

Abstract
We have previously shown that γδ T cells traffic to the CNS during EAE with concurrent increased expression of β2-integrins and production of IFN-γ and TNF-α. To extend these studies, we transferred bioluminescent γδ T cells to wild type mice and followed their movement through the acute stages of disease. We found that γδ T cells rapidly migrated to the site of myelin oligodendrocyte glycoprotein (MOG) peptide injection and underwent massive expansion. Within six days after EAE induction, bioluminescent γδ T cells were found in the spinal cord and brain, peaking in number between days ten and twelve and then rapidly declining by day fifteen. Reconstitution of γδ T cell−/− mice with γδ T cells derived from β2-integrin-deficient mice (CD11a, -b or -c) demonstrated that γδ T cell trafficking to the CNS during EAE is independent of this family of adhesion molecules. We also examined the role of γδ T cell-produced IFN-γ and TNF-α in EAE and found that production of both cytokines by γδ T cells was required for full development of EAE. These results indicate that γδ T cells are critical for the development of EAE and suggest a therapeutic target in demyelinating disease.
Keywords: EAE/MS, T cells, neuroimmunology, transgenic/knockout mice
Introduction
γδ T cells are one of several T cell subsets that contribute to the development of experimental autoimmune encephalomyelitis (EAE), a T cell-mediated autoimmune disease of the central nervous system that mimics many aspects of the human disease multiple sclerosis (MS) [1–3]. Cellular infiltration of the brain and spinal cord by several leukocyte subsets, including γδ T cells [4–8], is a characteristic feature of both EAE and MS [1, 3]. Although it has been appreciated for some time that γδ T cells produce cytokines that contribute to the pro-inflammatory milieu [9–15] and express adhesion molecules that may be critical for initial priming, trafficking to, and infiltration of the CNS [15–17], the significance of their contribution to demyelinating disease remains controversial.
γδ T cells are considered innate immune T cells by virtue of their limited T cell receptor repertoire, tissue-specific homing patterns, and recognition of non-traditional T cell antigens [18–21]. Activation of γδ T cells occurs on presentation of phosphoantigens, WC1 molecules or self-antigens by non-classical MHC molecules with cytokines and TLRs providing co-stimulation [22–30]. Regardless of the priming event(s), trafficking mechanisms employed by γδ T cells in demyelinating disease remain ill-defined. Studies have implicated VLA-4 as a participant in γδ T cell adhesion to endothelium, epithelium or fibroblasts, and in transmigration [16, 17, 31, 32], but none have implicated VLA-4 in migration of γδ T cells to the CNS. In contrast, γδ T cells express all four members of the β2-integrin family of adhesion molecules and expression increases through the course of MOG-induced EAE [15]. Importantly, deletion of three of the four β2-integrins (CD11a–c) results in significantly attenuated disease [33–36], implicating, but not directly proving, a role for these adhesion molecules in γδ T cell trafficking into the CNS during disease.
Cytokine-mediated modulation of demyelinating disease by γδ T cells, either in humans or animal models, although the subject of numerous studies, also remains controversial. Early studies examined for the contribution of γδ T cell-produced cytokines and chemokines after antibody-mediated depletion of γδ T cells [10, 11] and implicated these cells in the production of TNF-α, IFN-γ, IL-1, IL-6, IL-12 and several others. Although informative, these studies could not directly attribute cytokine production to γδ T cells as they used spinal cord homogenate to analyze cytokine levels. In addition, γδ T cell depletion was not monitored over the disease course in these studies raising the possibility of undefined levels of γδ T cell-mediated cytokine production as disease progressed. Other studies have shown that γδ T cells producing IFN-γ and IL-4 are significantly elevated during EAE [12]. Similar results were reported by Gao and colleagues [37], however in this study it was shown that in both the spleen and CNS of normal mice, there were substantial numbers of CD3+ and γδ T cells secreting both IFN-γ and IL-4. Since the CNS of normal mice is usually devoid of any lymphocyte subset, and particularly of γδ T cells, the meaning of these results is unclear. More recent studies have indicated that γδ T cells act in an antigen-independent fashion to modulate cytokine production (IL-12 and IFN-γ) and thus the early effector phase of the immune response in EAE [13, 14]. Ponomarev and colleagues have suggested that the immunomodulatory effect of γδ T cells in EAE is independent of their ability to produce IFN-γ [14].
In this report, we examine the trafficking of bioluminescent γδ T cells in the inductive and acute phases of EAE as well as the requirement for β2-integrins in trafficking. We observed that γδ T cells rapidly migrate to and expand at the site of MOG peptide injection. By day six after EAE induction, bioluminescent γδ T cells were found in the brain and spinal cord, however, by day fifteen the CNS was largely devoid of γδ T cells. Reconstitution of γδ T cell−/− mice with γδ T cells derived from β2-integrin-deficient mice demonstrated that γδ T cell trafficking to the CNS is independent of this family of adhesion molecules. In addition, we examined the role of IFN-γ and TNF-α production by γδ T cells with respect to the development of EAE. We found that production of both cytokines by γδ T cells was required for fulminant EAE and that disease was significantly delayed and attenuated when mice were reconstituted with IFN-γ−/− γδ T cells. These results indicate that γδ T cells are critical in setting the stage for the development of EAE and may offer an unforeseen therapeutic target in demyelinating disease.
Results
Trafficking of γδ T cells prior to disease onset and in acute EAE
We induced EAE in γδ T cell−/− mice reconstituted with T-lux γδ T cells and performed in vivo bioluminescent imaging to visualize trafficking of γδ T cells before onset of disease symptoms. γδ T cells were found predominantly in the gut but could also be seen in various lymph nodes, primarily the cervical lymph nodes almost immediately after transfer as seen in ventral imaging of the mice (Figure 1A, upper panel). By day three post-induction of active EAE, the γδ T cells began accumulating in the cervical lymph nodes and the spleen and underwent expansion. Dorsal imaging of the mice revealed that γδ T cells rapidly traffic to the site of MOG injection (Figure 1A, lower panel). By day three after induction, γδ T cells accumulated at both injection sites and underwent continuous expansion through day nine post-induction of EAE. Quantification of the images shows a significant increase in signal on the dorsal view of the mice when compared to the ventral view on days eight and nine of EAE suggesting little to no expansion of γδ T cells in the gut or other peripheral lymphoid tissues (Figure 1B). Quantification of bioluminescent signal solely from the injection sites indicates that the increased dorsal bioluminescent signal was due largely to expansion of the γδ T cells in this location (Figure 1C).
Figure 1.
