

Abstract
The gene from Streptomyces coelicolor A3(2) encoding CYP102B1, a recently discovered CYP102 subfamily which exists solely as a single P450 heme domain, has been cloned, expressed in Escherichia coli, purified, characterized, and compared to its fusion protein family members. Purified reconstitution metabolism experiments with spinach ferredoxin, ferredoxin reductase, and NADPH revealed differences in the regio- and stereoselective metabolism of arachidonic acid compared to that of CYP102A1, exclusively producing 11,12-epoxyeicosa-5,8,14-trienoic acid in addition to the shared metabolites 18-hydroxy arachidonic acid and 14,15-epoxyeicosa-5,8,11-trienoic acid. Consequently, in order to elucidate the physiological function of CYP102B1, transposon mutagenesis was used to generate an S. coelicolor A3(2) strain lacking CYP102B1 activity and the phenotype was assessed.
Streptomycetes produce a vast array of antibiotics applied in human and veterinary medicine and agriculture, as well as antiparasitic agents, herbicides, and pharmacologically active metabolites (e.g., immunosuppressants). Streptomycetes also catalyze numerous transformations of xenobiotics of industrial and environmental importance (1). The most significant of these biocatalytic reactions include aromatic and aliphatic hydroxylations, O and N dealkylations, N oxidation, and C-C coupling and fission catalyzed by heme-containing cytochrome P450 (CYP) enzyme systems (25). Streptomyces coelicolor A3(2) is the most-studied member of the genus in molecular genetic and biochemical investigations (1). Eighteen cytochrome P450, six ferredoxin, and four ferredoxin reductase genes were shown to be distributed across the linear chromosome, with nine P450 genes arranged in polycistronic organization with other genes (1, 14).
Arguably, the most biochemically and structurally characterized CYP to date is CYP102A1 (P450 BM3) from Bacillus megaterium. CYP102A1 was first isolated as a fatty acid monooxygenase in the laboratory of Armand Fulco more than 20 years ago (16). It was established as a catalytically self-sufficient monooxygenase consisting of a CYP heme domain fused at the carboxy terminus to a flavin mononucleotide (FMN)/flavin adenine dinucleotide (FAD)-containing flavin reductase domain in a single polypeptide chain. It has been proposed that such architecture affords the CYP domain optimal catalytic activity in the turnover of substrates. Indeed, of all the CYPs characterized functionally, CYP102A1 exhibits the highest P450 turnover frequency, with rates of >15,000 min−1 obtained in exogenous arachidonate hydroxylation (17, 18). Furthermore, CYP102A1-like P450 reductase fusion proteins have also been found in certain fungi, including Fusarium oxysporum (13) and Phanerochaete chrysosporium (3).
CYP102B1 was the first member of a new CYP102 subfamily discovered outside Bacillus spp. (1) and is intriguing since it exists and functions as a single CYP protein domain. Here we report the cloning, expression, and characterization of CYP102B1 and demonstrate that the enzyme has activity in metabolizing arachidonic acid but with very different product profiles and with enzymatic rates orders of magnitude lower than those of CYP102A1. To address the question of the contribution of CYP102B1 to S. coelicolor A3(2) physiology, a CYP102B1 transposon mutant was generated and isolated and the phenotype of the subsequent mutant strains was analyzed.
MATERIALS AND METHODS
General methods.
Reduced carbon monoxide (CO) difference spectra for quantification of cytochrome P450 content were measured and calculated according to the method described by Omura and Sato (19). Protein quantification was performed by using the bicinchoninic acid assay. Unless otherwise stated, all chemicals were supplied by Sigma Chemical Company (Poole, Dorset, United Kingdom). UV-visible absorption spectra of purified CYP102B1 were recorded using a Shimadzu UV-2401 scanning spectrophotometer as described previously (24, 26).
Cloning, gene synthesis, expression, and purification of CYP102A1 and CYP102B1.
The gene for CYP102B1 was commercially synthesized using codon optimization for efficient protein expression in Escherichia coli and incorporating eight histidine residues at the carboxy terminus to facilitate protein purification by nickel-nitrilotriacetic acid (Ni-NTA) chromatography (DNA2.0 Inc., Menlo Park, CA 94025) and inserted into the expression vector pET17b, producing the final construct CYP102B1syn-His8:pET17b. The expression construct was transformed into E. coli strain BL21(DE3)pLysD, in which expression of the T7 RNA polymerase gene is under the control of the lac promoter. To facilitate the production of correctly folded P450, CYP102B1 was coexpressed in the presence of the E. coli molecular chaperones GroES and GroEL as described previously (20). Three liters of E. coli heterologously expressing CYP102B1 was pelleted by centrifugation at 1,500 × g, and CYP102B1 was purified using an Ni2+-NTA affinity column (Qiagen), using methods similar to those applied to the purification of other S. coelicolor P450s (47, 49). CYP102A1 was expressed and purified as described previously (6).
