

Abstract
Vegetable fermentations rely on the proper succession of a variety of lactic acid bacteria (LAB). Leuconostoc mesenteroides initiates fermentation. As fermentation proceeds, L. mesenteroides dies off and other LAB complete the fermentation. Phages infecting L. mesenteroides may significantly influence the die-off of L. mesenteroides. However, no L. mesenteroides phages have been previously genetically characterized. Knowledge of more phage genome sequences may provide new insights into phage genomics, phage evolution, and phage-host interactions. We have determined the complete genome sequence of L. mesenteroides phage Φ1-A4, isolated from an industrial sauerkraut fermentation. The phage possesses a linear, double-stranded DNA genome consisting of 29,508 bp with a G+C content of 36%. Fifty open reading frames (ORFs) were predicted. Putative functions were assigned to 26 ORFs (52%), including 5 ORFs of structural proteins. The phage genome was modularly organized, containing DNA replication, DNA-packaging, head and tail morphogenesis, cell lysis, and DNA regulation/modification modules. In silico analyses showed that Φ1-A4 is a unique lytic phage with a large-scale genome inversion (∼30% of the genome). The genome inversion encompassed the lysis module, part of the structural protein module, and a cos site. The endolysin gene was flanked by two holin genes. The tail morphogenesis module was interspersed with cell lysis genes and other genes with unknown functions. The predicted amino acid sequences of the phage proteins showed little similarity to other phages, but functional analyses showed that Φ1-A4 clusters with several Lactococcus phages. To our knowledge, Φ1-A4 is the first genetically characterized L. mesenteroides phage.
Bacteriophages are the most abundant biological entities (estimated to be on the order of ≥1031) on the planet (9, 18). Phages are ubiquitous in nature and can influence the microbial ecology and genetics of bacteria. Because of their small (usually <60 kb) genomes, phages can provide an excellent model system for studying many biological processes, including DNA replication and genetic evolution. Despite this, many phages remain uncharacterized. Very little is known about phage diversity and phage-host interactions owing to the small number of sequenced phages. Furthermore, the existing phage sequence database is highly biased toward a limited spectrum of phage hosts, namely, Enterobacteriaceae, Bacillus, Staphylococcus, Pseudomonas, Vibrio cholerae, Lactococcus, Streptococcus thermophilus, and S. pyogenes. The majority of host species for sequenced phages are either pathogenic or dairy-related bacteria. Most of the newly sequenced phage genes have no assigned functions or matches in the GenBank database (7).
Vegetable fermentations rely on a variety of lactic acid bacteria (LAB). The proper succession of LAB directly determines the quality and safety of the final fermentation products. Leuconostoc mesenteroides initiates most vegetable fermentations. It converts the sugars in vegetables (primarily glucose and fructose) to lactic acid, acetic acid, ethanol, CO2, and other flavor compounds (22, 58, 59, 60, 61). Acid production lowers the pH of fermenting vegetables and inhibits the growth of many microorganisms, including pathogens. CO2 production promotes the establishment of an anaerobic environment which favors the growth of other LAB. The metabolites produced by L. mesenteroides largely determine the flavor characteristics of the final products. As fermentation proceeds, L. mesenteroides rapidly dies off. Other LAB, including Lactobacillus plantarum, take over and complete the fermentation.
It has been a widely held view that the disappearance of L. mesenteroides and the subsequent bacterial succession in sauerkraut fermentations are due to the inhibitory effect of acids that accumulate during fermentation (54, 61). Little is known about other factors that may play a role in bacterial succession. Recent studies have shown that phages are present in the vegetable fermentations (4, 47, 48, 74, 75). Because of the rapid lytic cycle of these phages, they may significantly impact starter cultures and bacterial succession in vegetable fermentations (56). Phages active against L. mesenteroides have been isolated and characterized (48); however, genome sequences have not been reported.
L. mesenteroides phage 1-A4 (designated Φ1-A4) is of particular interest. Φ1-A4 is a lytic phage that was repeatedly isolated during the initial stages of a commercial sauerkraut fermentation. As a result, Φ1-A4 may significantly influence the survival of L. mesenteroides and flavor development during sauerkraut fermentation. It was found that Φ1-A4 infects at least three different strains of L. mesenteroides (48), and therefore it may also promote genetic exchange and genetic diversity in microbial communities (34).
The objectives of this study were to determine and analyze the complete genome sequence of Φ1-A4, to experimentally identify the structural protein genes, and to compare the genome organization with that of related phages. To our knowledge, this study represents the first complete genomic and molecular characterization of Leuconostoc phage. The results from this study may provide new insights into our understanding of phage genetics. This study may aid the development of phage control technologies in vegetable and other fermentations that are susceptible to phage attack.
MATERIALS AND METHODS
Bacterial strain, phage, and media.
Phage Φ1-A4 and host L. mesenteroides 1-A4 were both isolated on the first day of fermentation in a 90-ton industrial sauerkraut fermentation tank (48). The phage and its host were maintained as separate glycerol stocks at −80°C for later use. L. mesenteroides 1-A4 was cultured statically in de Man-Rogosa-Sharpe (MRS) broth (Difco Laboratories, Detroit, MI) at 30°C. Φ1-A4 was propagated on L. mesenteroides 1-A4 in MRS broth supplemented with 10 mM CaCl2 at 30°C.
Phage lysate preparation.
Phage lysate was prepared as previously described by Lu et al. (46, 47). Briefly, early-log-phase host 1-A4 cells were inoculated with a Φ1-A4 stock at a multiplicity of infection of about 0.01 and then incubated at 30°C until complete cell lysis occurred (in about 6 h). The lysate was centrifuged at 4,000 × g for 10 min and filtered (0.22-μm pore size). The filtrate was treated with DNase I and RNase A. Phage particles were concentrated by polyethylene glycol precipitation and then purified by cesium chloride density gradient ultracentrifugation at 600,000 × g for 6 h at 15°C.
Phage DNA preparation.
Phage DNA was prepared from the ultracentrifuge-purified phage preparation by phenol-chloroform extraction as described previously (46, 47). Briefly, the CsCl-purified phage suspension was extracted with phenol and chloroform-isoamyl alcohol, followed by extraction with chloroform-isoamyl alcohol. Phage DNA was precipitated with sodium acetate and ethanol and then pelleted with a microcentrifuge. After being washed with 70% ethanol, the DNA pellet was air dried and then resuspended in Tris-EDTA buffer.
Phage genome sequencing and verification of the assembly.
Initial sequencing was done using plasmid libraries of short phage fragments. Φ1-A4 DNA was digested with MseI, and the resulting fragments were ligated into pUC18. Recombinant clones (n = 24) were sequenced using vector primer sequences. The sequences of 184 fragments were used to design additional primers. Sequencing reactions were then performed directly on uncut phage DNA using ThermoFidelase and Fimer technology (63, 67). This resulted in one linear fragment representing the completed phage genome. Trace assembly was done with Phred/Phrap software (21). Selected genome regions were verified by PCR using the primers listed in Table 1.
TABLE 1.
