled base of the unstructured probes. Thus, it is possible that the 0.3 M PBS buffer data in Figure 2 do not sample large enough metal–dye separations to see the decrease in emission intensity at long distances expected for SEF but that the 48-base beacon in Figure 3 does. Unfortunately, interpretation is complicated by potential differences in hybridization efficiency between the different beacon probes, which could not be accounted for experimentally in the same way as for unstructured probes (i.e., it was not possible to keep probe length constant and thus hold surface coverage and hybridization efficiency constant for the beacon probes, since they must have the dye on a terminal base). For the data shown in Figure 3, probe surface coverage was held approximately constant ((1.2–2.5) × 1011 probes/cm2) for all probe lengths to minimize differences in steric effects between the samples. Nonetheless, we cannot entirely rule out the possibility that the decreased intensity for the 48-base beacon arises from a smaller number of target binding events (i.e., unquenched probes) on the surface as compared to the 24- and 34-base probes, rather than a decrease in SEF as the separation increases. Regardless, the fact that the same trend was observed for all three buffers tested indicates that the optimal fluorophore label position in beacon probes is insensitive to hybridization efficiency, unlike the unstructured probes in Figure 2.
Some comments can be made on the differences between the results for the three buffers in Figure 3. Changes in intensity as a function of probe length are smallest in HS114 buffer, which gave lower hybridization efficiencies for traditional DNA binding experiments as described above. Formation of secondary structure, like hybridization to target strands, requires overcoming electrostatic repulsions. Thus, a more stringent buffer such as HS114 favors both less target binding (i.e., less fluorescence) and also less hairpin folding (i.e., more fluorescence). The fraction of fluorescence arising from unfolded probes should be lowest for the higher ionic strength buffers (0.3 M PBS and CAC); thus we can expect these buffers to best approximate a fully double-stranded response. Clearly, differences in intensity due to the effects of the hybridization buffer are more complicated for beacons than for nonhairpin sequences. Stringency conditions for optimizing hybridization of unstructured probes are not directly applicable to beacon probe assays, as the stem interactions must also be considered.
The fact that the 34-base beacon probe gave the highest intensity response regardless of the stringency of the hybridization buffer suggests that the fluorescence response of molecular beacon probes will be more straightforward than that for unstructured probes such as those in Figure 2. This is because, while the conformational flexibility of unstructured single-stranded DNA complicates the observed separation-dependence of emission (Scheme 2), beacon probes have limited conformational possibilities (quenched hairpins or fully extended duplexes; Scheme 3).29 This is critically important for bioassay applications, since the major factor responsible for the degree of hybridization on the surface in a bioassay will be solution target concentration, i.e., the quantity to be measured, rather than choice of buffer, which would be held constant under assay conditions.
We investigated the effect of target concentration (i.e., hybridization efficiency) for a 48-base beacon probe on the nanowire surface. Figure 4 shows the fluorescence intensity of each assay containing different concentrations of DNA target. As the concentration of target DNA was decreased, the fluorescence intensity also decreased, over several orders of magnitude in dynamic range. This suggests that the distance dependence effects did not unduly complicate the fluorescence response although the percent hybridization was changed. A common figure of merit for molecular beacon assays is the quenching efficiency, QE, defined as the ratio of fluorescence present when quenched versus unquenched, which is calculated as [1 − (signal in absence of target/signal with target)]%. In this assay, the quenching efficiency calculated at 1 μM target was 88% (see Figure 4 inset). The binding curve suggests that saturation may not have been achieved at 1 μM target DNA, the highest concentration tested. Thus, the 88% QE reported here might be improved at higher target concentrations. Since our focus here was on the separation dependence, we did not optimize assay conditions before acquiring the data in Figure 4. Nonetheless, there was a substantial change in the fluorescence between the target and no target samples, such that the presence or absence of target was apparent. The sensitivity of this assay is in the 100 pM to 10 nM range in its current form (<1 nM limit of detection based on average intensity for the no target control plus twice the standard deviation)58 and is limited by inefficient quenching. This is comparable to sensitivities reported by other groups for surface-bound molecular beacon assays.32,33,35 Typical quenching efficiencies for surface-bound beacons are in the 80 to 95% range,32,42,53,54 with the highest reported values >99% from Krauss and co-workers for beacons bound to planar Au.35 Libchaber and co-workers reported quenching efficiencies of 99.97% for solution-based beacon structures where organic quenchers were replaced by 1.4 nm Au clusters.30
Figure 4.