Arachidonic acid metabolism by CYP102A1 and CYP102B1.
Assays of CYP102A1 and CYP102B1 arachidonic acid metabolism were performed at 30°C for 20 min. Each reaction mixture contained either 0.01 μM CYP102A1 or 1 μM CYP102B1, 0.01 U/100 μl spinach ferredoxin reductase and 20 μM spinach ferredoxin, and 70 μM (70,000 cpm/nmol) [14C]arachidonic acid. Reactions were carried out in the following reaction buffer: 0.15 N KCl, 10 mM MgCl2, 50 mM Tris-HCl (pH 7.4), 2 mg/ml isocitrate, and 0.1 U/ml isocitrate dehydrogenase. NADPH was added to a final concentration of 1 mM to start the reaction. Briefly, the products of the catalytic turnover of each fatty acid were determined by reverse-phase, high-performance liquid chromatography (RP-HPLC) and comparison to authentic standards (8).
Transposon mutagenesis of CYP102B1.
To assess the role of CYP102B1 in the contribution of S. coelicolor A(3)2 physiology and potential roles in endogenous secondary and lipid metabolism, CYP102B1 was mutated in the S. coelicolor chromosome. To do so, a Tn5062 insertion (SCF43.1.A08 [http://strepdb.streptomyces.org.uk]) in CYP102B1 (SCO0801) carried on cosmid SCF43 was obtained from a library of mutated cosmids (2). The transposon-tagged cosmid was initially introduced into E. coli strain ET12567 containing pUZ8002 (4, 21) prior to its intergeneric conjugal transfer into S. coelicolor (12). Mutants that had undergone allelic replacement were selected through their resistance to apramycin, conferred by Tn5062, and sensitivity to kanamycin as a consequence of the loss of the nonreplicating cosmid. The nature of the mutation was subsequently confirmed by Southern hybridization using a Tn5062-specific probe.
Growth medium for wild-type and CYP102B1 mutant strains of S. coelicolor A3(2) and method for lipid analysis.
Phenotypic analysis of the CYP102B1 mutant and comparison to the wild type was undertaken on different solid and liquid media as described previously (12), as was antibiotic determination. Lipid-free minimal liquid medium was used for growth of both the wild type and strains of S. coelicolor A3(2), as described previously (12). The medium consisted of 0.2% (wt/vol) (NH4)2SO4, 0.5% (wt/vol) Difco Casamino Acids, 0.06% (wt/vol) MgSO4·7H2O, 5% (wt/vol) polyethylene glycol (PEG) 6000, Minor elements solution (consisting of 0.1% [wt/vol] [each] of ZnSO4·7H2O, FeSO4·7H2O, MnCl2·4H2O and CaCl2 anhydrous), 1% (wt/vol) glucose, and 0.02% (vol/vol) NaH2PO4-K2HPO4 buffer (0.1 M, pH 6.8). Following growth at 25°C and 150 rpm for 7 days, cells were harvested by centrifugation, and lipids were extracted using the Folch method as described previously (5). Lipids were analyzed by liquid chromatography/mass spectrometry (LC/MS) using a Finnigan MAT TSQ-7000 triple quadrupole mass spectrometer (Finnigan Corp., San Jose, CA) equipped with a standard electrospray ionization source outfitted with a 100-μm-inside-diameter deactivated fused capillary. The mass spectrometer was operated in full scan mode. The data were collected from m/z 300 to m/z 900 with a 1-s scan time.
RESULTS
Cloning, expression, and purification of CYP102B1.
Heterologous expression of CYP102B1syn-His8 in E. coli produced extremely low levels of protein, preventing its biochemical analysis. Hence, we established an efficient system for expression with the molecular chaperones GroES and GroEL, which have been shown to enhance the production of active and correctly folded human P450s (11). Consequently, expression of active CYP102B1syn-His8 was greatly enhanced, with levels of CYP reaching >250 nmol P450/liter culture after 44 h of culture at 15°C. Subsequent fractionations indicated that the overexpressed CYP102B1syn-His8 was soluble and capable of generating a spectral maximum at 451 nm in reduced CO difference analysis. CYP102B1syn-His8 was purified to near-homogeneity by a strategy similar to that described for other S. coelicolor P450s (24, 26), with subtle alterations. The specific content of CYP102B1syn-His8 was approximately 12.5 nmol/mg protein. The oxidized absolute spectrum of the purified CYP102B1syn-His8 showed a Soret band at 417 nm and α- and β-bands at 567 and 534 nm, while reduction with sodium dithionite resulted in a Soret peak at 413 nm. The Soret maximum obtained from the reduced CO difference spectrum was located at 451 nm (Fig. 1A).