Primer pairs used in PCR and the sizes of PCR products
| Primer sequence (5′ to 3′) |
Size (bp) |
||
|---|---|---|---|
| Forward | Reverse | Predicted | Estimated |
| AAATATCAGTAGCAACTACACCCGACAAGTCC | ATGAAACGAGAACGACAGCAACGTCTTA | 3,149 | 3,100 |
| AATACACAAACACAATCATCAACCCAGTTAG | ATGAAACGAGAACGACAGCAACGTCTTA | 1,340 | 1,350 |
| AAATATCAGTAGCAACTACACCCGACAAGTCC | TGAAGAGATGTTCCGTAATGAGACTGGTCA | 1,224 | 1,250 |
| TCCCACAATTCAAGTTCATTAATACTATCAG | ATTGTTGTGTTCATTATAGGCAAATAGC | 978 | 950 |
| AGCAGAGTTCATTGCAGTTGAGTTTACAAAGG | ATCATTCAAAGCAACGGCGTTCTTATTACTC | 910 | 900 |
| TTCTCAGCGTACATAAACTTAAGAACTTGG | TACGTACCTTTGGTCTCTGGAATATTGTATC | 886 | 875 |
| AATACACAAACACAATCATCAACCCAGTTAG | TATTGTACGTGCCATATTATTAGCCGTAATCC | 738 | 725 |
DNA sequence and bioinformatic analyses.
The finished phage genome sequence was subjected to an automated two-step annotation using GAMOLA software (global annotation of multiplexed on-site blasted DNA sequences engine [2]). First, the predicted open reading frames (ORFs) were analyzed using a gapped BLASTP (basic local alignment search tool for protein sequences) algorithm and the nonredundant protein database provided by the National Center for Biotechnology Information (November 2009). This revealed common unbiased protein similarities to all known protein sequences. Second, a customized database generated from known phage genomes (E. Altermann, unpublished data) was used to reveal phage-specific protein similarities. Additionally, the deduced amino acid sequences were analyzed by using the Pfam (Protein Family) (5), TIGRfam (The Institute for Genomic Research, family) (27), and COG (Clusters of Orthologous Groups of Proteins) (70) functional databases to detect conserved motifs in the proteins. The automated annotation was augmented by structural analyses including the prediction of Rho-independent terminator structures (20), transmembrane helices (41), and signal peptide sequences (6). Subsequently, a manual verification was performed based on the functional analyses, physical genome properties (e.g., the expected modular makeup of the phage genome), physicochemical attributes of the predicted ORFs, and their deduced amino acid sequences.
The Molecular BioComputing Suite (57) was used to calculate the molecular masses and isoelectric points of predicted proteins of Φ1-A4.
Analysis of structural proteins.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described by Lu et al. (46). Briefly, CsCl-purified phages in SDS-PAGE sample buffer were heated in a boiling water bath for 10 min and then applied to a NuPAGE precast gradient minigel (4 to 12% Bis-Tris; Invitrogen Corporation, Carlsbad, CA). After electrophoresis, the protein bands were transferred onto a polyvinylidene difluoride membrane (0.2-μm pore size) and then stained with 0.1% Amido Black. Visible protein bands were excised, and their N-terminal amino acid sequences (10 or 11 residues) were determined by ProSeq, Inc. (Boxford, MA). Based on the N-terminal amino acid sequence of each protein, the corresponding ORF was identified in the Φ1-A4 genome.
Functional analysis.
A total of 112 phage genomes were analyzed, and a functional genome tree was generated using compACTor (Altermann, unpublished software) and Mega4 (69). The genomes were chosen from representative phages of hosts assigned to the phylogenetic class of bacilli in GenBank. Briefly, a unique genomic distance matrix was calculated based on round-robin ORF comparisons and subsequently imported into Mega4 for visualization. The functional distribution was visualized using the unweighted-pair group method using average linkages (UPGMA) (68).
Nucleotide sequence accession number.
The final verified genome sequence and annotation of Φ1-A4 were submitted to the publicly accessible GenBank database under accession number GQ451696.
RESULTS AND DISCUSSION
Φ1-A4 belongs to the Siphoviridae family in the order Caudovirales (Fig. 1). The average burst size of Φ1-A4 is 24 particles per infective center. The latency period is about 30 min in MRS broth supplemented with 10 mM CaCl2 at 30°C based on a one-step growth kinetics curve (56). In addition to infecting its natural host, L. mesenteroides 1-A4, Φ1-A4 was also found to infect two other L. mesenteroides strains, 1-B8 and 1-E10 (48). To our knowledge, this is the first report of genomic sequencing and characterization of an L. mesenteroides phage at the molecular level.
FIG. 1.

Transmission electron micrograph of phage Φ1-A4.
Genomic features of Φ1-A4.
The genome sequence of Φ1-A4 was determined in two steps. Plasmid libraries were used first to obtain initial phage sequences for primer design, and in the second step, several rounds of direct genomic sequencing were performed that yielded one linear contig (see Materials and Methods for details). The resulting genome sequence and the predicted gene model were verified by PCR amplification for accuracy of sequence assembly. The measured sizes of PCR products agreed well with the corresponding predicted sizes (Table 1), which confirmed the unusual genome structure of Φ1-A4. Furthermore, the repeats flanking the predicted origin of DNA replication were amplified and resequenced.
Φ1-A4 has a linear, double-stranded DNA genome consisting of 29,508 bp with a G+C content of 36%, which is very close to the G+C content (37%) of L. mesenteroides subsp. mesenteroides ATCC 8293 (53), which is closely related to the natural host of Φ1-A4 (L. mesenteroides 1-A4). The origin of DNA replication and a cos site were identified in the genome (Fig. 2 and 3). Two adjacent runoff sequencing reactions obtained during Sanger sequencing of the phage genome identified the cos site boundaries (Fig. 3a and c). Manual enhancement of the electropherograms resulted in the identification of the 22-nucleotide (nt) cos site sequence (GGTTAATAGTAGTCTTTTTTAA) (Fig. 3b). The presence of a cos site indicated that the phage DNA may be circularized upon entry into the host. The origin of DNA replication was identified based on its similarities in structure to other phage replication origins (3, 38). Briefly, a noncoding region was identified harboring an AT-rich domain (50 to 60 nt, >95%) flanked by direct and indirect repeat structures. In Φ1-A4, the noncoding region spans over 410 nt and contains a 55-nt AT-rich domain (98.19%) flanked by nine different types of direct and indirect repeats.
FIG. 2.

Genome atlas of phage Φ1-A4. The linear map was created using Genewiz, developed by Pederson et al. (60a; Center for Biological Sequence Analysis), and software developed in house. The legend at the bottom describes the individual panels corresponding to their respective designations (A to G). (A) Gapped BLASTP results obtained using the nonredundant database. (B) Gapped BLASTP results obtained using the custom phage genome database. In both panels A and B, blue regions represent unique proteins in Φ1-A4 and highly conserved features are red. The color intensity corresponds to the level of similarity. Individual E values were grouped into trust interval levels as depicted in the legend. (C) G+C content deviation: green shading, low-GC regions; orange shading, high-GC islands. (D) Annotation shows the absolute positions of functional features as indicated. (E) ORF orientation. ORFs in the sense orientation (ORF +) are blue; ORFs oriented in the antisense direction (ORF −) are red. The origin of DNA replication is indicated by a red inverted triangle. The cos site, depicting the phage genome boundaries, is indicated by two purple inverted triangles on the genome line (gray line). (F) COG classification. COG families were assembled into five major groups: 1, information storage and processing; 2, cellular processes and signaling; 3, metabolism; 4, poorly characterized; 5, ORFs with uncharacterized COGs or no COG assignment. (G) GC skew. ORFs with predicted functional annotation are shown at the top of panel A with bars indicating their absolute sizes. The color coding corresponds to predicted functional phage modules. Red, DNA replication; purple, DNA packaging; dark blue, phage head; sky blue, tail structure; green, lysis; greenish yellow, DNA regulation and modification; black, not assigned to a specific phage module.