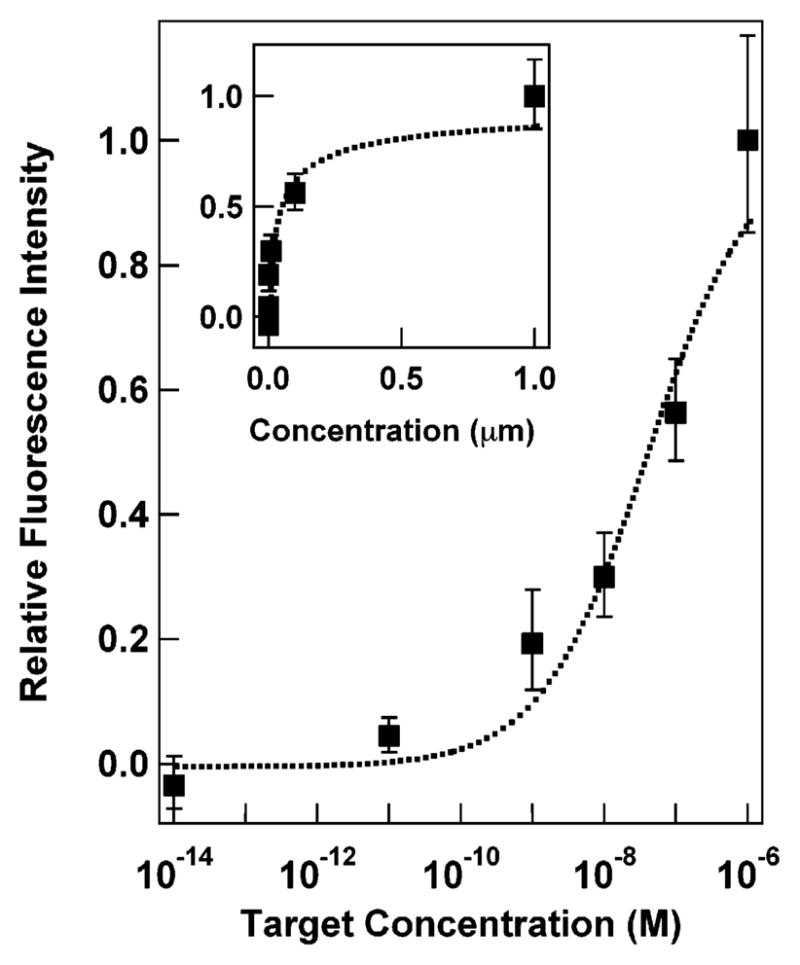
Mean fluorescence response of a nanowire-based molecular beacon assay as a function of DNA target concentration, plotted on a log scale. Inset shows the same data plotted on a linear scale. Dotted lines are Sips isotherm fits, with Ka = 2.7 × 107 and a = 0.6. Background signal from a no target control has been subtracted from each data point. Error bars represent the 95% confidence interval.
Fluorescence Patterning Phenomena
Some patterning of fluorescence intensity on the Au/Ag striped nanowires is observable in Figure 1. Indeed, we often observe patterned fluorescence intensities on the nanowires. We have previously reported fluorescence patterning in sandwich immunoassays, where the dye molecules were bound to the secondary antibodies (i.e., relatively far from the metal surface). In those earlier experiments, the fluorescence patterning we observed correlated with the reflectivities of the underlying metals.38 Thus, although the dyes were located in the near field of the metal nanowires, useful predictions of patterning could be made simply by treating the wire as a variably reflective mirror (i.e., shorter wavelengths of excitation and emission were more efficiently reflected from Ag as compared with Au, in accordance with the wavelength-dependent bulk reflectivities of these metals). A key difference between those experiments and the experiments described here is the magnitude and variability of the dye–surface separation. Here, rather than holding the separation constant at ~15 nm, we are varying it between ~3.6 nm and ~16 nm, depending on the labeled probe position (0.34 nm per base plus the 6 carbon spacer). Additionally, while the surface attachment chemistry was insensitive to metal identity in the immunoassay data, thiols could be expected to give higher coverages on clean Ag as compared to Au59,60 or lower coverage on partially oxidized Ag. We expect that the surface density of DNA is dominated by steric and electrostatic repulsions between the surface-bound oligonucleotides to a greater extent than the identity of the underlying metal. However, differences in DNA coverage, if present, could influence patterning (either directly due to a greater number of dye molecules or indirectly due to a difference in hybridization efficiency and therefore less extension from the surface). Our prior work has demonstrated that storage in ethanol or citrate-containing PBS prevents Ag oxidation.61 All of the experiments reported here were performed under conditions designed to avoid Ag oxidation (i.e., wires were used immediately after coating in DNA or stored in citrate PBS buffer until use); thus, extensive Ag oxidation can be ruled out. Surface coverage experiments for thiolated oligonucleotides on nanowires having different lengths of Ag gave coverages of (4 ± 1.5) × 1012 and (4 ± 1.7) × 1012 beacon probes per cm2, respectively, indicating that any differences in coverage on the two metals are smaller than our experimental error.