FIG. 1.
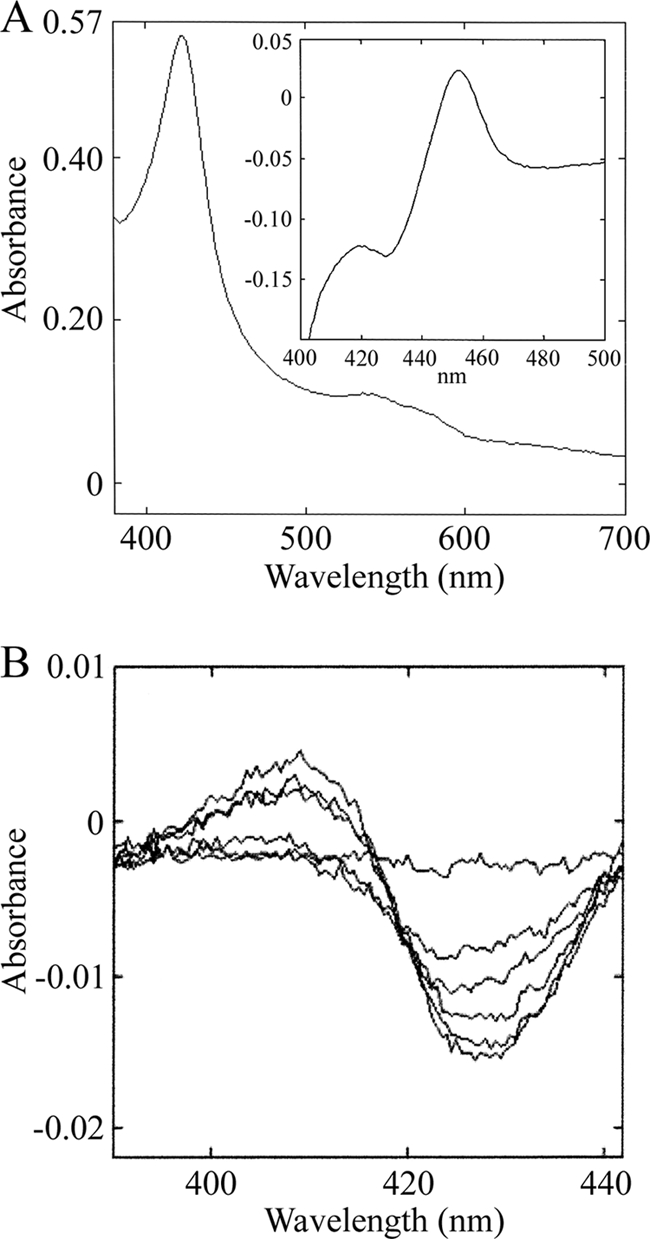
Expression of recombinant CYP102B1 and spectral changes induced by binding of arachidonic acid. (A) UV-visible spectra of purified CYP102B1. Absorption spectra in resting (ferric) and carbon monoxide (CO)-bound (inset) forms are shown. All spectra were recorded at ambient temperature in 50 mM potassium phosphate buffer, pH 7.4. (B) CYP102B1 arachidonate-induced difference spectra. Various concentrations of arachidonic acid (10 μM, 20 μM, 30 μM, 40 μM, and 50 μM dissolved in CH3OH) were added to a solution of CYP102B1 (0.5 μM) in 50 mM potassium phosphate buffer (pH 7.4) containing 20% (vol/vol) glycerol.
Spectral characterization of fatty acid binding.
Binding of fatty acids to CYP102A1 induces a shift in equilibrium of the heme iron low-spin state toward the high-spin form, leading to changes in the Soret region (18). To determine whether fatty acids bound CYP102B1, spectral binding titrations were performed with saturated (palmitic and stearic acids) and unsaturated (oleic and arachidonic acids) forms. Only arachidonic acid showed substrate-induced difference spectra (Fig. 1B). Plots of absorption changes versus the arachidonate concentration were hyperbolic, and the apparent dissociation constant, the Kd value, was determined. A clear observation is that CYP102B1 binds arachidonic acid moderately, with a Kd value calculated to be 33.3 μM, whereas CYP102A1 binds arachidonate more tightly, with a Kd value of 3.6 μM (18). Furthermore, palmitate, stearate, and oleate show negligible binding to CYP102B1, in contrast to CYP102A1.