FIG. 3.

Determination of the cos site. Two runoff Sanger electropherograms on linear phage DNA prepared from infectious particles identified the cos site boundaries. (a and c) Runoff electropherograms. (b) Manually enhanced traces identified the cos sequence. The figure is drawn to scale, and individual boxes are placed in relation to their expected sequence positions. Trace files were visualized using Chromas (http://www.technelysium.com.au/).
Bioinformatic analysis of the Φ1-A4 genome revealed 50 possible ORFs. The ORFs are numbered consecutively starting from the ORF immediately downstream of the origin. Twenty-two ORFs were located on one strand, and 28 ORFs were on the complementary strand (Table 2 and Fig. 2). Of the 50 ORFs, 48 apparently initiated translation with the ATG start codon, 1 initiated with TTG, and 1 initiated with GTG (Table 2). Forty-six of the 50 ORFs were preceded by potential Shine-Dalgarno sequences (64) complementary to the 3′ end of the 16S rRNA gene of L. mesenteroides ATCC 8293 (5′-CACCTCCTTTCT-3′). The consensus sequence (AGGAGG) of Shine-Dalgarno elements is generally conserved and was chosen as the recognition sequence for ribosome binding site (RBS) prediction (64). An in silico restriction site analysis of the nucleotide sequence agreed well with the experimentally determined restriction pattern (Fig. 4).
TABLE 2.
ORFs and genetic features of L. mesenteroides Φ1-A4a
| ORFb | Putative RBSe |
Database search resultsi for predicted product |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Startc | Endd | 3′-AGGAGG…f | Start codon | Size (aa) | Massg (kDa) | pIh | Predicted functionj | Organism matched | E valuek | Referencel | |
| 1 | 361 | 1713 | AGGAGG | ATG | 450 | 51.95 | 5.22 | DNA helicase | S. pneumoniae SP6-BS73 | 1e-52 | ZP_01821431.1 |
| 2 | 1710 | 2612 | ATG | 300 | 35.00 | 7.81 | Phage-related adenine-specific DNA methyltransferase | Streptococcus phage 9429.1 | 8e-86 | YP_596295.1 | |
| 3 | 2614 | 3369 | TGGTGAA | ATG | 251 | 29.07 | 5.43 | DNA primase/polymerase | |||
| 4 | 3448 | 3996 | AGGAGAA | ATG | 182 | 20.80 | 8.67 | DNA polymerase | L. mesenteroides subsp. cremoris ATCC 19254 | 3e-78 | ZP_03913787.1 |
| 5 | 4005 | 5840 | ACGAGAA | ATG | 611 | 69.27 | 6.39 | DNA polymerase | Listeria phage P40 | 4e-42 | YP_002261447.1 |
| 6 | 5896 | 6468 | AGGAGAA | ATG | 190 | 21.63 | 4.93 | Cons. hypo. | |||
| 7 | 6521 | 7153 | GGGAGGA | ATG | 210 | 24.01 | 5.57 | Hydrolase (tRNA 3′ endonuclease) | Listeria phage P40 | 8e-18 | YP_002261444.1 |
| 8 | 7164 | 7490 | AGGAGG | ATG | 108 | 12.40 | 4.81 | Terminase small subunit, deduced from position | |||
| 9 | 7552 | 7710 | ATG | 52 | 5.94 | 11.62 | Terminase small subunit, deduced from position | ||||
| 10 | 7694 | 9340 | AGGAGG | ATG | 548 | 62.91 | 5.02 | Terminase large subunit | S. pneumoniae prophage lambda Sa2 | 2e-72 | ZP_01828913.1 |
| 11 | 9353 | 10474 | AGGAGAA | ATG | 373 | 42.91 | 4.89 | Portal protein, amino acid sequence verified | E. coli phage HK97 | 4.5e-4 | This work and NP_037699 |
| 12 | 10434 | 11153 | AGGAGAA | ATG | 239 | 26.18 | 4.43 | Phage prohead protease | A. aphrophilus NJ8700 caudovirus phage | 1e-04 | YP_003006998.1 |
| 13 | 11205 | 12170 | AGGAGAA | ATG | 321 | 34.98 | 4.89 | Major capsid protein | Lactococcus phage Q54 | 2e-04 | YP_762590.1 |
| 14 | 12310 | 12582 | AGGAGGTG | ATG | 90 | 10.35 | 4.33 | Cons. hypo. | |||
| 15 | 12572 | 12850 | AGGAGG | ATG | 92 | 10.59 | 10.06 | Put. phage tail protein | |||
| 16 | 12850 | 13167 | GGGAGG | ATG | 105 | 12.39 | 4.44 | Cons. hypo. | |||
| 17 | 13164 | 13493 | AGGTGT | ATG | 109 | 12.32 | 10.92 | Cons. hypo. | |||
| 18 | 13542 | 14123 | AGGAGAA | ATG | 193 | 21.25 | 4.55 | Major tail protein, amino acid sequence verified | This work | ||
| 19 | 14197 | 16866 | AGGAAATG | ATG | 889 | 90.98 | 5.34 | Phage tail TMP, amino acid sequence verified | B. longum DJO10A | 1e-74 | This work and YP_001955090.1 |
| 20 | 16930 | 18126 | TGGAGGA | ATG | 398 | 45.48 | 4.71 | Cons. hypo. | |||
| 21 | 18129 | 19127 | AGGAGA | ATG | 332 | 36.69 | 4.49 | Baseplate, partial amino acid sequence match | This work | ||
| 22 | C19335 | C19736 | AGGAGAA | ATG | 133 | 15.74 | 4.75 | Cons. hypo. | |||
| 23 | 19816 | 20583 | AGGAGA | ATG | 255 | 27.84 | 5.17 | Tail protein, receptor binding, amino acid sequence verified | Oenococcus oeni phage fOg44 | 0.015 | This work and CAD19145.1 |
| 24 | C20616 | C20987 | AGGAGA | ATG | 123 | 14.00 | 5.72 | Holin | G. adiacens ATCC 49175 | 7e-07 | ZP_05738230.1 |
| 25 | C21054 | C21188 | AGGAGA | ATG | 44 | 5.50 | 10.37 | Cons. hypo. | |||
| 26 | C21185 | C21349 | TGGAGA | ATG | 54 | 6.39 | 8.34 | Unknown | |||
| 27 | C21349 | C21579 | TTGAGG | ATG | 76 | 9.01 | 8.94 | Cons. hypo. | |||
| 28 | C21579 | C21815 | AGGAAAA | ATG | 78 | 9.18 | 6.31 | Unknown | |||
| 29 | C21805 | C22020 | ATAGGA | ATG | 71 | 8.93 | 6.94 | Unknown | |||