While studying different length DNA beacon probes on striped metal nanowires, we noticed that fluorescence patterning varied with dye–metal separation (Figure 1). Figure 5 explores this effect further: reflectance and corresponding fluorescence images are shown for Ag/Au striped nanowires coated with internally rhodamine labeled, thiolated sequences 8D through 48D and hybridized to complementary DNA in 0.3 M PBS buffer. Reflectance and fluorescence linescans for a representative wire from those samples with appreciable fluorescence intensity are shown. Strong fluorescence patterning is observed for rhodamine after the 12th and 24th base positions, with intensities on average ~3-fold brighter on the silver segments than the gold. At a distance of 36 bases, the rhodamine dye shows less distinct patterning, barely discernible on some wires in this sample. Rhodamine positioned 48 bases from the surface also shows very little patterning. Although small (i.e., less than the standard deviation of our coverage measurements) differences in DNA surface density on Au vs Ag segments cannot be entirely ruled out, and if present could give rise to a slight patterning, the changes in fluorescence patterning observed here cannot be explained by differences in coverage. The overall coverage is the same for sequences 8D through 48D, as are any slight differences in coverage on the different metals. Thus, we ascribe these changes in fluorescence patterning to separation-dependent electromagnetic interactions between the dye molecules and the metal surface.
Figure 5.

Reflectance and corresponding fluorescence images of nanowires coated with 5′ thiolated oligonucleotides internally labeled with rhodamine red-X dye at different base positions. Linescans correspond to the wires indicated by arrows. The left axis corresponds to the black trace (reflectance), and the red trace corresponds to the right axis (fluorescence).
More striking changes in fluorescence patterning were observed for greater changes in dye–metal separation. Figure 6 shows reflectance and corresponding fluorescence images and linescans for half gold–half silver nanowires coated with TAMRA-labeled DNA sequences of various lengths after hybridization to complementary strands. The Ag portion, identified based on its greater intensity in the 430 nm reflectance image, has been positioned to the left in each image for ease of comparison. All experiments in this figure were conducted under identical conditions (0.3 M PBS buffer). The shortest length DNA (24 bases) shows fluorescence patterning that matches the reflectance patterning of the nanowires, where silver is brighter and gold is darker. This result is qualitatively similar to what was seen in Figure 5 for the internal rhodamine labels after the 24th base position, despite the change to the TAMRA dye. To achieve greater metal–dye separations, we compared this result with intensities for 34- and 48-base molecular beacon probes with 5′ thiols and 3′ TAMRA. Probes with secondary structure were used in order to maximize the extension from the surface upon hybridization. For beacon probes 34 bases in length, approximately uniform fluorescence is observed on Au and Ag segments. Beacon probes 48 bases long exhibited a reversal in fluorescence patterning, such that fluorescence intensities from TAMRA on the Au segments were brighter than those on Ag segments of the wires. The observed changes in surface patterning arose primarily from changes in the emission intensity for TAMRA on the Ag segments, while emission on the Au segments was relatively insensitive to metal–dye separation.
Figure 6.

Linescans and corresponding reflectance (top) and fluorescence (bottom) images of half Ag/half Au nanowires coated with 5′ thiol, 3′ TAMRA probe oligonucleotides of different lengths. The 24-base probe did not have secondary structure; however the 34- and 48-base probes were molecular beacons with self-complementarity at the 5′ and 3′ ends. Unlabeled complementary DNA was present in all cases to extend the probes from the surface. The black trace (reflectance) corresponds to the axis on the left, and the red trace (fluorescence), to the axis on the right.