Arachidonic acid metabolism by CYP102A1 and CYP102B1.
The turnover of arachidonic acid by CYP102A1 and CYP102B1 was determined by RP-HPLC using [14C]arachidonic acid as a substrate, and the identities of the products were established by comparison of retention times with authentic standards and by gas chromatography-mass spectrometry (GCMS). As expected, in our experiments CYP102A1 catalyzed the metabolism of arachidonic acid with efficient hydroxylation rates of >15,000 min−1 (18), with two major products being observed: 18-hydroxy arachidonic acid (18-OH AA) (Fig. 2A) (retention time, 14.2 min) and 14,15-epoxyeicosa-5,8,11-trienoic acid (14,15-EET) (Fig. 2A) (retention time, 20.5 min). Analysis of catalytic activity for CYP102B1 revealed marked differences in the ratio of products produced and the production of a new product compared to results for CYP102A1, 11,12-epoxyeicosa-5,8,14-trienoic acid (11,12-EET) (Fig. 2A) (retention time, 22 min). 18-OH AA and 14,15-EET eluted at identical retention times in all experiments. CYP102B1 produced similar amounts of 11,12-EET and 14,15-EET, as well as 18-OH AA, with a catalytic rate of 20 ± 3 min−1, whereas CYP102A1 produced approximately 60% 18-OH AA and 40% 14,15-EET, with a catalytic rate of 3,200 ± 400 min−1. The production of 11,12-EET by CYP102B1 is unique to this CYP102 family member, since no CYP102A has been shown to produce this molecule from arachidonic acid (Fig. 2B).
FIG. 2.

Arachidonic acid hydroxylation by CYP102A1 and CYP102B1 in heterologous electron transfer systems. (A) The positions of arachidonic acid and major products are shown in each case as indicated. The concentrations of the individual protein components used were 1 μM CYP102A1 and 1 μM CYP102B1, 1.0 μM spinach ferredoxin reductase, and 1.6 μM spinach ferredoxin. Experiments were performed in triplicate, and in all control experiments (without CYP or without reductase), no product formation was observed (data not shown). (B) Metabolic pathways for the catalysis of arachidonic acid by CYP102A1 and CYP102B1.
CYP102B1 is not required for normal cell growth and secondary metabolite production.
A CYP102B1 transposon mutant (insertion SCF43.1.A08; http://strepdb.streptomyces.org.uk) was generated, and lipids from the mutant strain were extracted, analyzed, and compared to the lipids from the parent strain. The location of the Tn5062 insertion within the CYP102B1 gene was 463 bp from the 5′ end (following amino acid G154, 285 residues prior to the conserved cysteine located at amino acid position 440) and 1,120 bp from the 3′ end of the gene, i.e., 5′ to the part of the gene encoding the heme binding domain (Fig. 3A). The heme-binding domain is located at 433FGTGARACIG442. Since this domain is essential for any CYP catalytic activity, no functional CYP102B1 was produced in the mutant.