| 30 | C22020 | C22142 | GAGAGG | ATG | 40 | 4.43 | 9.24 | Unknown | |||
| 31 | C22139 | C22639 | AGGAGAA | ATG | 166 | 18.81 | 9.38 | Phage-related hydrogenase | L. mesenteroides subsp. mesenteroides ATCC 8293 | 1e-46 | YP_819431.1 |
| 32 | C22660 | C22869 | AGGAGAA | ATG | 69 | 8.14 | 4.10 | Unknown | |||
| 33 | C22945 | C23115 | AAGAGGA | ATG | 56 | 6.09 | 8.60 | Unknown | |||
| 34 | C23112 | C23357 | AGGAGAA | ATG | 81 | 9.76 | 5.88 | Unknown | |||
| 35 | C23473 | C24354 | AGGAGGA | ATG | 293 | 31.60 | 5.92 | Phage-related amidase, lysin | E. faecalis phage phiEF24C | 4e-41 | YP_001504118.1 |
| 36 | C24356 | C24640 | TGGTGG | GTG | 94 | 10.61 | 8.94 | Holin | |||
| 37 | C24696 | C25061 | AGGAGA | ATG | 121 | 13.14 | 4.13 | Cons. hypo. | |||
| 38 | C25073 | C25807 | AGGAGGA | ATG | 244 | 26.08 | 5.85 | Baseplate | L. lactis phage r1t | 0.022 | NP_695073.1 |
| 39 | C25873 | C26190 | AGGAGG | ATG | 105 | 11.91 | 9.11 | Unknown | |||
| 40 | C26192 | C26401 | TTG | 69 | 8.04 | 4.93 | Unknown | ||||
| 41 | C26395 | C26670 | AGGAAGTG | ATG | 91 | 10.63 | 4.59 | Cons. hypo. | |||
| 42 | C26667 | C26918 | AGGAGA | ATG | 83 | 9.71 | 4.33 | Cons. hypo. | |||
| 43 | C26911 | C27099 | AGGAGA | ATG | 62 | 7.58 | 6.94 | Unknown | |||
| 44 | C27086 | C27220 | GGGAGAA | ATG | 44 | 5.35 | 4.66 | Put. baseplate | |||
| 45 | C27368 | C27538 | AGGAAA | ATG | 56 | 6.50 | 5.31 | Repressor, CopG | Nocardioides sp. strain JS614 | 0.26 | YP_919229.1 |
| 46 | C27947 | C28066 | ATG | 39 | 4.76 | 10.38 | Unknown | ||||
| 47 | C28103 | C28405 | AGGATA | ATG | 100 | 11.64 | 7.84 | Phage HNH endonuclease | L. casei prophage lambda Ba04 | 4e-07 | YP_001987049.1 |
| 48 | C28443 | C28634 | AGGAGAA | ATG | 63 | 7.47 | 4.06 | Unknown | |||
| 49 | C28636 | C29076 | AGGAGC | ATG | 146 | 16.95 | 8.66 | Phage-related protein | E. faecalis phage phiEF24C | 3e-12 | YP_001504167.1 |
| 50 | C29073 | C29447 | AGGAGGA | ATG | 124 | 14.87 | 8.60 | Endodeoxyribonuclease RusA | |||
See the text for details.
ORFs were numbered sequentially starting from the ORF immediately downstream of the predicted origin of DNA replication.
Genes located on the complementary strand are indicated by the letter C.
The end position includes the stop codon.
The sequence shown matches at least 60% of the consensus RBS (AGGAGG) sequence.
RBS consensus sequence identified downstream of promoters.
Molecular masses were calculated with the Molecular Biocomputing Suite (57).
Isoelectric points were calculated with the Molecular Biocomputing Suite.
Database searches based on homologies of deduced amino acid sequences to the nonredundant amino acid database provided by NCBI were performed with the gapped BlastP algorithm. The best phage-related hits are included here.
Abbreviations: Cons. hypo., conserved hypothetical; Put., putative.
Probabilities derived from BLAST scores for obtaining a match by chance.
Accession number or source.
FIG. 4.
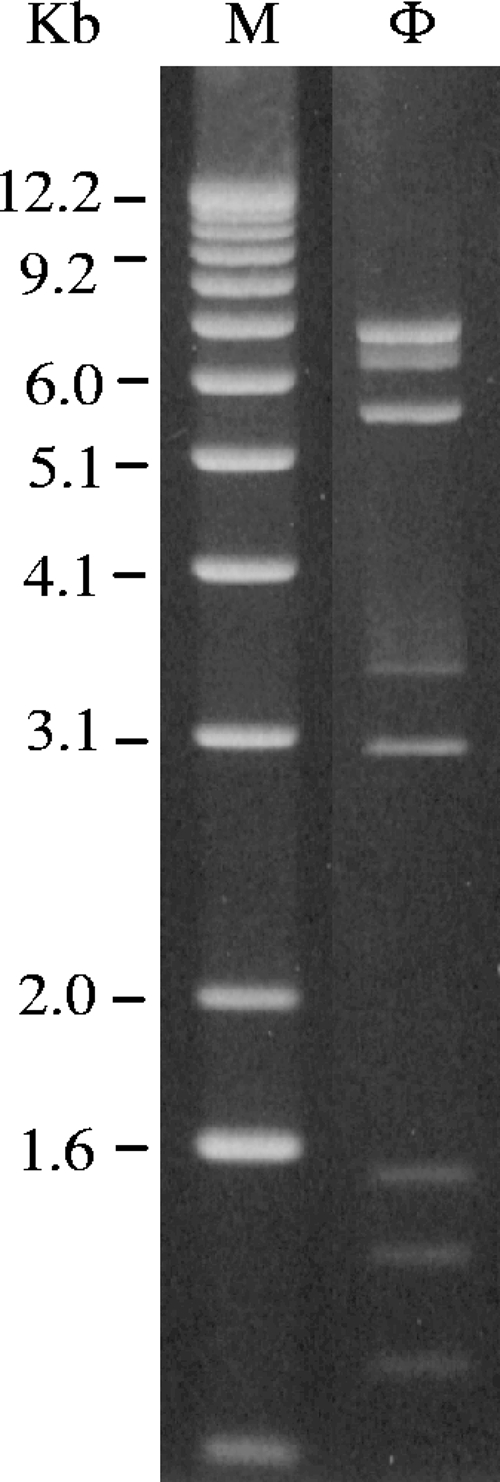
HindIII restriction digestion analysis of Φ1-A4 DNA. Lane M, 1-kb DNA ladder; lane Φ, Φ1-A4.
A total of 26 (52%) ORFs showed homology with previously characterized genes or were functionally annotated based on their respective positions in the phage genome. In many cases, homology to sequences of phages infecting gram-positive bacteria was found (Table 2). Twelve ORFs exhibited similarities to uncharacterized database entries (conserved hypotheticals), and 12 other ORFs had no homology to sequenced genes.
The phage genome was found to be modularly organized, consisting of DNA replication, DNA-packaging, head and tail morphogenesis, cell lysis, and gene regulation modules (Fig. 2). In silico analyses revealed that Φ1-A4 contains a large-scale genome inversion (∼30% of the genome) compared to other phages, including the lysis module, part of the structural module, and the likely cos site. The predominant genome of phages with hosts phylogenetically closely related to Φ1-A4 is linear, with nearly all of the genes located on the sense strand (for example, Leuconostoc phage KM20 and L. mesenteroides ATCC 8293 prophage, Fig. 5). The genome of Φ1-A4 has a significant number of genes on the antisense strand, which is similar to the organization of Lactococcus phage genomes (Fig. 5). Large-scale genome inversions can significantly alter transcription patterns. We have identified putative promoter structures in the genome sequence of Φ1-A4 that are likely to initiate the transcription of the inverted genome region (not shown). While we cannot exclude the possibility that the apparent inverted sequences are the result of recombination with another phage, we found no evidence to support this hypothesis. Both sections of the genome of Φ1-A4 show the same GC content and codon usage. Further analyses will be carried out when the genome sequences of other Leuconostoc phages (or phages isolated from the same ecosystem) become available.