The observation of fluorescence reversal in the TAMRA-labeled beacon probe experiments, as compared with just a reduction in the degree to which fluorescence was brighter on Ag vs Au in the rhodamine-labeled unstructured probe experiments, could be due to differences in the dye and/or to the greater metal–dye separations probed in the beacon experiment. Unfortunately, we were unable to purchase rhodamine-labeled beacon probes to directly test which of these effects was dominant. However, the excitation and emission wavelengths for rhodamine (588 and 608 nm, respectively) are at longer wavelengths than those for TAMRA (559 and 583 nm). Our previous studies of patterned fluorescence using sandwich immunoassays on Au/Ag striped nanowires showed that chromophores having excitation and emission wavelengths at which the underlying metal was more reflective appeared brighter in the fluorescence microscope image.38 Ag metal is somewhat more reflective than Au in the yellow and equally reflective in the red. Therefore, based on reflectivity arguments,38 TAMRA is expected to give either the same or lower relative intensities on Au segments as compared to rhodamine, rather than the higher intensity on Au which is observed here. Thus, while dye-specific properties cannot be ruled out as the cause of these differences, we interpret the stronger apparent distance-dependence of the TAMRA-labeled beacon probes (Figure 6) as arising from the greater average surface separation for the dye molecules responsible for emission in this experiment as compared to those lacking secondary structure (Figure 5).
The nature of the metal–fluorophore interaction depends not only on the separation between them, and the identity of the metal, but also on the identity of the dye. For example, low quantum yield dyes have been reported to exhibit particularly impressive surface enhanced fluorescence on colloidal silver surfaces.6,62 In the case of molecular beacon probes, additional properties may become important. For example, efficient quenching of the dye may benefit from weak adhesion between the dye and the metal surface. The size of the dye and electrostatic considerations may also come into play, potentially impacting the ability of the hairpin structures to fold and/or unfold. We compared four different fluorescent dyes with a range of excitation/emission frequencies, extinction coefficients, and quantum yields (Table 2). In these experiments, 5′ thiolated beacon probes with the dyes at their 3′ end were attached to nanowires composed of half Ag and half Au. The chemical structures of these dyes are also different, with greater positive charge and less condensed structures for the two cyanine dyes as compared with the FAM and TAMRA.
Table 2.
Properties of Fluorescent Dyes Studied
| dye | λex (nm) | λem (nm) | ελmax (M−1 cm−1) | quantum yield |
|---|---|---|---|---|
| 6-FAM | 495 | 520 | 75 000 | 0.9 |
| Cy 3 | 550 | 564 | 150 000 | 0.1 |
| TAMRA | 559 | 583 | 89 000 | 0.7 |
| Cy 5 | 648 | 668 | 250 000 | 0.2 |
We compared 34-base molecular beacon-style probes with TAMRA, Cy3, Cy5, and 6-FAM fluorophores, after exposure to unlabeled complementary DNA to extend the probe stands. Figures 6 (middle panel) and 7 show results for each of these dyes, on half Au/half Ag nanowires. Several differences between the dyes can be observed. Cy3 gave the highest peak intensity, with a sharp spike of fluorescence near the Au/Ag interface at the center of the wire and smaller spikes at both ends. Cy5 also showed sharp spikes of intense emission corresponding to the Au/Ag interface, the edges of the wire, and a defect in the Ag segment that appears dark in the reflectance image. In contrast, TAMRA and 6-FAM appear to be less sensitive to edges and surface defects in the wires. These regions of intense fluorescence response most likely arise from SEF at regions of heightened electromagnetic fields.1–5 This interpretation is consistent with the positions of these regions (edges and pits, which have greater roughness). SEF is expected to be greater for dyes having low quantum yields,6,62 which could explain the higher sensitivity for surface roughness exhibited by the two cyanine dyes as compared with 6-FAM and TAMRA.63
In addition to fluorescence patterns caused by SEF, emission intensities for dyes on Ag and Au portions of the wires can be compared. TAMRA and Cy3 exhibited roughly equal intensities on Au and Ag segments of the nanowires, such that one segment could not be readily distinguished from the other in the fluorescence images. On the other hand, 6-FAM dye was much brighter on Ag than Au, and Cy5 was somewhat brighter on Au than Ag. These differences correlate with differences in excitation and emission wavelengths of the dyes. 6-FAM is the shortest wavelength dye, while TAMRA and Cy3 are similar in color, and Cy5 was the longest wavelength dye investigated here. However, since the data in Figures 5 and 6 show metal–dye separation can alter the relative intensities on Au and Ag segments, it is clear that more than excitation and emission wavelengths for the dyes are involved in the patterning. Other factors may include differences in the distance dependence of electromagnetic field distribution above the two metals.