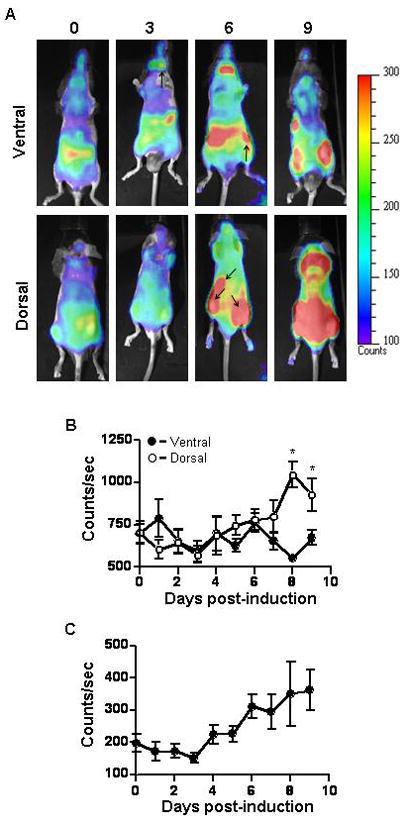
Bioluminescent imaging of early trafficking events of γδ T cells during active EAE. γδ T cell deficient mice were reconstituted with T-lux γδ T cells as described in materials and methods. EAE was induced and imaging was performed as described in materials and methods daily from day 0 to 9. (A) Each panel shows a representative ventral or dorsal image from one mouse at the indicated time points. Pseudo-color scale is shown to emphasize individual organ structures and areas in which γδ T cells have accumulated. Arrows indicate cervical and inguinal lymph nodes on ventral images. Arrows on dorsal images indicate spleen and the two sites of MOG peptide immunization. Quantification of bioluminescent signal from (B) the whole body in ventral (n = 5, filled circles) and dorsal (n = 5, open circles) images and (C) the sites of injection in the dorsal images are shown in counts/sec. The dorsal images had significantly (*p < 0.05) more bioluminescent signal on days 8 and 9 of EAE as measured by Anova. Shown are the mean ± SEM.
To further characterize the trafficking patterns of γδ T cells during EAE, we performed ex vivo imaging of spinal cord and brains from γδ T cell−/− mice reconstituted with T-lux γδ T cells at several time points after disease induction. γδ T cells were seen initially in the sacral region of the spinal cord at day 6 post-induction of EAE and, as disease progressed, the cells were found through out the spinal cord (Figure 2A). Similarly brain infiltration was observed starting at day six post-induction of EAE and, by day nine, γδ T cells fully infiltrated the brain (Figure 2B). γδ T cell infiltration into the brain peaks at day 12 post-induction of EAE, since by day 15, γδ T cells could not be detected by bioluminescent imaging (Figure 2B and data not shown). These results are supported by flow cytometric data in which spinal cords were isolated at multiple time points post EAE induction and assessed for γδ T cell infiltration (Figure 2C). The expansion of γδ T cells peaked between days 10 and 12 post induction of EAE and these cells are essentially absent from the CNS by day 15 and at later time points ([15], data not shown) We observed a peak in γδ T cell infiltration between days 10 and 12 that matches the bioluminescent imaging data followed by a rapid decline.
Figure 2.
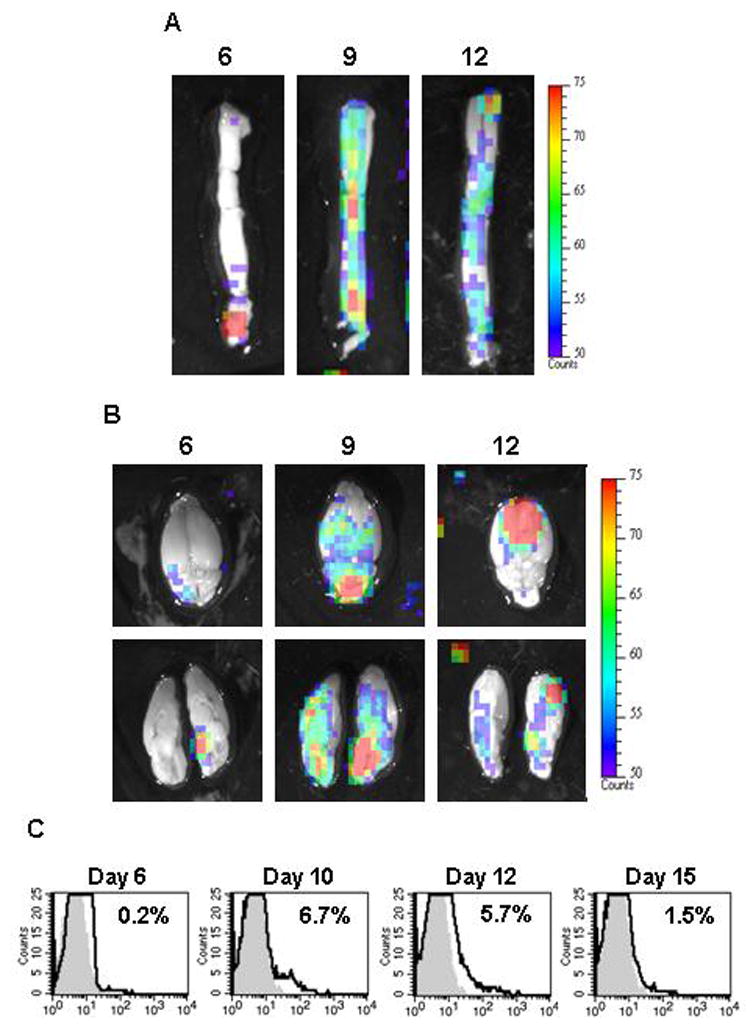
Bioluminescent imaging of γδ T cells in the brain and spinal cord during EAE. Mice deficient in γδ T cells were reconstituted with T-lux γδ T cells as described in materials and methods. Active EAE was induced in reconstituted mice and ex vivo bioluminescent images of the spinal cord (A, n = 3) and brain (B, n = 3) at indicated days were taken. The lower panel in B is the same brain sectioned at midline to show bioluminescent γδ T cells in the parenchyma and cerebellum. Representative images with a pseudo-color scale are shown to emphasize areas in which γδ T cells have accumulated. (C) Representative histograms show γδ (bold line) T cells in the spinal cord of EAE-induced mice 6, 10, 12, 15 days post immunization. Percentages of positive cells are indicated in each histogram. Cells were pooled from at least 4 mice per experiment and data shown are representative of at least 3 independent experiments. Day 6 (n=5) are from one experiment. The gray area represents control staining.