FIG. 5.

pACT alignment of Φ1-A4 against phages of its functional cluster (F-Cluster 1 in Fig. 7), as well as against Leuconostoc phage KM20 and L. mesenteroides ATCC 8293 prophage. Based on the FGD algorithm, predicted phage ORFeomes were subjected to an all-versus-all analysis. Results were stored in pMSP data files (http://www.sanger.ac.uk/Software/ACT/v7/manual/start.html#COMPARISON-FILE-FORMAT) and visualized using the Artemis comparison tool (10). Red lines show direct similarities between amino acid sequences of predicted ORFs, and blue lines represent inverted similarity hits. Color intensities are proportional to the respective increasing levels of sequence similarity. Phage genomes are represented by two gray lines (showing sense and antisense strands), and light blue arrowed boxes indicate individual predicted ORFs, their relative lengths, and their orientations. Genomes are drawn to scale.
No evidence of a lysogeny module was found in the Φ1-A4 genome. However, ORF22 exhibited very weak BLASTP similarities to the integrase subunit predicted in the Solibacter usitatus genome (GenBank accession no. CP000473). A two-iteration PSI-BLAST supported the initial finding. No significant functional Pfam or TIGRfam domains were identified. Only a very weak Pfam match (E value, 0.67) to SNF5, as well as a weak Pfam match to INI1, was detected. It is possible that ORF22 represents the remnants of an integration module or that it encodes a unique phage-specific protein of unknown function.
DNA replication module.
Downstream of the putative origin of DNA replication (Fig. 2) are five genes (labeled in red) encoding proteins that may be involved in phage DNA replication. The predicted gene product of ORF1 (gpORF1) shows strong similarity to the putative DNA helicase of Streptococcus pneumoniae SP6-BS73 (Table 2) and that of Lactobacillus reuteri (data not shown). The derived protein product of ORF2 displays homology to the adenine-specific DNA methyltransferase from Streptococcus phage 9429.1. Adenine-specific DNA methyltransferase is often involved in chromosomal site-specific DNA modification systems. In prokaryotes, the major role of DNA methylation is to protect host DNA against degradation by restriction enzymes (12). ORF3 encodes a protein homologous to various hypothetical bacterial proteins that have been shown to be bifunctional, having both DNA primase and polymerase activities (44). This suggests that the ORF3 protein is possibly involved in DNA synthesis. The predicted protein products of ORF4 and ORF5 exhibit sequence similarities to the hypothetical DNA polymerases from L. mesenteroides subsp. cremoris ATCC 19254 and to the putative DNA polymerase from Listeria phage P40 (Table 2) and Lactobacillus phage SalI (15), suggesting that these two proteins may be required for DNA replication.
The predicted product of ORF7 was found to be homologous to tRNA 3′ endonucleases from Listeria phage P40 and Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842. tRNA 3′ endonuclease is a hydrolase that belongs to the metallo-β-lactamase superfamily. Members of this family are ribonucleases that process the 3′ end of tRNA precursors by cleaving the 3′ trailing sequence, thereby preparing the pre-tRNA for the addition of a CCA tail (72).
DNA-packaging module.
The protein specified by ORF10 exhibited similarity (Table 2) to a putative terminase large subunit from prophage lambda Sa2 harbored by Streptococcus pneumoniae SP14-BS69 (71). Terminases are enzymes involved in phage DNA packaging into proheads (11). These enzymes function as heteromultimers consisting of a large subunit and small subunits. In tailed phages, the small subunits of the terminase are responsible for specific DNA binding and the large subunit of the terminase mediates cleavage of the phage DNA into genome size units to be packaged into the prohead (19). Generally, the genes encoding terminase small subunits are located upstream of the terminase large subunit gene. Since the gene positions of ORF8 and ORF9 are upstream of the terminase large-subunit gene (Fig. 2), the products derived from these two ORFs are likely to be the small terminase subunits. Experimental data are needed to confirm if these two proteins are involved in phage DNA packaging.
Head and tail morphogenesis modules.
Immediately downstream of the DNA-packaging module are the morphogenesis modules, including the head and tail modules. SDS-PAGE with a 4 to 12% Bis-Tris gradient gel revealed several structural protein bands (Fig. 6). Five bands were collected for the determination of N-terminal amino acid sequences (10 or 11 amino acids at the N terminus). Based on the N-terminal sequences, ORF11, ORF18, ORF19, ORF21, and ORF23 from the Φ1-A4 genome were identified as the genes encoding the proteins extracted from the bands. An N-terminal methionine was absent from four of the five protein sequences (Fig. 6). Processing of the N-terminal methionine during protein maturation has been observed for many phages and occurs via host methionine aminopeptidase activity (45, 50).
FIG. 6.

Analysis of phage Φ1-A4 structural proteins. After the purification of Φ1-A4 by CsCl density gradient centrifugation, the phage-containing band was analyzed by 4 to 12% SDS-PAGE. Lane M, molecular mass marker; lane Φ, proteins from Φ1-A4. The N-terminal amino acid sequences of the five structural proteins were determined. The position of the N-terminal sequence in the corresponding Φ1-A4 ORF and its proposed function are indicated.
The phage head morphogenesis module (dark blue in Fig. 2) includes ORFs encoding the experimentally determined minor head protein (also a putative portal protein), a putative prohead protease, and a putative head protein. The head genes are clustered together and precede the tail genes. The 42.9-kDa protein revealed on the SDS-PAGE (Fig. 6) has an N-terminal amino acid sequence (SQFDDINTRI) identical to residues 2 to 11 of the predicted gpORF11. This protein shows homology to the portal protein of Escherichia coli phage HK97 (35) (Table 2). Portal proteins typically function as dodecamers of a single polypeptide with an average molecular mass of 40 to 90 kDa. They form a hole (or portal) that enables DNA passage into the phage head during packaging (43).
gpORF12 showed similarities to the prohead proteases of the tailed phages of Aggregatibacter aphrophilus and Burkholderia thailandensis (Table 2) and, to a lesser degree, to the phage prohead peptidase of Enterococcus faecium phage HK97 (32). Prohead proteases are involved in the processing of the prohead protein (31). The prohead protease gene is generally located next to the gene for the prohead protein that is processed. In some cases, a prohead protease gene may be fused to the prohead protein gene. Immediately downstream of ORF12 is ORF13. gpORF13 showed homology to a major capsid protein of lactococcal phage Q54 (23). Based on the location of its gene in the genome, the putative major head protein is likely to be the protein processed by prohead protease. The cleavage of the head protein by the prohead protease generates the scaffold protein which aids prohead construction (28, 29).
Downstream of the head morphogenesis module is the tail morphogenesis module (sky blue in Fig. 2) including genes encoding a putative tail protein, four experimentally identified tail proteins (major tail protein, “tape measure” protein [TMP], baseplate protein, and receptor binding protein), and two putative baseplate proteins.