FIG. 3.
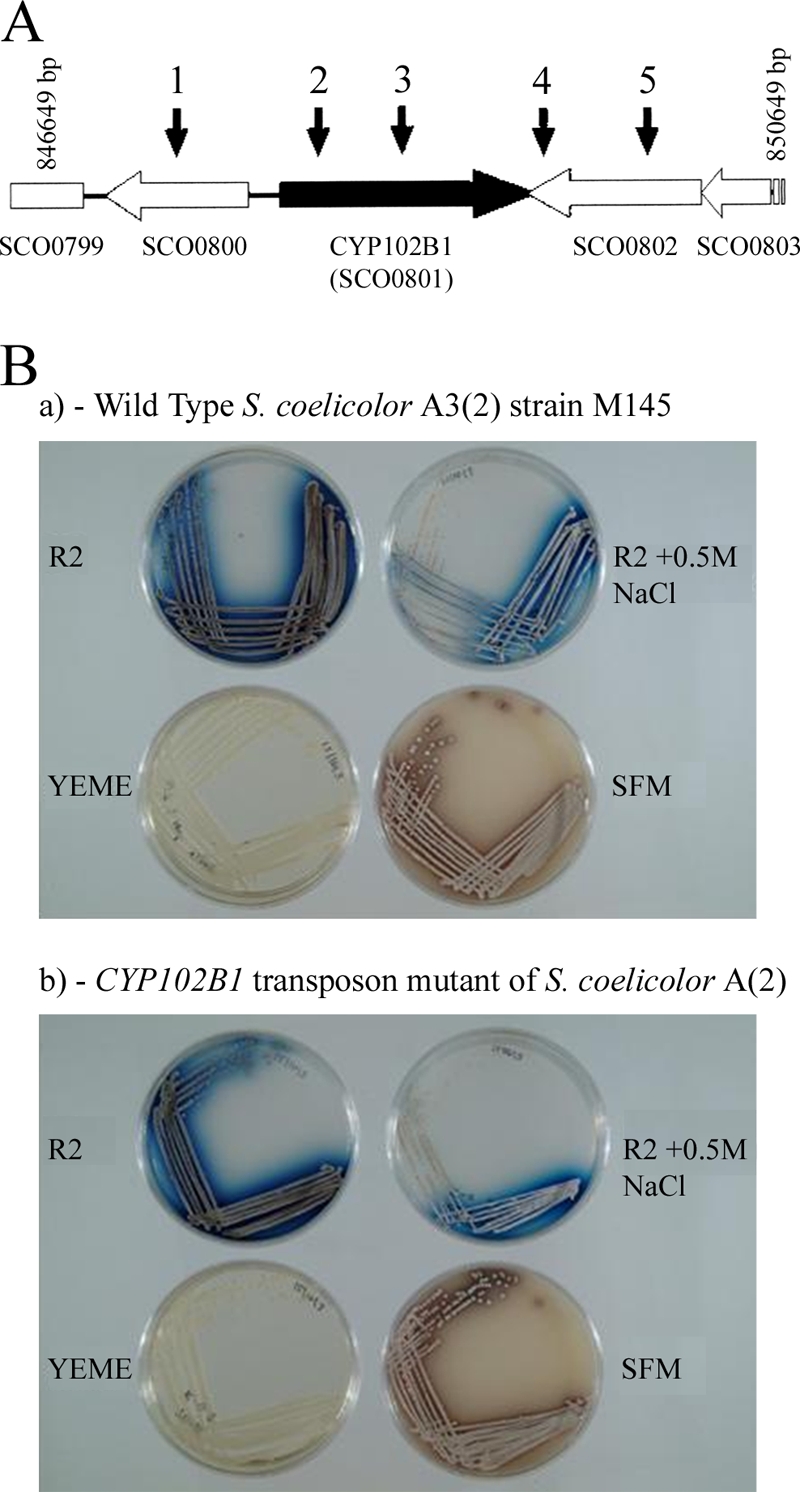
(A) Structure of the CYP102B1 gene and position of the transposon insertions. Arrows above the CYP102B1 gene region indicate the location of representative Tn5062 insertions. (B) CYP102B1 is not essential for growth in S. coelicolor A(2). a, wild type M145 S. coelicolor A3(2); b, CYP102B1 transposon mutant of S. coelicolor A(2). Cultures were incubated for 7 days at 30°C. R2 agar medium containing 10% (wt/vol) sucrose, 1% (wt/vol) glucose, 0.5% (wt/vol) yeast extract, and 0.01% (wt/vol) Casamino Acids; YEME agar medium containing 34% (wt/vol) sucrose, 1% (wt/vol) glucose, 0.3% (wt/vol) yeast extract, 0.3% (wt/vol) malt extract, and 0.5% (wt/vol) Bacto-peptone; and SFM agar medium containing 2% (wt/vol) soya flour and 2% (wt/vol) mannitol were used for growth of both the wild-type and CYP102B1 strains of S. coelicolor A3(2), as described previously (41).
No phenotypic differences were observed between the mutant and the M145 parental strain in comparing both antibiotic production and cell development when they were grown under different solid medium conditions (Fig. 3B). Similarly, B. subtilis CYP102A2 and CYP102A3 were previously shown to be nonessential (9). Given the overwhelming evidence in the literature documenting fatty acid metabolism by members of the CYP102 family, particularly CYP102A1, and our own data establishing CYP102B1 as a fatty acid hydroxylase, we were led to investigate the lipid profiles of both the parent and mutant strains.
Using wild-type and CYP102B1 mutant whole-cell lipid extracts from 1 liter of stationary-phase cultures, a variety of previously identified and new conjugated lipids were observed. These included phospholipids, glycolipids, lipoamino acids, glycerides, and fatty acids. Careful examination of the lipid profiles from both the parent and CYP102B1 mutant lipid extracts revealed differences in the lipids produced between strains (Fig. 4A to C). Although there were quantitative differences in the lipids isolated between experiments, the qualitative differences observed in the lipid profiles were always consistent. Due to the complexity of the many new classes of previously unidentified S. coelicolor fatty acids and lipids which are observed, including a range of hydroxyl fatty acids, deciphering the basis of these changes is not possible at this time. Given that there are clear alterations in the lipid profile following disruption of the CYP102B1 gene, a role for the gene product in lipid biochemical pathways is strengthened. However, further work in the long term will be required to decipher these alterations.