Finally, we note that, in related experiments reported by Sha et al., the effect of dye selection on quenching efficiency for nanowire beacon bioassays was investigated. Of the dyes tested (Cy5, FITC, Rhodamine 6G, Texas Red, and TAMRA), Texas Red gave the best performance, with 94% QE. TAMRA gave 89% QE and was chosen over Texas Red for further studies due to its commercial availability.37 Dye performance in our experiments gave comparable results, with Cy3 giving the highest overall intensity, 6-FAM giving the lowest overall intensity, and TAMRA giving the best QE.
Conclusions
The promise of metallic surfaces and nanostructures, including barcoded metal nanowires, for fluorescence-based bioanalysis led us to investigate factors that influence emission from surface-bound dyes. Emission from internal rhodamine dye in 52-base DNA on striped metal nanowires revealed that quenching was significant up to the 12th base position. When dyes were incorporated after the 24th to 48th base position, much brighter fluorescence was observed. The shape of the intensity–separation response was sensitive to the hybridization efficiency of the surface-bound probes for complementary DNA (i.e., percent double-stranded), dictated by the hybridization buffer used. Samples with ~20% hybridization efficiency showed increased emission with increased separation from the metal surface, with samples with ~40% hybridization efficiency showing an increase up to 24 bases, and a leveling off or slight decrease in intensity at greater separations. These differences result from the greater metal–dye separation for double-stranded as compared to single-stranded DNA (i.e., the average separation was greater for the samples with twice as much double-stranded DNA). Increasing the surface coverage of fluorescently tagged DNA decreased the fluorescence intensity, due to steric inhibition of hybridization, which led to increased quenching because less of the DNA was in its fully extended double-stranded form.
In contrast, the separation-dependent response of dye-labeled oligonucleotides designed to form haripin secondary structures analogous to solution-phase molecular beacons were relatively insensitive to the hybridization buffer. The formation of secondary structure in the absence of complementary DNA leads to efficient quenching of emission. This means that any fluorescence observed should arise from probes which have bound complementary strands and results in less complicated target response curves for probes having secondary structure than might be expected for metal-bound, chromophore-labeled probes otherwise.
Selection of striped metal nanowires, composed of identifiable patterns of Ag and Au segments, enabled us to perform many of these experiments in a multiplexed fashion for direct, quantitative comparisons, i.e., between different dye labeling positions. In addition, differences in emission intensity from chromophores on the Ag vs Au segments of the nanowires could be readily compared. We observed differences in fluorescence patterning as a function of metal–chromophore separation, with emission from TAMRA-labeled DNA changing from brighter on Ag for 24-base probes to brighter on Au for 48-base probes. While our results do not suggest substantial surface enhancement of fluorescence under the conditions of our experiments, anomalously bright emission noted from the ends, metal–metal interfaces, and surface defects does suggest some SEF at these sites of greater nanoscale roughness.
This research provides insight into the importance of DNA conformation and hybridization efficiency on performance in fluorescence-based experiments on metal surfaces and points to the benefits of encoded nanowires for multiplexed surface characterization. Our findings are relevant to the design of bioassays at metal surfaces, particularly those based on hybridization to surface-bound probe DNA strands. Differences between the fluorescence responses of probe sequences designed to avoid versus encourage the formation of secondary structure should be taken into account in assay design and interpretation.
Figure 7.

Linescans with corresponding reflectance (top) and fluorescence (bottom) images of nanowires coated with 5′ thiol, 3′ fluorophore, 34-base molecular beacon probes, for three fluorophores. The black traces (reflectance) correspond to the left axis, and the colored traces (fluorescence) correspond to the right axis.