Trafficking of LFA-1−/− γδ T cells in EAE
To determine if LFA-1 was critical to the trafficking of γδ T cells during EAE, in vivo bioluminescent images of actively induced γδ T cell deficient mice reconstituted with either wild type T-lux or LFA-1−/−/T-lux γδ T cells were taken at various time points post-EAE induction (Figure 3A). Ventral images showed no difference in localization of wild type and LFA-1−/− γδ T cells in the gut and the cervical lymph nodes of mice with active EAE (data not shown). Dorsal imaging demonstrated that wild type and LFA-1−/− γδ T cells had localized and expanded at the site of immunization by day seven post-induction of active EAE (Figure 3A, upper panel), similar to the results shown in figure 1. By day nine post-induction, and continuing through day eleven, wild type γδ T cells expanded throughout the dorsal region of the mouse and began to localize in the brachial lymph nodes and the brain. In contrast, at the same time points LFA-1−/− γδ T cells left the site of immunization and did not continue expanding (Figure 3A, lower panel). Quantitation of bioluminescent signal from the images verified that wild type and LFA-1−/− γδ T cell localization and expansion was similar at the site of injection on day seven but thereafter LFA-1−/− γδ T cell bioluminescent signaled decreased while the wild type signal remained high until day 13 of EAE (Figure 3B). These data suggest that γδ T cells require LFA-1 for retention at a priming site and that LFA-1 is required, directly or indirectly, for γδ T cell co-stimulation.
Figure 3.
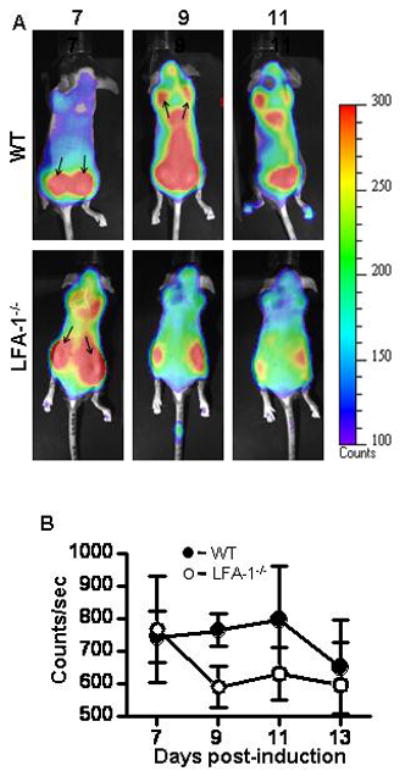
Bioluminescent imaging of wild type and LFA-1−/−γδ T cells during EAE. γδ T cell deficient mice were reconstituted with T-lux and LFA-1−/−/T-lux γδ T cells as described in materials and methods. EAE was induced and imaging was performed as described in materials and methods. (A) Representative dorsal images from individual mice are shown from the indicated time points. A Pseudo-color scale is shown to highlight organ structures and areas in which the γδ T cells have accumulated. Arrows indicate sites of immunization and brachial lymph nodes. (B) Bioluminescence from the whole body of dorsal images was quantitated (WT filled circles, n = 4 and LFA-1−/− open circles, n = 4). Shown are the mean ± SEM.
γδ T cells do not require β2-integrins for the development of EAE
It has been previously shown that γδ T cell−/− mice develop significantly delayed and attenuated disease compared to wild type mice (Spahn et al., 1999) and we confirmed these original observations. In our hands EAE in γδ T cell−/− mice was significantly delayed (p=0.04, Wilcoxon sign rank test) and attenuated (p<0.0001, Wilcoxon sign rank test) (Figure 4A, Table 1). Furthermore, when γδ T cell−/− mice are reconstituted with wild type γδ T cells EAE mirrored that of wild type mice. Reconstituted γδ T cell−/− mice developed EAE with a clinical severity closely approximating wild type disease (CDI 54.8 vs. 59.8, respectively) (Figure 4A, Table 1). To determine if expression of β2-integrins on γδ T cells is critical to the development of clinical disease we reconstitutedγδ T cell−/− mice with CD11a−/−, CD11b−/−, or CD11c−/− γδ T cells, induced EAE and monitored mice for disease symptoms. We found that reconstitution with γδ T cells deficient in the various β2-integrin α-chains resulted in disease severity comparable to reconstitution with wild type γδ T cells. A representative example of the EAE disease course after reconstitution with CD11a−/−γδ T cells is shown in Figure 4B. Similar results were obtained on reconstitution with CD11b−/− and CD11c−/−γδ T cells (data not shown). Overall EAE parameters for reconstitution with CD11a-CD11c−/−γδ T cells are shown in Table 2. These data indicate that β2-integrins on γδ T cell are not critical to the development of EAE, at least in the experimental setting we employed. These results contrast sharply with the role of β2-integrins on αβ T cells in EAE [33–36].
Figure 4.
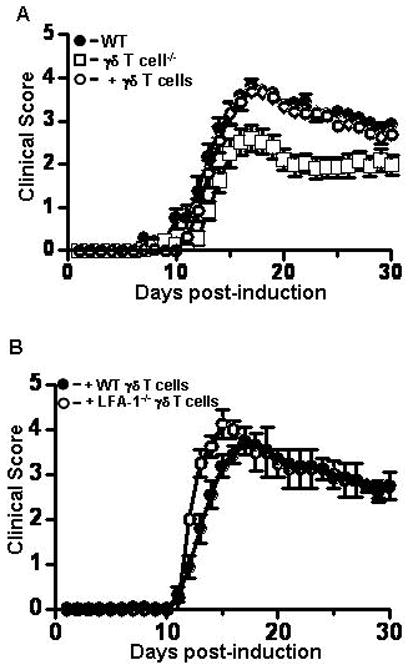
The clinical course of active EAE in γδ T cell−/− mice reconstituted with wild type or LFA-1−/−γδ T cells is comparable to that of control mice. (A) γδ T cell deficient mice were reconstituted with wild type γδ T cells and active EAE was induced in reconstituted (open circles, n = 19), γδ T cell−/− (open squares, n = 22), and control mice (filled circles, n = 20). Clinical disease was monitored for 30 days as described in materials and methods. Mice deficient in γδ T cells had a significantly reduced clinical disease course compared to control mice (p<0.0001, Wilcoxon signed rank test), while there was no significant difference in EAE between control and γδ T cell−/− mice reconstituted with wild type γδ T cells. (B) γδ T cell−/− mice were reconstituted with wild type γδ T cells or LFA-1−/−γδ T cells and EAE was induced. The course of disease in wild type (filled circles, n=4) and LFA-1−/−γδ T cell (open circles, n=4) reconstituted mice was not significantly different. Shown are the mean ± SEM from three or more experiments. Where not visible errors bars are contained within the symbol.
Table 1.
EAE in γδ T cell deficient mice reconstituted with wildtype γδ T cells
| Onseta | CDIb | Incidencec | |
|---|---|---|---|
| Wild type (n=20) | 12.d | 59.8 | 100% |
| γδ TCR−/− (n=22) | 13.4d | 36.8 | 91% |
| γδ TCR−/− + wild type γδ T cells (n=25) | 13.1d | 54.8 | 100% |
Disease onset is the first of 2 consecutive days with a clinical score of 2 or more.
Cumulative disease index is the mean sum of daily clinical scores from day 0 to 30.
Incidence is defined as the percent of mice that displayed any signs of clinical disease.
Table 2.