FIG. 4.

(A) Positive-ion electrospray mass spectra (m/z 500 to m/z 700) of CH3Cl-methanol extracts of lipids from wild-type Streptomyces coelicolor A3(2) (top) or the CYP102B1 mutant (bottom). High-resolution mass spectra were determined by direct liquid infusion using a Waters, Inc., Synapt Q-TOF instrument operating in reflectron time-of-flight (TOF) mode at resolution 9,000 (peak width at half-height). Samples were infused at 10 μl/min in chloroform-acetonitrile-isopropanol-water (30:35:30:5) with 5 mM ammonium acetate. Lipid species in wild-type S. coelicolor differing from those of the mutant are identified by the “#” symbol (top); conversely, changes in the CYP102B1 mutant from the wild-type are denoted by “*” (bottom). The lipids differing between the wild-type and mutant strains were not readily identifiable by searching the Lipid MAPS database (http://www.lipidmaps.org/index.html). (B) Positive-ion electrospray mass spectra (m/z 700 to m/z 900) of CH3Cl-methanol extracts of lipids from wild-type Streptomyces coelicolor A3(2) (top) or the CYP102B1 mutant (bottom). Lipid species in wild-type S. coelicolor differing from those of the mutant are identified by the “#” symbol (top); conversely, changes in the CYP102B1 mutant from the wild type are denoted by “*” (bottom). The lipids differing between the two organisms were not readily identifiable by searching the Lipid MAPS database (http://www.lipidmaps.org/index.html). (C) Negative-ion electrospray mass spectra (m/z 580 to m/z 910) of CH3Cl-methanol extracts of lipids from wild-type Streptomyces coelicolor A3(2) (top) or the CYP102B1 mutant (bottom). High-resolution mass spectra were determined by direct liquid infusion using a Waters, Inc., Synapt Q-TOF instrument operating in reflectron TOF mode at resolution 9,000 (peak width at half-height). Samples were infused at 10 μl/min in chloroform-acetonitrile-isopropanol-water (30:35:30:5) with 5 mM ammonium acetate. Lipid species in wild-type S. coelicolor differing from those of the mutant are identified by “#” (top); conversely, changes in the CYP102B1 mutant from the wild type are denoted by “*” (bottom). The lipids differing between the two organisms were not readily identifiable by searching the Lipid MAPS database (http://www.lipidmaps.org/index.html).
DISCUSSION
To date, 27 members of the CYP102 family have been uncovered and named. First, it is important to note that all CYP102 genes have been found exclusively in soil-dwelling bacteria, suggesting that they confer a metabolic advantage to these particular organisms in this environment. Second, 14 belong to the CYP102A subfamily. CYP102A6 was identified in the nitrogen-fixing bacterium Bradyrhizobium japonicum, which is the only Gram-negative bacterium to date to possess a CYP102 gene. The remaining 13 CYP102s belong in different subfamilies (B to J). Further, the majority of CYP102s are fusion proteins with a P450 heme domain and FMN/FAD-containing reductase domain, with only S. coelicolor CYP102B1, Streptomyces avermitilis CYP102B2, Rhodococcus sp. RHA1 CYP102B3, and Streptomyces scabies CYP102B4 existing as a P450 domain alone in a single polypeptide chain (15). Only three CYP102 P450s have been biochemically analyzed to date, CYP102A1, A2, and A3 (10). Even comparing these three members in the same subfamily, differences in catalytic activity were reported, with CYP102A2 and A3 from Bacillus subtilis showing high selectivity for long-chain unsaturated and branched fatty acids compared with results for B. megaterium CYP102A1 (10).
In S. coelicolor, CYP102B1 is not associated genetically with other genes unlinked in the chromosome. This is also true for CYP102B2 in S. avermitilis. To assess the contribution of CYP102B1 to the life cycle of S. coelicolor, we isolated a strain carrying a transposon mutation of this gene. Earlier work using deletion of CYP102A2 and CYP102A3 individually or combined has shown that they are not essential to the life cycle of B. subtilis (9). Disruption of CYP102B1 in S. coelicolor had no effect on the ability of the organism to grow and differentiate compared to the wild type, ruling out roles for CYP102B1 in cell development. We then explored the mutation of CYP102B1 for secondary metabolite production and the production of the two pigmented antibiotics actinorhodin and undecylprodigiosin in particular. There was no difference in the generation and yield of the two antibiotics on comparing the mutant with the wild type. With no phenotypic differences observed in a comparison of the wild-type and CYP102B1 mutant S. coelicolor strains, one cannot exclude enzymatic redundancy of CYP102B1 in the genome of S. coelicolor, but given the high selectivity and substrate specificity for P450s in general and CYP102s in particular, this would be highly unlikely.