Acknowledgments
This work was funded by the National Institutes of Health (R01 EB000268), the National Science Foundation (NIRT CCR-0303976), and Pennsylvania State University. CDK also acknowledges support from a Beckman Foundation Young Investigator Award, a Sloan Fellowship, and a Dreyfus Teacher-Scholar Award.
References
- 1.Moskovits M. Rev Mod Phys. 1985;57:783–826. [Google Scholar]
- 2.Chance RR, Prock A, Silbey R. Adv Chem Phys. 1978;37:1–65. [Google Scholar]
- 3.(a) Lakowicz JR. Anal Biochem. 2001;298:1–24. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lakowicz JR. Anal Biochem. 2005;337:171–194. doi: 10.1016/j.ab.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meitiu H. Prog Surf Sci. 1984;17:153–320. [Google Scholar]
- 5.Neumann T, Johansson ML, Kambhampati D, Knoll W. Adv Funct Mater. 2002;12:575–586. [Google Scholar]
- 6.Weitz DA, Garoff S, Gersten JI, Nitzan A. J Chem Phys. 1983;78:5324–5338. [Google Scholar]
- 7.Corni S, Tomasi J. J Chem Phys. 2003;118:6481–6494. [Google Scholar]
- 8.(a) Sokolov K, Chumanov G, Cotton TM. Anal Chem. 1998;70:3898–3905. doi: 10.1021/ac9712310. [DOI] [PubMed] [Google Scholar]; (b) Chumanov G, Sokolov K, Gregory BW, Cotton TMJ. J Phys Chem. 1995;99:9466–9471. [Google Scholar]
- 9.Whitmore PM, Robata HJ, Harris CB. J Phys Chem. 1982;77:1560–1568. [Google Scholar]
- 10.Yokota H, Saito K, Yanagida T. Phys Rev Lett. 1998;80:4606–4609. [Google Scholar]
- 11.Knobloch H, Brunner H, Leitner A, Aussenegg F, Knoll W. J Phys Chem. 1993;98:10093–10095. [Google Scholar]
- 12.(a) Huang X, Lee KT, George TF. J Phys Chem. 1986;85:567–572. [Google Scholar]; (b) Glass AM, Liao PF, Bergman JG, Olson DH. Opt Lett. 1980;5:368–370. doi: 10.1364/ol.5.000368. [DOI] [PubMed] [Google Scholar]; (c) Wokaun A, Lutz HP, King AP, Wild UP, Ernst RR. J Phys Chem. 1983;79:509–514. [Google Scholar]; (d) Amos RM, Barnes WL. Phys Rev B. 1999;59:7708–7714. [Google Scholar]; (e) Alivisatos AP, Waldeck DH, Harris CB. J Chem Phys. 1985;82:541–547. [Google Scholar]; (f) Zhang J, Malicka J, Gryczynski I, Lakowicz JR. J Chem Phys B. 2005;109:7643–7648. doi: 10.1021/jp0490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aslan K, Lakowicz JR, Geddes CD. Anal Bioanal Chem. 2005;382:926–933. doi: 10.1007/s00216-005-3195-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kummerlen J, Leitner A, Brunner H, Aussenegg FR, Wokaun A. Mol Phys. 1993;80:1031–1046. [Google Scholar]
- 15.Tarcha PJ, DeSaja-Gonzalez J, Rodriguez-Llorente S, Aroca R. Appl Spectrosc. 1999;53:43–48. [Google Scholar]
- 16.Schneider G, Decher G, Nerambourg N, Praho R, Werts MHV, Blanchard-Desce M. Nano Lett. 2006;6:530–536. doi: 10.1021/nl052441s. [DOI] [PubMed] [Google Scholar]
- 17.Tovmachenko OG, Graf C, van den Heuvel DJ, van Blaaderen A, Gerritsen HC. Adv Mater. 2006;18:91–95. [Google Scholar]
- 18.Hong B, Kang KA. Biosens Bioelectron. 2006;21:1333–1338. doi: 10.1016/j.bios.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Stranik O, McEvoy HM, McDonagh C, MacCraith BD. Sens Actuators, B. 2005;107:148–153. [Google Scholar]
- 20.Zhang J, Lakowicz JR. J Phys Chem B. 2006;110:2387–2392. doi: 10.1021/jp055370u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liebermann T, Knoll W, Sluka P, Herrmann R. Colloids Surf, A. 2000;169:337–350. [Google Scholar]