EAE in γδ T cell-deficient mice reconstituted with β2-integrin deficient γδ T cells
| Onseta | CDIb | Incidencec | |
|---|---|---|---|
| γδ TCR−/− + wild type γδ T cells (n=4) | 12.5d | 55.6 | 100% |
| γδ TCR−/− + CD11a−/− (n=4) | 11.7d | 59.3 | 100% |
| γδ TCR−/− + wild type γδ T cells (n=6) | 13.8d | 47 | 100% |
| γδ TCR−/− + CD11b−/− (n=9) | 11.5d | 48 | 100% |
| γδ TCR−/− + wild type γδ T cells (n=8) | 14.4 | 47.8 | 100% |
| γδ TCR−/− + CD11c−/− (n=5) | 13.6d | 49.5 | 100% |
Disease onset is the first of 2 consecutive days with a clinical score of 2 or more.
Cumulative disease index is the mean sum of daily clinical scores from day 0 to 30.
Incidence is defined as the percent of mice that displayed any signs of clinical disease.
IFN-γ and TNF-α produced by γδ T cells is critical for full development of EAE
Several studies have shown that γδ T cells produce IFN-γ and TNF-α in the spinal cord during EAE [12–15, 37]. To determine if this production is critical to the development of EAE we reconstituted γδ T cell−/− mice with IFN-γ−/− or TNF-α−/− γδ T cells. Reconstitution with IFN-γ−/− γδ T cells resulted in a significantly reduced (p<0.0001, Wilcoxon sign rank test) and delayed (p=0.04, unpaired T-test) clinical disease course compared to reconstitution with wild type γδ T cells (Figure 5A). The cumulative disease index and incidence rate was markedly lower in the IFN-γ−/− reconstituted mice than in mice reconstituted with wild type γδ T cells (Table 3). Reconstitution with TNF-α−/− γδ T cells also failed to induce fulminant EAE (Figure 5B, p<0.0004, Wilcoxon sign rank test), with a similar reduction in cumulative disease index and incidence rate compared to mice reconstituted with wild type γδ T cells (Table 3). These data indicate that IFN-γ and TNF-α production by γδ T cells is critical for the development of severe EAE, particularly in the chronic phase of disease.
Figure 5.
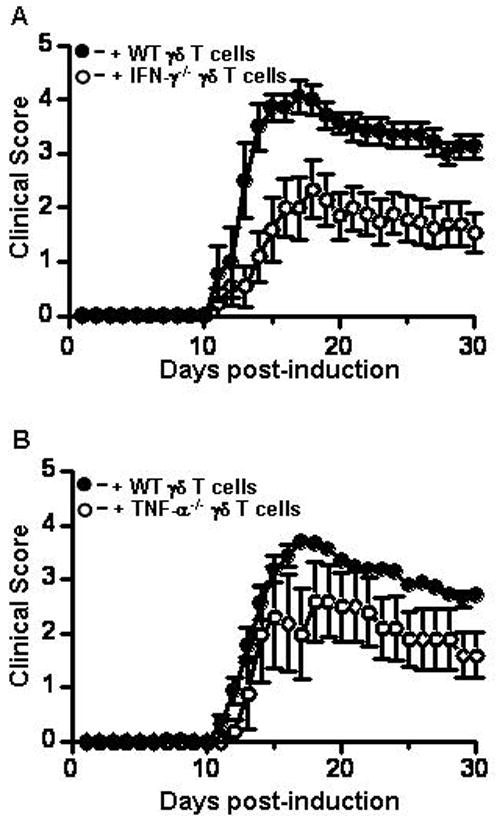
IFN-γ and TNF-α produced by γδ T cells is critical to clinical disease severity in active EAE. (A) γδ T cell−/− mice were reconstituted with wild type (n = 7, filled circles), or IFN-γ−/− γδ T cells (n=9, open circles), active EAE was induced, and signs of disease monitored for 30 days as described in materials and methods. Disease onset was significantly delayed (p=0.046; unpaired t-test) and overall severity was reduced (p=0.0001; Wilcoxon sign rank test) on reconstitution with IFN-γ−/− γδ T cells (B) Same as A except γδ T cell−/− mice were reconstituted with wild type (filled circles, n=5) or TNF-α−/− γδ T cells (open circles, n=5) and followed by active EAE induction. Disease severity was significantly lower in γδ T cell−/− mice reconstituted with TNF-α−/− γδ T cells (p=0.0004. Wilcoxon sign rank test). Shown are the mean ± SEM from two independent experiments. Where not visible errors bars are contained within the symbol.
Table 3.
EAE in γδ T cell deficient mice reconstituted with cytokine deficient γδ T cells
| Onseta | CDIb | Incidencec | |
|---|---|---|---|
| γδ TCR−/− + wild type γδ T cells (n=5) | 14.6d | 52.6 | 100% |
| ©δ TCR−/− + TNF-α−/− (n=5) | 14.3d | 36.4 | 80% |
| γδ TCR−/− + wild type γδ T cells (n=7) | 13d | 62.2 | 100% |
| γδ TCR−/− + IFN-γ−/− (n=9) | 14.7d | 31.45 | 78% |
Disease onset is the first of 2 consecutive days with a clinical score of 2 or more.
Cumulative disease index is the mean sum of daily clinical scores from day 0 to 30.
Incidence is defined as the percent of mice that displayed any signs of clinical disease.
Discussion
The results we report here indicate that γδ T cells play a critical role early in the development of EAE. We visualized γδ T cell trafficking in vivo during active EAE using bioluminescence and observed that γδ T cells rapidly accumulated at the site of MOG peptide immunization after disease induction and underwent massive expansion between days three and six. Based on imaging, γδ T cells did not migrate to and expand in the spleen and other secondary lymphoid organs to any significant degree, with the exception of the cervical lymph nodes at day three post-induction of EAE and the brachial lymph nodes at day nine. These data demonstrate that γδ T cells, like CD4+ T cells [38–41], drain to cervical nodes and raise the possibility that brachial lymph nodes may also serve as a draining lymph node for the CNS, at least for γδ T cells. Expansion of γδ T cells continued at the site of MOG injection up to day eleven post-induction of EAE, however, by day thirteen there was marked decline in expansion. Interestingly, we observed similar but delayed kinetics of γδ T cell trafficking to the CNS. γδ T cells entered the sacral region of the spinal cord and the cerebellum of the brain by day six, trafficked throughout the parenchyma of the spinal cord and brain peaking between days ten and twelve and then rapidly declined by day fifteen in all tissues. These kinetics are consistent with γδ T cells playing an important role in demyelinating disease well before clinical signs of disease and when few CD4 and CD8 T cells have reached the CNS [15], most likely in priming events important for acute disease development.