The product of ORF15 was found to have some similarity (E value, 1.2) to a tail protein of Lactococcus lactis phage TP901-1 (IPR011855) in the TIGRfam database. ORF15 is located between the major head and major tail genes (Fig. 2). In general, the genes located in this region are involved in the formation and connection of the head and tail structures and in DNA packaging (8).
The smallest structural protein observed by SDS-PAGE (Fig. 6) had a predicted molecular mass of 21.3 kDa. This protein appears to be the major phage protein, based on the amount of the protein observed in the SDS-PAGE gel. This protein is encoded in the tail protein gene region (Fig. 2) and displayed homology (although it was weak) to a phage major tail protein from the Phi13 family of Streptococcus pneumoniae CDC1873-00 (data not shown). The N-terminal sequence (MYDTRKITHGS) of the protein was identical to predicted residues 1 to 11 of the Φ1-A4 gpORF18.
A protein with a predicted molecular mass of 91 kDa was the largest structural protein observed on SDS-PAGE (Fig. 6). The N-terminal amino acid sequence of the 91-kDa protein was ANTTSYMLKF, matching predicted amino acids 2 through 11 of the protein encoded by ORF19. The protein sequence was similar to that of the Mu-like phage minor tail protein in Bifidobacterium longum DJO10A (42) (Table 2). In many Mu-like phages, the gene(s) encoding tail fibers is relatively long. The predicted protein product of ORF19 contains a TMP domain that is only found in phages and prophages. These types of proteins determine phage tail length. In phage lambda, the TMP is used as a template for tail polymerization and remains inside the tail tube (37). The length of the phage tail is directly proportional to the size of the TMP (36). TMPs are found in almost all Siphoviridae phages (62). Like most TMPs (800 to 5,006 amino acids long [55]), the 91-kDa protein (889 amino acids long) of Φ1-A4 is the largest predicted protein in the Φ1-A4 genome (Table 2) and is encoded in the putative tail protein gene region (Fig. 2). These features suggested that this protein is most likely to be a TMP.
The N-terminal amino acid sequence (ADTRTIYLHT) determined for the 36.7-kDa protein (Fig. 6) partially matched amino acids 2 through 11 (ADTSSIYLHT) of gpORF21. Residues 4 and 5 of gpORF21 were SS instead of RT. This result is possibly due to miscalled amino acids during the sequencing of this low-abundance protein. The protein appears to be a baseplate protein, based on its weak TIGRfam match to the baseplate assembly protein in L. lactis phage P2 (IPR013046). Kondou et al. (40) showed that baseplate protein is essential for phage Mu assembly and the generation of viable phages.
The N-terminal sequence of the 27.8-kDa protein (Fig. 6) is TLNTNEIVYT, matching predicted amino acids 2 through 11 of gpORF23. This protein may be responsible for host specificity because of the similarity (found by PSI-BLAST analysis) to a receptor binding protein in Lactococcus phage Q41 (accession number AAT81505.1). The protein also displayed sequence similarity to a hypothetical protein (ORF232) of Oenococcus oeni phage fOg44 (Table 2). This may suggest that the hypothetical protein of phage fOg44 is also involved in host range determination.
Figure 2 shows that the putative tail morphogenesis module was interspersed with ORFs belonging to the cell lysis module and other ORFs with unknown functions. This is not usually observed in other phages. This may have resulted from the large genome inversion as described above. Two ORFs, ORF38 and ORF44, are downstream of the lysis module and possibly encode baseplate proteins. The product of ORF38 displays some similarity to the structural protein from L. lactis phage r1t (73) (Table 2). It also shows weak similarity to the receptor binding protein from Lactococcus phage TP901-1 and to the putative tail-host specificity protein in Lactococcus phage P335 (E value, 1.4). Receptor binding proteins or tail-host specificity proteins are usually located on the baseplate. Based on Pfam database analysis, gpORF44 showed weak similarity to the baseplate structural protein domain.
Host cell lysis module.
The cell lysis module consists of ORF24, ORF35, and ORF36, encoding a putative endolysin and two holins, respectively (Table 2). In double-stranded DNA phages, the holin-lysin dual-lysis system is responsible for bacterial lysis and phage progeny release (17). Unexpectedly, this module is embedded in the tail morphogenesis module.
The predicted protein from ORF35 shows a strong sequence similarity to the putative N-acetylmuramoyl-l-alanine amidase of Enterococcus faecalis phage phiEF24C (Table 2). The protein appears to be a phage endolysin belonging to the amidase family. The amidase family includes zinc amidases that have N-acetylmuramoyl-l-alanine amidase activity. This enzyme domain cleaves the amide bond between N-acetylmuramoyl and l-amino acids (preferentially d-lactyl-l-Ala) in bacterial cell walls. The structure is known for phage T7 and indicates that two of the conserved histidines are zinc binding (13). Surprisingly, the putative endolysin gene in Φ1-A4 was flanked by two predicted holin genes (ORF24 upstream and ORF36 downstream, Fig. 2).
The protein specified by ORF36 displays similarity to a hypothetical protein from Leuconostoc citreum KM20. The protein contains a consensus sequence (through hidden Markov model sequence analysis) which represents a putative phage holin from a number of phage and prophage regions of gram-positive bacteria. Like other holins, gpORF36 is small (94 amino acids) with stretches of hydrophobic sequence and is encoded adjacent to the endolysin gene. The derived protein from ORF24 shows homology with the product of the holin gene which was found in the genomes of Granulicatella adiacens ATCC 49175 (Table 2) and Streptococcus pyogenes MGAS10394. However, unlike other holin genes, ORF24 is not adjacent to an endolysin gene. This has not been observed in other phage genomes. It is noteworthy that gpORF36 was not found to contain transmembrane domains, while gpORF24 had three transmembrane domains, similar to other holins (76). Further in vitro experiments are needed to elucidate whether ORF24 or ORF36 (or both) encodes a functional holin.
DNA regulation and modification module.
The regulation and modification module contains ORF45, ORF47, and ORF50 (greenish color in Fig. 2). ORF45 encodes a protein that shows weak similarity to the transcriptional repressor, CopG, found in Nocardioides sp. JS614 (Table 2). The putative function of ORF45 as a regulator of transcription was supported by COG and Pfam database matches (data not shown). CopG (also known as RepA) is responsible for the regulation of plasmid copy number in Nocardioides. It binds to the repAB promoter and controls the synthesis of the plasmid replication initiator protein RepB. Many bacterial proteins that regulate transcription bind DNA through a helix-turn-helix (HTH) motif. CopG displays a fully defined HTH motif structure; however, it is not involved in DNA binding but is apparently responsible for the maintenance of the intrinsic dimeric functional structure and cooperativity (1, 25).
The product of ORF47 shows similarity to the HNH endonuclease of prophage lambda Ba04 in Lactobacillus casei BL23 (Table 2) and prophage L54a in Staphylococcus aureus subsp. aureus COL (24). The HNH motif was originally identified in the subfamily of HNH homing endonucleases, which initiate the process of the insertion of mobile genetic elements into specific sites (14). HNH endonucleases cleave phosphodiester bonds and are found in bacteria and viruses (16, 26, 66). This family includes pyocins, colicins, and anaredoxins.
Based on the Pfam database analysis, gpORF44 showed similarity (E value, 7.3e-04) to the endodeoxyribonuclease RusA. RusA functions as a Holliday junction resolvase (key enzyme in DNA recombination). The RusA protein of E. coli can resolve Holliday intermediates and correct the defects in genetic recombination and DNA repair associated with inactivation of RuvAB or RuvC (51).