- 22.Perez-Luna VH, Yang S, Rabinovich EM, Buranda T, Sklar LA, Hampton PD, Lopez GP. Biosens Bioelectron. 2002;17:71–78. doi: 10.1016/s0956-5663(01)00260-3. [DOI] [PubMed] [Google Scholar]
- 23.Maxwell DJ, Taylor JR, Nie S. J Am Chem Soc. 2002;124:9606–9612. doi: 10.1021/ja025814p. [DOI] [PubMed] [Google Scholar]
- 24.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 25.Heyduk T, Heyduk E. Nat Biotechnol. 2002;20:171–175. doi: 10.1038/nbt0202-171. [DOI] [PubMed] [Google Scholar]
- 26.Fang X, Li JJ, Tan W. Anal Chem. 2002;72:3280–3285. doi: 10.1021/ac991434j. [DOI] [PubMed] [Google Scholar]
- 27.Alberts DP, Parman JM, Goddard NL. Biophys J. 2003;84:3212–3217. doi: 10.1016/S0006-3495(03)70045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antony T, Thomas T, Sigal LH, Shirahata A, Thomas TJ. Biochemistry. 2001;40:9387–9395. doi: 10.1021/bi010397z. [DOI] [PubMed] [Google Scholar]
- 29.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Proc Natl Acad Sci USA. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubertret B, Calame M, Libchaber AJ. Nat Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- 31.(a) Fang X, Liu X, Schuster S, Tan W. J Am Chem Soc. 1999;121:2921–2922. [Google Scholar]; (b) Brown L, Cummins J, Hamilton A, Brown T. Chem Commun. 2000:621–622. [Google Scholar]
- 32.Liu X, Tan W. Anal Chem. 1999;71:5054–5059. doi: 10.1021/ac990561c. [DOI] [PubMed] [Google Scholar]; (b) Liu X, Farmerie W, Schuster S, Tan W. Anal Biochem. 2000;283:56–63. doi: 10.1006/abio.2000.4656. [DOI] [PubMed] [Google Scholar]; (c) Yao G, Tan W. Anal Biochem. 2004;331:216–223. doi: 10.1016/j.ab.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Steemers F, Ferguson JA, Walt DR. Nat Biotechnol. 2000;18:91–94. doi: 10.1038/72006. [DOI] [PubMed] [Google Scholar]; (b) Epstein JR, Leung APK, Lee KH, Walt DR. Biosens Bioelectron. 2003;18:541–546. doi: 10.1016/s0956-5663(03)00021-6. [DOI] [PubMed] [Google Scholar]
- 34.Rang H, Li J, Liu H, Liu Q, Mei Q, Wang Y, Zhu J, He N, Lu Z. Nucleic Acids Res. 2002;30:e61. doi: 10.1093/nar/gnf061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du H, Strohsahl CM, Camera J, Miller BL, Krauss TD. J Am Chem Soc. 2005;127:7932–7940. doi: 10.1021/ja042482a. [DOI] [PubMed] [Google Scholar]
- 36.Du H, Disney MD, Miller BL, Krauss TD. J Am Chem Soc. 2003;125:4012–4013. doi: 10.1021/ja0290781. [DOI] [PubMed] [Google Scholar]
- 37.Sha MY, Yamanaka M, Walton ID, Norton SM, Stoermer RL, Keating CD, Natan MJ, Penn SG. NanoBiotechnology. 2005;1:327–335. doi: 10.1385/NBT:1:4:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicewarner-Pena SR, Carado AJ, Shale KE, Keating CD. J Phys Chem B. 2003;107:7360. [Google Scholar]
- 39.Nicewarner-Pena SR, Freeman RG, Reiss BD, He L, Pena DJ, Walton ID, Cromer R, Keating CD, Natan MJ. Science. 2001;294:137–141. doi: 10.1126/science.294.5540.137. [DOI] [PubMed] [Google Scholar]
- 40.Keating CD, Natan MJ. Adv Mater. 2003;15:451–454. [Google Scholar]
- 41.Lakowicz JR, Shen B, Gryczynski Z, D’ Auria S, Gryczynski I. Biochem Biophys Res Commun. 2001;286:875–879. doi: 10.1006/bbrc.2001.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malicka J, Gryczynski I, Fang J, Kusba J, Lakowicz JR. J Fluorescence. 2002;12:439–447. doi: 10.1023/A:1021370111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malicka J, Gryczynski I, Fang J, Kusba J, Lakowicz JR. Anal Biochem. 2003;315:160–169. doi: 10.1016/S0003-2697(02)00710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malicka J, Gryczynski I, Maliwal BP, Fang J, Lakowicz JR. Biopolymers (Biospectroscopy) 2003;72:96–104. doi: 10.1002/bip.10301. [DOI] [PubMed] [Google Scholar]