The dramatic expansion of γδ T cells at the site of MOG peptide injection raises questions regarding priming events for these cells in demeylinating disease. Proteins from mycobacterium tuberculosis (MT), a component of the adjuvant used to induce EAE, are potent antigens for γδ T cells [42]. More recently it has been shown that the expansion of γδ T cells seen after the induction of experimental autoimmune uveitis can be attributed to the MT in the adjuvant and the pertussis toxin injected at the time of immunization [30]. Taken together, these observations suggest that the MT and pertussis toxin used to induce EAE may be in large part responsible for the localization, activation, and proliferation of the γδ T cells, at the site of immunization. However, the rapid entry of γδ T cells into the CNS may suggest that some of the γδ T cells expanding at the immunization site are antigen-specific and directed toward the MOG35–55 peptide, but this remains to be shown. Antigen-specific CD4+ T cells have also been shown to migrate to MOG peptide injection sites [43]. However in this model system MOG-specific T cells were adoptively transferred into mice with an ongoing Mycobacterium bovis infection making it difficult to determine if CD4+ T cells normally traffic to the MOG emulsion. In our hands, naive αβ T cells did not traffic to the injection site (data not shown) suggesting that the trafficking observed by Sewell and colleagues is activation-dependent or, given the nature of their model system, that γδ T cells are unique among T cells subsets in trafficking to the MOG injection site.
The β2-integrin family of adhesion molecules plays an important role in the development of EAE [33–36]. Deletion of CD11a, CD11b, or CD11c results in significantly delayed and attenuated disease due, in part, to reduced trafficking of T cells to the CNS. It has recently been shown that β2-integrins are differentially expressed on αβ and γδ T cells throughout EAE [15], but it is unclear how deletion of any one of the β2-integrins on γδ T cells contributes to disease development and progression. To answer this question we performed reconstitution experiments in which γδ T cells, deficient in CD11a, CD11b, or CD11c, were transferred to γδ T cell−/− mice. To our surprise all of the β2-integrin mutant γδ T cells were able to restore disease severity comparable to that seen in reconstitutions using wild type γδ T cells. These data indicate that the disease phenotypes seen in β2- integrin-deficient mice during EAE are independent of γδ T cell-mediated expression of β2-integrins. Previous studies have demonstrated that anti-LFA-1 antibodies prevented the adhesion of γδ T cells to endothelial monolayers in vitro [16, 17]. Such studies do not however sufficiently replicate in vivo events during EAE and specific targeting of γδ T cells with anti-LFA-1 antibodies during disease would be technically challenging. Our bioluminescent data clearly demonstrated that LFA-1−/−γδ T cells trafficked to the sites of MOG injection and expanded in a fashion comparable to that of wild type γδ T cells. Despite these initial similarities, LFA-1−/−γδ T cells were either not retained at the MOG injections sites or had altered proliferation kinetics compared to wild type γδ T cells based on the rapid clearance (compared to wild type γδ T cells) from injections site. Although these data are consistent with activation deficits in LFA-1−/−γδ T cells, this seems unlikely since the clinical course of disease was not changed relative to wild type γδ T cells. We cannot rule out the possibility that loss of a given β2-intergrin on γδ T cells is compensated for by other family members and/or that other adhesion molecules are utilized by γδ T cells for migration to priming sites and to the CNS. Our results suggest, a likely candidate for γδ T cell trafficking to the CNS may be VLA-4 based on studies in several inflammatory model systems [16, 17, 32, 44–46]
IFN-γ is produced in the CNS during EAE by infiltrating T cells and thought to help drive early stages of the disease [47, 48]. From a therapeutic point of view, IFN-γ is considered central to pathogenic and inflammatory mechanisms based on the results of clinical trials in which IFN-γ exacerbated disease [49, 50]. However, IFN-γ may also help to regulate inflammation since IFN-γ−/− mice develop exacerbated, sometimes fatal EAE, during the chronic phase of disease [51–54]. Previous studies have suggested that IFN-γ production by γδ T cells is not critical to disease development and progression; γδ T cells were required solely for induction of IFN-γ by CD4 and CD8 T cells [14]. Evidence supporting this potential mechanism was derived from quantitation of mRNA levels for IFN-γ in bone marrow chimeric mice reconstituted with γδ T cells derived from wild type and IFN-γ−/− mice. Interpretation of these studies is complicated by the fact that IFN-γ protein levels were not assessed, nor was the disease course monitored to allow correlation to IFN-γ production. In contrast, we found that the production of IFN-γ by γδ T cells is critical to EAE development. Reconstitution studies using congenic C57BL/6 mice (as opposed to mixed B6.129 × B10.PL backgrounds) clearly demonstrated that IFN-γ production by γδ T cells contributes to disease onset and development. We cannot determine from our studies if γδ T cell-mediated IFN-γ production modulated IFN-γ production by αβ T cells, but previous studies from our laboratory and others have demonstrated that before and during the acute phase of EAE, the majority of γδ T cells (70% or more) produce IFN-γ, while αβ T cells produce significantly less [14, 15]. It is unclear if IFN-γ and TNF-α produced by γδ T cells during EAE modulates trafficking of γδ T cells during disease. However, local production of these cytokines may alter adhesion molecule expression in a manner that affects T cell trafficking to the CNS. These data suggest that IFN-γ production by γδ T cells may be central to initial inflammatory events during EAE.
TNF-α has also been shown to be an important cytokine in MS and EAE pathogenic mechanisms. TNF-α−/− mice are protected from EAE and transgenic mice expressing TNF-α in the CNS develop spontaneous neurodegenerative disease characterized by cellular inflammation and neuronal damage [55–58]. These studies did not establish cellular subset production of TNF-α or correlate the corresponding functional role of TNF-α to that cell type in EAE. Likewise, in studies where γδ T cells were depleted by antibody treatment, TNF-α levels were markedly reduced, but which cells were producing TNF-α and how production by those cells modulated the disease course was not addressed [11]. To overcome the technical limitations of γδ T cell depletion, we performed reconstitution studies in which γδ T cell−/− mice were given TNF-α−/− γδ T cells and observed that TNF-α production by γδ T cells modulated the chronic phase of EAE. The disease phenotype in these reconstitutions was more severe than that observed for IFN-γ−/− γδ T cell reconstitutions suggesting that TNF-α production by γδ T cells is not as crucial as IFN-γ to disease development and progression. Nonetheless, γδ T cells in the spinal cord during EAE are a significant source of TNF-α production, perhaps even the most significant source of this cytokine on a cellular basis [15]. These results raise the possibility that TNF-α additively or synergistically acts in concert with IFN-γ to modulate early disease development.