In biotechnology, there is much potential for application of CYPs demonstrable in past use, such as the 11β-hydroxylation of Reichstein S in the biosynthesis of hydrocortisone and the formation of dicarboxylic acids from alkanes (22). Such reactions are catalyzed by fungal fermentations and P450 monooxygenases. Consequently, several laboratories have used isolated P450 enzymes, and CYP102A1 in particular, to investigate novel enzymatic activities with applied use. For example, site-directed mutagenesis of CYP102A1 led to an enzyme with activity toward testosterone and the drugs dextromethorphan and amodiaquine (23), while random mutagenesis led to a CYP102A1 mutant with alkane hydroxylase activity (7). Herein, we have characterized a new member of the CYP102 family which has unique stereo- and regiospecific catalysis toward a known CYP102A1 substrate and structurally consists only of the heme domain, whereas all CYP102s known to date comprise a fusion between the heme domain and a redox partner. It will be of interest in the future to understand the key molecular features, including specific amino acid residues, underpinning these enzymatic differences and to generate a fusion protein of the CYP102B1 heme domain with a suitable reductase partner in order to exploit this enzyme fully for biotechnological purposes. Such work will be undertaken in our laboratories in the future.
Acknowledgments
Support was provided by National Institutes of Health grants R01GM69970 and P30ES00267 and by a Leverhulme Trust Research Fellowship and a Royal Society Travel Award.
We appreciate the advice and experimental support of Jorge Capdevila and S. Wei.
Footnotes
Published ahead of print on 22 January 2010.
REFERENCES
- 1.Bentley, S. D., K. F. Chater, A. M. Cerdeño-Tárraga, G. L. Challis, N. R. Thomson, et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141-147. [DOI] [PubMed] [Google Scholar]
- 2.Bishop, A., S. Fielding, P. Dyson, and P. Herron. 2004. Systematic mutagenesis of a streptomycete genome: a link between osmoadaptation and antibiotic production. Genome Res. 14:893-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doddapaneni, H., R. Chakraborty, and J. S. Yadav. 2005. Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: evidence for gene duplications and extensive gene clustering. BMC Genomics 6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flett, F., V. Mersinias, and C. P. Smith. 1997. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA restricting streptomycetes. FEMS Microbiol. Lett. 155:223-229. [DOI] [PubMed] [Google Scholar]
- 5.Folch, J., M. Lees, and G. H. S. Stanley. 1957. Preparation of lipid extracts from brain tissue. J. Biol. Chem. 226:497-509.13428781 [Google Scholar]
- 6.Girvan, H. M., T. N. Waltham, R. Neeli, H. F. Collins, K. J. McLean, N. S. Scrutton, D. Leys, and A. W. Munro. 2006. Flavocytochrome P450 BM3 and the origin of CYP102 fusion species. Biochem. Soc. Trans. 34:1173-1177. [DOI] [PubMed] [Google Scholar]
- 7.Glieder, A., E. T. Farinas, and F. H. Arnold. 2002. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20:1135-1139. [DOI] [PubMed] [Google Scholar]
- 8.Graham-Lorence, S., G. Truan, J. A. Peterson, J. R. Falck, S. Wei, C. Helvig, and J. H. Capdevila. 1997. An active site substitution, F87V, converts cytochrome P450 BM-3 into a regio- and stereoselective (14S,15R)-arachidonic acid epoxygenase. J. Biol. Chem. 272:1127-1135. [DOI] [PubMed] [Google Scholar]
- 9.Gustafsson, M. C., C. N. Palmer, C. R. Wolf, and C. von Wachenfeldt. 2001. Fatty-acid-displaced transcriptional repressor, a conserved regulator of cytochrome P450 102 transcription in Bacillus species. Arch. Microbiol. 176:459-464. [DOI] [PubMed] [Google Scholar]
- 10.Gustafsson, M. C., O. Roitel, K. R. Marshall, M. A. Noble, S. K. Chapman, A. Pessegueiro, A. J. Fulco, M. R. Cheesman, C. von Wachenfeldt, and A. W. Munro. 2004. Expression, purification, and characterization of Bacillus subtilis cytochromes P450 CYP102A2 and CYP102A3: flavocytochrome homologues of P450 BM3 from Bacillus megaterium. Biochemistry 43:5474-5487. [DOI] [PubMed] [Google Scholar]
- 11.Kagawa N., H. Hori, M. R. Waterman, and S. Yoshioka. 2004. Characterization of stable human aromatase expressed in E. coli. Steroids 69:235-243. [DOI] [PubMed] [Google Scholar]
- 12.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom.