Functional analysis.
A functional distribution analysis of multiple genome sequences was performed using compACTor. The functional genome tree (Fig. 7) was constructed based on the ORFs from 112 phages. The hosts for the 112 phages include lactobacilli, lactococci, streptococci, enterococci, listeriae, bacilli, and clostridia. Φ1-A4 clusters with several Lactococcus phages (functional cluster I [FC-I], 49.9 distance units [du]) and was part of another node in a separate group containing predominantly Bacillus phages (FC-II, 56.07 du, Fig. 7). Within FC-I, four separate groups could be observed (17.6, 18.6, and 49.9 du apart) and individual groups appeared to be tightly clustered.
FIG. 7.

Functional genome distribution of 112 bacteriophages. A total of 112 bacteriophage genomes were analyzed using compACTor. A distance matrix was calculated and subsequently imported into Mega4. The functional history was inferred using the UPGMA. The optimal tree with a branch length sum of 2,115.25304686 is shown. The tree is drawn to scale.
As previously described in the literature, L. lactis 936-type phages show extensive genetic similarities, and it has been speculated that these phages have evolved from the same ancestral core genome (30, 52). The slight differences (0.47 du) within this subgroup supported the notion of a group of highly related phages. Based on the compACTor analysis, levels of amino acid sequence similarities within FC-I were visualized in ACT (10) (Fig. 5). As expected, 936-type phages revealed a high level of sequence similarity and gene synteny in their late-gene modules (packaging, morphogenesis, and lysis); however, a greater level of diversity was observed for early and middle genes (replication).
A second group within FC-I (0.42 du, Fig. 7) comprised prolate-head c2-like lactococcal phages (49). Both phages c2 (33) and bIL67 (65) showed extensive genome similarity (Fig. 5). Interestingly, the c2-like group exhibited similarity in their structural module to L. lactis phage Q54 (23). In contrast, Q54 revealed a weaker similarity and gene synteny to 936-type phages (data not shown). The apparent differences in the ORFs of Q54 from 936- and c2-like phages were also illustrated by a functional distance of 18.6 du. With a distance of 49.9 du, Leuconostoc phage Φ1-A4 represented a distinct member of FC-I. Only few ORFs showed amino acid sequence similarities to the other three subgroups (Fig. 5). Based on the similar functional distances between FC-I, FC-II, and Φ1-A4 of 56.07 and 49.9 du, respectively, it is tempting to speculate that Φ1-A4 might represent a separate functional cluster (FC-III) within this larger node.
Interestingly, Φ1-A4 did not cluster with the two prophages harbored by L. mesenteroides ATCC 8293 (53) and L. citreum KM20 (39), respectively (Fig. 7). Instead, Φ1-A4 clustered with several Lactococcus phages. The two Leuconostoc prophages show significant sequence similarity and gene synteny within their structural modules. They formed a distinct functional cluster (Fig. 7), and they were grouped together with several Lactobacillus phages. In contrast, Φ1-A4 only displayed similarity to the predicted large terminase subunits of the two other phages (Fig. 5).
Overall, extensive genetic shuffling is apparent among the phages from different hosts, supporting the widely held view that tailed phages are genetic mosaics arising by the exchange of functional modules within a diverse genetic pool. The weak amino acid similarities among these phages are indicated by the long branches (∼200 du).
Conclusion.
To our knowledge, this study represents the first complete genome sequence and genetic characterization of an L. mesenteroides phage. Bioinformatic analysis revealed that Φ1-A4 is a unique lytic phage compared to phages infecting related species of LAB. Approximately 30% of the Φ1-A4 genome is inverted compared to the genome organization of other sequenced phages. The endolysin gene was flanked by two holin genes. The tail morphogenesis module was interspersed with cell lysis genes. The overall amino acid sequences of the phage proteins had little similarity to other sequenced phages. Functional analyses showed that Φ1-A4 clusters with several Lactococcus phages. The results of this study may provide new insights that deepen our understanding of phage genetics and phage-host interactions in dynamic ecosystem such as vegetable fermentations.
Acknowledgments
This study was partially supported by the Kennesaw State University Incentive Fund and by Pickle Packers International Inc., Washington, DC.
We thank Roger McFeeters for helpful discussions and Sandra Parker for excellent secretarial assistance.
Footnotes
Published ahead of print on 29 January 2010.
REFERENCES
- 1.Acebo, P., M. Garcia de Lacoba, G. Rivas, J. M. Andreu, M. Espinosa, and G. del Solar. 1998. Structural features of the plasmid pMV158-encoded transcriptional repressor CopG, a protein sharing similarities with both helix-turn-helix and beta-sheet DNA binding proteins. Proteins 32:248-261. [DOI] [PubMed] [Google Scholar]
- 2.Altermann, E., and T. R. Klaenhammer. 2003. GAMOLA: a new local solution for sequence annotation and analyzing draft and finished prokaryotic genomes. OMICS 7:161-169. [DOI] [PubMed] [Google Scholar]
- 3.Altermann, E., J. R. Klein, and B. Henrich. 1999. Primary structure and features of the genome of the Lactobacillus gasseri temperate bacteriophage (phi)adh. Gene 236:333-346. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou, R., S.-S. Yoon, F. Breidt, H. P. Fleming, and T. R. Klaenhammer. 2002. Characterization of six Leuconostoc fallax bacteriophages isolated from an industrial sauerkraut fermentation. Appl. Environ. Microbiol. 68:5452-5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bateman, A., E. Birney, L. Cerruti, R. Durbin, L. Etwiller, S. R. Eddy, S. Griffiths-Jones, K. L. Howe, M. Marshall, and E. L. Sonnhammer. 2002. The Pfam protein families database. Nucleic Acids Res. 30:276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783-795. [DOI] [PubMed] [Google Scholar]
- 7.Breitbart, M., P. Salamon, B. Andresen, J. M. Mahaffy, A. M. Segall, D. Mead, F. Azam, and F. Rohwer. 2002. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U. S. A. 99:14250-14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brøndsted, L., S. Ostergaard, M. Pedersen, K. Hammer, and F. K. Vogensen. 2001. Analysis of the complete DNA sequence of the temperate bacteriophage TP901-1: evolution, structure, and genome organization of lactococcal bacteriophages. Virology 283:93-109. [DOI] [PubMed] [Google Scholar]
- 9.Brüssow, H., and R. W. Hendrix. 2002. Phage genomics: small is beautiful. Cell 108:13-16. [DOI] [PubMed] [Google Scholar]
- 10.Carver, T. J., K. M. Rutherford, M. Berriman, M. A. Rajandream, B. G. Barrell, and J. Parkhill. 2005. ACT: the Artemis comparison tool. Bioinformatics 21:3422-3423. [DOI] [PubMed] [Google Scholar]
- 11.Catalano, C. E. 2000. The terminase enzyme from bacteriophage lambda: a DNA-packaging machine. Cell. Mol. Life Sci. 57:128-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng, X. 1995. Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct. 24:293-318. [DOI] [PubMed] [Google Scholar]