γδ T cells are innate-like lymphocyes and it has been proposed that they may provide a link between the innate and adaptive immune response. It has recently been shown that activated murine γδ T cells can present MOG35–55 peptide to naive CD4 T cells [30]. γδ T cells also interact with dendritic cells driving their maturation through TNF-α and increasing IL-12 production by dendritic cells through IFN-γ-mediated mechanisms [13, 59–61]. Antibody depletion of γδ T cells in EAE results in delayed cytokine and chemokine production in the CNS [10, 11], possibly by hindering the generation of the adaptive immune response normally augmented by γδ T cells. Our data suggest that local production of IFN-γ and/or TNF-α is important for dendritic cell maturation or programming which aides in initiating an immune response to myelin antigens. Concurrently, γδ T cells are also entering the CNS, and in situ production of IFN-γ and TNF-α may activate APCs in the CNS and prime the microenvironment for CD4+ and CD8+ T cell recruitment and activation. In this scenario, γδ T cells play a critical role in disease initiation that has largely been overlooked both mechanistically and therapeutically. Deletion of γδ T cells in demyelinating disease may be advantageous in that the αβ T cell compartment would remain unaffected, thereby reducing the risk of therapeutically-induced immunodeficiencies.
Materials and Methods
Mice
Tcrdtm1Mom mice, deficient in γδ T cells, were obtained from Jackson laboratories. TNF-α−/− and IFN-γ−/− mice were generous gifts from Drs. David Chaplin and Alan Zajac (Department of Microbiology, University of Alabama at Birmingham), respectively. CD11a−/−, CD11b−/−, and CD11c−/− mice have been previously described [62–64]. For all studies, we used the β2-integrin deficient mice at an N16 backcross onto C57BL/6. The luciferase transgenic mouse line (T-lux), expressing firefly luciferase under the control of the human CD2 promoter, was generated in the C57BL/6 background as previously described [65]. T-lux mice express luciferase activity in all CD3+ cells and bioluminescence generated by this enzyme is directly proportional to the number of cells expressing the gene, allowing real-time assessment of T cell proliferation and migration in vivo. For some experiments, LFA-1 deficient T-lux transgenic mice were generated by intercrossing the CD11a−/− and T-lux mice. Inbred T-lux transgenic or non-transgenic C57BL/6 mice were used as controls for all experiments. All studies were performed with approval from the UAB IACUC.
Bioluminescent Imaging
Mice were subjected to bioluminescent imaging as previously described [65]. Briefly, mice were anesthetized with isofluorane and placed in a light-tight chamber. The photographic (gray-scale) reference image was obtained at 10 minutes after D-luciferin injection; the bioluminescent image was collected immediately thereafter. Images were obtained with a CCD camera cooled to −120°C, using the IVIS Imaging System (Xenogen Corp., Alameda, CA) with the field of view set at 10 cm height. The photographic images were taken at a 0.2 second exposure, 8 f/stop, 2 binning (resolution), and an open filter. The bioluminescent images used exposures of 600 seconds, 1f/stop, 16 binning and open filter. The bioluminescent and gray-scale images were overlaid using Living Image software (Xenogen Corp.). Igor image analyses software (Wavemetrics, Lake Oswego, OR) was employed to obtain a pseudocolor image representing bioluminescence intensity (blue, least intense; red, most intense). The total counts were normalized to image acquisition. Ex vivo images were obtained by removal of brain and spinal cord and imaging at 3X magnification after treatment with the Luciferase Assay system (Promega). Localization of T-lux T cells in lymphoid tissue was verified by ex vivo imaging performed on representative mice. Expansion of T-lux T cells has been previously verified in studies demonstrating that cell number is directly proportional to bioluminescent signal [65].
Flow cytometry and intracellular cytokine staining
Cells obtained from draining lymph nodes, spleens, and spinal cords were incubated with anti-CD16/32 (FcR block, eBioscience) to prevent nonspecific staining. Cells were incubated for 30 minutes in the dark at 4°C with anti-γδ TCR FITC (GL3, BD Pharmingen). All antibodies were diluted in FACS buffer (1X PBS, 2% FCS, 0.1% NaN3). Immunofluorescence analyses were performed using a FACSCalibur and CellQuest software (BD Biosciences).
Reconstitution
γδ T cells were isolated from spleen, lymph nodes, and thymus of wild type, T-lux, CD11a−/−, LFA-1−/−/T-lux, CD11b−/−, CD11c−/−, TNF-α−/−, and IFN-γ−/− mice using the magnetic bead isolation kits from Miltenyi Biotech. γδ T cells (5 × 105, > 90% pure) were injected retro-orbitally into γδ T cell−/− mice. Active EAE was induced as described below two days after reconstitution.
Active EAE
For active EAE, control, γδ T cell deficient, and reconstituted mice were immunized with MOG peptide35–55 (Biosynthesis, Lewisville, TX) as described [33]. Onset and progression of EAE were monitored daily using a standard clinical scale ranging from 0 to 6 as follows: 0, asymptomatic; 1, loss of tail tone; 2, flaccid tail; 3, incomplete paralysis of one or two hind limbs; 4, complete hind limb paralysis; 5, moribund; and 6, death. Only mice with a score of at least a 2 (flaccid tail) for more than 2 consecutive days were judged to have EAE. For each animal a cumulative disease index was calculated from the sum of the daily clinical scores observed between day0 and day 30.
Statistics
Statistical significance for the bioluminescent images was measured using Anova with Tukey’s post test. Clinical disease courses were analyzed using Wilcoxon Signed Rank test and disease onset was analyzed using the Student’s t-test.
Acknowledgments
The authors acknowledge the support of the Multi-Modality Imaging Facility and Dr. Kari Dugger for help with the imaging. This work was supported by grants from the National Multiple Sclerosis Society (RG 3437-B-9) to SRB and DCB and from the National Institutes of Health (T32 AI07051) to SSS and JEW.
Abbreviations in this paper
- EAE
experimental autoimmune encephalomyelitis
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- CDI
cumulative disease index
Footnotes
Conflicts of Interest
The authors have no financial conflicts to disclose.