- 45.Malicka J, Gryczynski I, Lakowicz JR. Biochem Biophys Res Commun. 2003;306:213–218. doi: 10.1016/S0006-291X(03)00935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malicka J, Gryczynski I, Gryczynski Z, Lakowicz JR. Anal Chem. 2003;75:6629–6633. doi: 10.1021/ac034881e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malicka J, Gryczynski Z, Lakowicz JR. Anal Biochem. 2003;315:57–66. doi: 10.1016/S0003-2697(02)00702-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rant U, Arinaga K, Fujita S, Yokoyama N, Abstreiter G, Tornow M. Langmuir. 2004;20:10086–10092. doi: 10.1021/la0492963. [DOI] [PubMed] [Google Scholar]
- 49.Bloomfield VA, Crothers DM, Tinoco I., Jr . Nucleic Acids: Structures, Properties, and Functions. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 50.Walton ID, Norton SM, Balasingham A, He L, Oviso DF, Jr, Gupta D, Raju PA, Natan MJ, Freeman RG. Anal Chem. 2002;74:2240–2247. doi: 10.1021/ac020073w. [DOI] [PubMed] [Google Scholar]
- 51.Zhou XH, Gao S. Stat Med. 1997;16:783–790. doi: 10.1002/(sici)1097-0258(19970415)16:7<783::aid-sim488>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 52.Demers LM, Mirkin CA, Mucic RC, Reynolds RA, III, Letsinger RL, Elghanian R, Viswanadham G. Anal Chem. 2000;72:5535–5541. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]
- 53.Wehry EL. Molecular Fluorescence and Phosphorescence Spectrometry. In: Settle FA, editor. Instrumental Techniques for analytical chemistry. Prentice Hall; Upper Saddle River, NJ: 1997. pp. 507–539. [Google Scholar]
- 54.(a) Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Science. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]; (b) Jin R, Wu G, Li Z, Mirkin CA, Schatz GC. J Am Chem Soc. 2003;125:1653–1654. doi: 10.1021/ja021096v. [DOI] [PubMed] [Google Scholar]
- 55.(a) Herne TM, Tarlov MJ. J Am Chem Soc. 1997;119:8916–8920. [Google Scholar]; (b) Steel AB, Levicky TM, Herne TM, Tarlov MJ. Biophys J. 2000;79:975–981. doi: 10.1016/S0006-3495(00)76351-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) Shchepinov MS, Case-Green SC, Southern EM. Nucleic Acids Res. 1997;25:1155–1161. doi: 10.1093/nar/25.6.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Southern E, Mir K, Shchepinov M. Nat Genet. 1999;21:5–9. doi: 10.1038/4429. [DOI] [PubMed] [Google Scholar]
- 57.Nicewarner-Peña SR, Raina S, Goodrich GP, Fedoroff NV, Keating CD. J Am Chem Soc. 2002;124:7314–7323. doi: 10.1021/ja0177915. [DOI] [PubMed] [Google Scholar]
- 58.Harris DC. Quantitative Chemical Analysis. 4. W. H. Freeman and Company; New York: 1995. [Google Scholar]
- 59.Herrwerth S, Eck W, Reinhardt S, Grunze M. J Am Chem Soc. 2003;125:9359–9366. doi: 10.1021/ja034820y. [DOI] [PubMed] [Google Scholar]
- 60.Ulman A. Chem Rev. 1996;96:1533–1554. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 61.Stoermer RL, Sioss JA, Keating CD. Chem Mater. 2005;17:4356–4361. [Google Scholar]
- 62.Maliwal BP, Malicka J, Gryczynski I, Gryczynski Z, Lakowicz JR. Biopolymers (Biospectroscopy) 2003;70:585–594. doi: 10.1002/bip.10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. [accessed May 27, 2006]; www.idtdna.com.