- 13.Kitazume, T., N. Takaya, N. Nakayama, and H. Shoun. 2000. Fusarium oxysporum fatty-acid subterminal hydroxylase (CYP505) is a membrane-bound eukaryotic counterpart of Bacillus megaterium cytochrome P450BM3. J. Biol. Chem. 275:39734-39740. [DOI] [PubMed] [Google Scholar]
- 14.Lamb, D. C., T. Skaug, H.-L. Song, C. J. Jackson, L. Podust, M. R. Waterman, D. B. Kell, D. E. Kelly, and S. L. Kelly. 2002. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem. 277:24000-24005. [DOI] [PubMed] [Google Scholar]
- 15.Lamb, D. C., H. Ikeda, D. R. Nelson, J. Ishikawa, T. Skaug, C. J. Jackson, S. Omura, M. R. Waterman, and S. L. Kelly. 2002. Cytochrome P450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2). Biochem. Biophys. Res. Commun. 307:610-619. [DOI] [PubMed] [Google Scholar]
- 16.Narhi, L. O., and A. J. Fulco. 1986. Characterization of a catalytically self-sufficient 119,000-Dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 261:7160-7169. [PubMed] [Google Scholar]
- 17.Narhi, L. O., and A. J. Fulco. 1987. Identification and characterization of two functional domains in cytochrome P-450BM-3, a catalytically self-sufficient monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 262:6683-6690. [PubMed] [Google Scholar]
- 18.Noble, M. A., C. S. Miles, S. K. Chapman, D. A. Lysek, A. C. MacKay, G. A. Reid, R. P. Hanzlik, and A. W. Munro. 1999. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J. 339:371-379. [PMC free article] [PubMed] [Google Scholar]
- 19.Omura, T., and R. Sato. 1964. The carbon monoxide binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239:2379-2385. [PubMed] [Google Scholar]
- 20.Rupasinghe, S., M. A. Schuler, N. Kagawa, H. Yuan, L. Lei, B. Zhao, S. L. Kelly, M. R. Waterman, and D. C. Lamb. 2006. The cytochrome P450 gene family CYP157 does not contain EXXR in the K-helix reducing the absolute conserved P450 residues to a single cysteine. FEBS Lett. 280:6338-6342. [DOI] [PubMed] [Google Scholar]
- 21.Ryding, N. J., G. H. Kelemen, C. A. Whatling, K. Flardh, M. J. Buttner, and K. F. Chater. 1998. A developmentally regulated gene encoding a repressor-like protein is essential for sporulation in Streptomyces coelicolor A3(2). Mol. Microbiol. 29:343-357. [DOI] [PubMed] [Google Scholar]
- 22.Urlacher, V., and R. D. Schmid. 2002. Biotransformations using prokaryotic P450 monooxygenases. Curr. Opin. Biotechnol. 13:557-564. [DOI] [PubMed] [Google Scholar]
- 23.Van Vugt-Lussenburg, B. M., M. C. Damsten, D. M. Maasdijk, N. P. Vermeulen, and J. N. Commandeur. 2006. Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450 BM3. Biochem. Biophys. Res. Commun. 346:810-818. [DOI] [PubMed] [Google Scholar]
- 24.Zhao, B., F. P. Guengerich, A. Bellamine, D. C. Lamb, M. Izumikawa, L. Lei, L. M. Podust, M. Sundaramoorthy, J. A. Kalaitzis, L. M. Reddy, S. L. Kelly, B. S. Moore, D. Stec, M. Voehler, J. R. Falck, T. Shimada, and M. R. Waterman. 2005. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem. 280:11599-11607. [DOI] [PubMed] [Google Scholar]
- 25.Zhao, B., and M. R. Waterman. 2007. Novel properties of P450s in Streptomyces coelicolor. Drug Metab. Rev. 39:343-352. [DOI] [PubMed] [Google Scholar]
- 26.Zhao, B., X. Lin, L. Lei, D. C. Lamb, S. L. Kelly, M. R. Waterman, and D. E. Cane. 2008. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2). J. Biol. Chem. 283:8183-8189. [DOI] [PMC free article] [PubMed] [Google Scholar]