References
- 1.Sospedra M, Martin R. Immunology of multiple sclerosis *. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 2.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 3.Gold L, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 4.Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, Ottoboni L, Ciabini D, Caramia MD, Bernardi G, Laudanna C, Scarpini E, McEver RP, Butcher EC, Borsellino G, Constantin G. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood. 2003;101:4775–4782. doi: 10.1182/blood-2002-10-3309. [DOI] [PubMed] [Google Scholar]
- 5.Selmaj K, Brosnan CF, Raine CS. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+ oligodendrocytes in multiple sclerosis. Proc Natl Acad Sci U S A. 1991;88:6452–6456. doi: 10.1073/pnas.88.15.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A. 1992;89:4588–4592. doi: 10.1073/pnas.89.10.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Droogan AG, Crockard AD, Hawkins SA, McNeill TA. Gamma delta T cell distribution in cerebrospinal fluid and peripheral blood of patients with multiple sclerosis. J Neurol Sci. 1994;126:172–177. doi: 10.1016/0022-510x(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 8.Hvas J, Oksenberg JR, Fernando R, Steinman L, Bernard CC. Gamma delta T cell receptor repertoire in brain lesions of patients with multiple sclerosis. J Neuroimmunol. 1993;46:225–234. doi: 10.1016/0165-5728(93)90253-u. [DOI] [PubMed] [Google Scholar]
- 9.Murzenok PP, Matusevicius D, Freedman MS. gamma/delta T cells in multiple sclerosis: chemokine and chemokine receptor expression. Clin Immunol. 2002;103:309–316. doi: 10.1006/clim.2001.5213. [DOI] [PubMed] [Google Scholar]
- 10.Rajan AJ, Asensio VC, Campbell IL, Brosnan CF. Experimental autoimmune encephalomyelitis on the SJL mouse: effect of gamma delta T cell depletion on chemokine and chemokine receptor expression in the central nervous system. J Immunol. 2000;164:2120–2130. doi: 10.4049/jimmunol.164.4.2120. [DOI] [PubMed] [Google Scholar]
- 11.Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol. 1998;160:5955–5962. [PubMed] [Google Scholar]
- 12.Jensen MA, Dayal A, Arnason BG. Cytokine secretion by deltagamma and alphabeta T cells in monophasic experimental autoimmune encephalomyelitis. J Autoimmun. 1999;12:73–80. doi: 10.1006/jaut.1998.0263. [DOI] [PubMed] [Google Scholar]
- 13.Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol. 2004;173:682–694. doi: 10.4049/jimmunol.173.1.682. [DOI] [PubMed] [Google Scholar]
- 14.Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol. 2004;173:1587–1595. doi: 10.4049/jimmunol.173.3.1587. [DOI] [PubMed] [Google Scholar]
- 15.Smith SS, Barnum SR. Differential expression of b2-integrins and cytokine production between gd and ab T cells in experimental autoimmune encephalomyelitis. J Leukocyte Biol. 2008;83:71–79. doi: 10.1189/jlb.0407263. [DOI] [PubMed] [Google Scholar]
- 16.Galea P, Brezinschek R, Lipsky PE, Oppenheimer-Marks N. Phenotypic characterization of CD4−/alpha beta TCR+ and gamma delta TCR+ T cells with a transendothelial migratory capacity. J Immunol. 1994;153:529–542. [PubMed] [Google Scholar]
- 17.Nakajima S, Roswit WT, Look DC, Holtzman MJ. A hierarchy for integrin expression and adhesiveness among T cell subsets that is linked to TCR gene usage and emphasizes V delta 1+ gamma delta T cell adherence and tissue retention. J Immunol. 1995;155:1117–1131. [PubMed] [Google Scholar]
- 18.Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H. Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS Lett. 2003;544:4–10. doi: 10.1016/s0014-5793(03)00483-6. [DOI] [PubMed] [Google Scholar]
- 19.Hayday A, Tigelaar R. Immunoregulation in the tissues by gammadelta T cells. Nat Rev Immunol. 2003;3:233–242. doi: 10.1038/nri1030. [DOI] [PubMed] [Google Scholar]
- 20.Girardi M, Hayday AC. Immunosurveillance by gammadelta T cells: focus on the murine system. Chem Immunol Allergy. 2005;86:136–150. doi: 10.1159/000086658. [DOI] [PubMed] [Google Scholar]
- 21.Hayday AC, Pennington DJ. Key factors in the organized chaos of early T cell development. Nat Immunol. 2007;8:137–144. doi: 10.1038/ni1436. [DOI] [PubMed] [Google Scholar]
- 22.Wijngaard PL, MacHugh ND, Metzelaar MJ, Romberg S, Bensaid A, Pepin L, Davis WC, Clevers HC. Members of the novel WC1 gene family are differentially expressed on subsets of bovine CD4−CD8− gamma delta T lymphocytes. J Immunol. 1994;152:3476–3482. [PubMed] [Google Scholar]
- 23.Hanby-Flarida MD, Trask OJ, Yang TJ, Baldwin CL. Modulation of WC1, a lineage-specific cell surface molecule of gamma/delta T cells augments cellular proliferation. Immunology. 1996;88:116–123. doi: 10.1046/j.1365-2567.1996.d01-649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szymanska B, Rajan AJ, Gao YL, Tronczynska E, Brosnan CF, Selmaj K. Evidence for gammadelta T cells with a restricted Vgamma6 junctional region in the normal mouse central nervous system. J Neuroimmunol. 1999;100:260–265. doi: 10.1016/s0165-5728(99)00204-0. [DOI] [PubMed] [Google Scholar]
- 25.Wilson IA, Stanfield RL. Unraveling the mysteries of gammadelta T cell recognition. Nat Immunol. 2001;2:579–581. doi: 10.1038/89718. [DOI] [PubMed] [Google Scholar]
- 26.Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 27.Aydintug MK, Roark CL, Chain JL, Born WK, O’Brien RL. Macrophages express multiple ligands for gammadelta TCRs. Mol Immunol. 2008;45:3253–3263. doi: 10.1016/j.molimm.2008.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thedrez A, Sabourin C, Gertner J, Devilder MC, Allain-Maillet S, Fournie JJ, Scotet E, Bonneville M. Self/non-self discrimination by human gammadelta T cells: simple solutions for a complex issue? Immunol Rev. 2007;215:123–135. doi: 10.1111/j.1600-065X.2006.00468.x. [DOI] [PubMed] [Google Scholar]
- 29.O’Brien RL, Roark CL, Jin N, Aydintug MK, French JD, Chain JL, Wands JM, Johnston M, Born WK. gammadelta T-cell receptors: functional correlations. Immunol Rev. 2007;215:77–88. doi: 10.1111/j.1600-065X.2006.00477.x. [DOI] [PubMed] [Google Scholar]
- 30.Cheng L, Cui Y, Shao H, Han G, Zhu L, Huang Y, O’Brien RL, Born WK, Kaplan HJ, Sun D. Mouse gammadelta T cells are capable of expressing MHC class II molecules, and of functioning as antigen-presenting cells. J Neuroimmunol. 2008 doi: 10.1016/j.jneuroim.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohagheghpour N, Bermudez LE, Khajavi S, Rivas A. The VLA-4/VCAM-1 molecules participate in gamma delta cell interaction with endothelial cells. Cell Immunol. 1992;143:170–182. doi: 10.1016/0008-8749(92)90014-g. [DOI] [PubMed] [Google Scholar]
- 32.Watkins AD, Hatfield CA, Fidler SF, Winterrowd GE, Brashler JR, Sun FF, Taylor BM, Vonderfecht SL, Conder GA, Holgate ST, Chin JE, Richards IM. Phenotypic analysis of airway eosinophils and lymphocytes in a Th-2-driven murine model of pulmonary inflammation. Am J Respir Cell Mol Biol. 1996;15:20–34. doi: 10.1165/ajrcmb.15.1.8679219. [DOI] [PubMed] [Google Scholar]
- 33.Bullard DC, Hu X, Adams JE, Schoeb TR, Barnum SR. p150,95 (CD11c/CD18) expression is required for the development of experimental autoimmune encephalomyelitis. Amer J Path. 2007;170:2001–2008. doi: 10.2353/ajpath.2007.061016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:6327–6333. doi: 10.4049/jimmunol.175.10.6327. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Kai H, Chang F, Shibata K, Tahara-Hanaoka S, Honda S, Shibuya A, Shibuya K. A critical role of LFA-1 in the development of Th17 cells and induction of experimental autoimmune encephalomyelytis. Biochem Biophys Res Commun. 2007;353:857–862. doi: 10.1016/j.bbrc.2006.12.104. [DOI] [PubMed] [Google Scholar]
- 36.Dugger KJ, Zinn KR, Weaver C, Bullard DC, Barnum SR. Effector and suppressor roles for LFA-1 during the development of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2009;206:22–27. doi: 10.1016/j.jneuroim.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao YL, Rajan AJ, Raine CS, Brosnan CF. gammadelta T cells express activation markers in the central nervous system of mice with chronic-relapsing experimental autoimmune encephalomyelitis. J Autoimmun. 2001;17:261–271. doi: 10.1006/jaut.2001.0547. [DOI] [PubMed] [Google Scholar]
- 38.Weller RO, Engelhardt B, Phillips MJ. Lymphocyte targeting of the central nervous system: a review of afferent and efferent CNS-immune pathways. Brain Pathol. 1996;6:275–288. doi: 10.1111/j.1750-3639.1996.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 39.de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, Ravid R, Rensing S, Boon L, t Hart BA, Laman JD. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002;169:5415–5423. doi: 10.4049/jimmunol.169.10.5415. [DOI] [PubMed] [Google Scholar]
- 40.Mohindru M, Kang B, Kim BS. Functional maturation of proteolipid protein(139–151)-specific Th1 cells in the central nervous system in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;155:127–135. doi: 10.1016/j.jneuroim.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 41.Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, Lafaille JJ. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol. 2008;181:4648–4655. doi: 10.4049/jimmunol.181.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller D, Pakpreo P, Filla J, Pederson K, Cigel F, Malkovska V. Increased gamma-delta T-lymphocyte response to Mycobacterium bovis BCG in major histocompatibility complex class I-deficient mice. Infect Immun. 1995;63:2361–2366. doi: 10.1128/iai.63.6.2361-2366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sewell DL, Reinke EK, Co DO, Hogan LH, Fritz RB, Sandor M, Fabry Z. Infection with Mycobacterium bovis BCG diverts traffic of myelin oligodendroglial glycoprotein autoantigen-specific T cells away from the central nervous system and ameliorates experimental autoimmune encephalomyelitis. Clin Diagn Lab Immunol. 2003;10:564–572. doi: 10.1128/CDLI.10.4.564-572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avdalovic M, Fong D, Formby B. Adhesion and costimulation of proliferative responses of human gamma delta T cells by interaction of VLA-4 and VLA-5 with fibronectin. Immunol Lett. 1993;35:101–108. doi: 10.1016/0165-2478(93)90077-f. [DOI] [PubMed] [Google Scholar]
- 45.Kennedy JD, Hatfield CA, Fidler SF, Winterrowd GE, Haas JV, Chin JE, Richards IM. Phenotypic characterization of T lymphocytes emigrating into lung tissue and the airway lumen after antigen inhalation in sensitized mice. Am J Respir Cell Mol Biol. 1995;12:613–623. doi: 10.1165/ajrcmb.12.6.7766426. [DOI] [PubMed] [Google Scholar]
- 46.Behr-Perst SI, Munk ME, Schaberg T, Ulrichs T, Schulz RJ, Kaufmann SH. Phenotypically activated gammadelta T lymphocytes in the peripheral blood of patients with tuberculosis. J Infect Dis. 1999;180:141–149. doi: 10.1086/314844. [DOI] [PubMed] [Google Scholar]
- 47.Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–2768. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- 48.Juedes AE, Hjelmstrom P, Bergman CM, Neild AL, Ruddle NH. Kinetics and cellular origin of cytokines in the central nervous system: insight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Immunol. 2000;164:419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- 49.Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 50.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 51.Duong TT, Finkelman FD, Singh B, Strejan GH. Effect of anti-interferon-gamma monoclonal antibody treatment on the development of experimental allergic encephalomyelitis in resistant mouse strains. J Neuroimmunol. 1994;53:101–107. doi: 10.1016/0165-5728(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 52.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 53.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 54.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 55.Probert L, Akassoglou K, Kassiotis G, Pasparakis M, Alexopoulou L, Kollias G. TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol. 1997;72:137–141. doi: 10.1016/s0165-5728(96)00184-1. [DOI] [PubMed] [Google Scholar]
- 56.Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158:438–445. [PubMed] [Google Scholar]
- 57.Sean Riminton D, Korner H, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Challenging cytokine redundancy: inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor-deficient, mice. J Exp Med. 1998;187:1517–1528. doi: 10.1084/jem.187.9.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K, Lassmann H, Fontana A. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain. 2000;123(Pt 10):2005–2019. doi: 10.1093/brain/123.10.2005. [DOI] [PubMed] [Google Scholar]
- 59.Ismaili J, Olislagers V, Poupot R, Fournie JJ, Goldman M. Human gamma delta T cells induce dendritic cell maturation. Clin Immunol. 2002;103:296–302. doi: 10.1006/clim.2002.5218. [DOI] [PubMed] [Google Scholar]
- 60.Conti L, Casetti R, Cardone M, Varano B, Martino A, Belardelli F, Poccia F, Gessani S. Reciprocal activating interaction between dendritic cells and pamidronate-stimulated gammadelta T cells: role of CD86 and inflammatory cytokines. J Immunol. 2005;174:252–260. doi: 10.4049/jimmunol.174.1.252. [DOI] [PubMed] [Google Scholar]
- 61.Dieli F, Caccamo N, Meraviglia S, Ivanyi J, Sireci G, Bonanno CT, Ferlazzo V, La Mendola C, Salerno A. Reciprocal stimulation of gammadelta T cells and dendritic cells during the anti-mycobacterial immune response. Eur J Immunol. 2004;34:3227–3235. doi: 10.1002/eji.200425368. [DOI] [PubMed] [Google Scholar]
- 62.Lu H, Smith CW, Perrard J, Bullard D, Tang L, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ren B, McCrory MA, Pass C, Bullard DC, Ballantyne CM, Xu Y, Briles DE, Szalai AJ. The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J Immunol. 2004;173:7506–7512. doi: 10.4049/jimmunol.173.12.7506. [DOI] [PubMed] [Google Scholar]
- 64.Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163:5029–5038. [PubMed] [Google Scholar]
- 65.Azadniv M, Dugger K, Bowers WJ, Weaver CT, Crispe IN. Imaging CD8+ T cell dynamics in vivo using a transgenic luciferase reporter. Int Immunol. 2007;19:1165–1173. doi: 10.1093/intimm/dxm086. [DOI] [PubMed] [Google Scholar]