- 13.Cheng, X., X. Zhang, J. W. Pflugrath, and F. W. Studier. 1994. The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 91:4034-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevalier, B. S., and B. L. Stoddard. 2001. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 29:3757-3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claesson, M. J., Y. Li, S. Leahy, C. Canchaya, J. P. van Pijkeren, A. M. Cerdeño-Tárraga, J. Parkhill, S. Flynn, G. C. O'Sullivan, J. K. Collins, D. Higgins, F. Shanahan, G. F. Fitzgerald, D. van Sinderen, and P. W. O'Toole. 2006. Multireplicon genome architecture of Lactobacillus salivarius. Proc. Natl. Acad. Sci. U. S. A. 103:6718-6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalgaard, J. Z., A. J. Klar, M. J. Moser, W. R. Holley, A. Chatterjee, and I. S. Mian. 1997. Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res. 25:4626-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniel, A., P. E. Bonnen, and V. A. Fischetti. 2007. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 189:2086-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danovaro, R., A. Dell'Anno, A. Trucco, M. Serresi, and S. Vanucci. 2001. Determination of virus abundance in marine sediments. Appl. Environ. Microbiol. 67:1384-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duffy, C., and M. Feiss. 2002. The large subunit of bacteriophage lambda's terminase plays a role in DNA translocation and packaging termination. J. Mol. Biol. 316:547-561. [DOI] [PubMed] [Google Scholar]
- 20.Ermolaeva, M. D., H. G. Khalak, O. White, H. O. Smith, and S. L. Salzberg. 2000. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 301:27-33. [DOI] [PubMed] [Google Scholar]
- 21.Ewing, B., L. Hillier, M. C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8:175-185. [DOI] [PubMed] [Google Scholar]
- 22.Fleming, H. P., R. F. McFeeters, and E. G. Humphries. 1988. A fermentor for study of sauerkraut fermentation. Biotechnol. Bioeng. 31:189-197. [DOI] [PubMed] [Google Scholar]
- 23.Fortier, L.-C., A. Bransi, and S. Moineau. 2006. Genome sequence and global gene expression of Q54, a new phage species linking the 936 and c2 phage species of Lactococcus lactis. J. Bacteriol. 188:6101-6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gill, S. R., D. E. Fouts, G. L. Archer, E. F. Mongodin, R. T. Deboy, J. Ravel, I. T. Paulsen, J. F. Kolonay, L. Brinkac, M. Beanan, R. J. Dodson, S. C. Daugherty, R. Madupu, S. V. Angiuoli, A. S. Durkin, D. H. Haft, J. Vamathevan, H. Khouri, T. Utterback, C. Lee, G. Dimitrov, L. Jiang, H. Qin, J. Weidman, K. Tran, K. Kang, I. R. Hance, K. E. Nelson, and C. M. Fraser. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomis-Rüth, F. X., M. Solà, P. Acebo, A. Parraga, A. Guasch, R. Eritja, A. Gonzalez, M. Espinosa, G. del Solar, and M. Coll. 1998. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 17:7404-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorbalenya, A. E. 1994. Self-splicing group I and group II introns encode homologous (putative) DNA endonucleases of a new family. Protein Sci. 3:1117-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haft, D. H., J. D. Selengut, and O. White. 2003. The TIGRFAMs database of protein families. Nucleic Acids Res. 31:371-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heath, H. E., L. S. Heath, J. D. Nitterauer, K. E. Rose, and G. L. Sloan. 1989. Plasmid-encoded lysostaphin endopeptidase resistance of Staphylococcus simulans biovar staphylolyticus. Biochem. Biophys. Res. Commun. 160:1106-1109. [DOI] [PubMed] [Google Scholar]
- 29.Heilmann, C., M. Hussain, G. Peters, and F. Götz. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 24:1013-1024. [DOI] [PubMed] [Google Scholar]
- 30.Hejnowicz, M. S., M. Golebiewski, and J. Bardowski. 2009. Analysis of the complete genome sequence of the lactococcal bacteriophage bIBB29. Int. J. Food Microbiol. 131:52-61. [DOI] [PubMed] [Google Scholar]
- 31.Helgstrand, C., W. R. Wikoff, R. L. Duda, R. W. Hendrix, J. E. Johnson, and L. Liljas. 2003. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 Å resolution. J. Mol. Biol. 334:885-899. [DOI] [PubMed] [Google Scholar]
- 32.Hendrix, R. W., and R. Polly. 2005. Bacteriophage HK97: assembly of the capsid and evolutionary connections. Adv. Virus Res. 64:1-14. [DOI] [PubMed] [Google Scholar]
- 33.Jarvis, A. W., M. W. Lubbers, T. P. Beresford, L. J. Ward, N. R. Waterfield, L. J. Collins, and B. D. Jarvis. 1995. Molecular biology of lactococcal bacteriophage c2. Dev. Biol. Stand. 85:561-567. [PubMed] [Google Scholar]
- 34.Jensen, E. C., H. S. Schrader, B. Rieland, T. L. Thompson, K. W. Lee, K. W. Nickerson, and T. A. Kokjohn. 1998. Prevalence of broad-host-range lytic bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl. Environ. Microbiol. 64:575-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juhala, R. J., M. E. Ford, R. L. Duda, A. Youlton, G. F. Hatfull, and R. W. Hendrix. 2000. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 299:27-51. [DOI] [PubMed] [Google Scholar]
- 36.Katsura, I. 1987. Determination of bacteriophage lambda tail length by a protein ruler. Nature 327:73-75. [DOI] [PubMed] [Google Scholar]
- 37.Katsura, I., and R. W. Hendrix. 1984. Length determination in bacteriophage lambda tails. Cell 39:691-698. [DOI] [PubMed] [Google Scholar]
- 38.Keppel, F., O. Fayet, and C. Georgopoulos. 1988. Strategies of bacteriophage DNA replication, p. 145-262. In R. Calendar (ed.), The bacteriophages. Plenum Press, New York, NY.
- 39.Kim, J. F., H. Jeong, J.-S. Lee, S.-H. Choi, M. Ha, C.-G. Hur, J.-S. Kim, S. Lee, H.-S. Park, Y.-H. Park, and T. K. Oh. 2008. Complete genome sequence of Leuconostoc citreum KM20. J. Bacteriol. 190:3093-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondou, Y., D. Kitazawa, S. Takeda, Y. Tsuchiya, E. Yamashita, M. Mizuguchi, K. Kawano, and T. Tsukihara. 2005. Structure of the central hub of bacteriophage Mu baseplate determined by X-ray crystallography of gp44. J. Mol. Biol. 352:976-985. [DOI] [PubMed] [Google Scholar]
- 41.Krogh, A., B. Larsson, G. von Heijne, and E. L. L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567-580. [DOI] [PubMed] [Google Scholar]
- 42.Lee, J. H., V. N. Karamychev, S. A. Kozyavkin, D. Mills, A. R. Pavlov, N. V. Pavlova, N. N. Polouchine, P. M. Richardson, V. V. Shakhova, A. I. Slesarev, B. Weimer, and D. J. O'Sullivan. 2008. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genomics 9:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leiman, P. G., S. Kanamaru, V. V. Mesyanzhinov, F. Arisaka, and M. G. Rossmann. 2003. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 60:2356-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipps, G., A. O. Weinzierl, G. von Scheven, C. Buchen, and P. Cramer. 2004. Structure of a bifunctional DNA primase-polymerase. Nat. Struct. Mol. Biol. 11:157-162. [DOI] [PubMed] [Google Scholar]
- 45.Lowther, W. T., and B. W. Matthews. 2000. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta 1477:157-167. [DOI] [PubMed] [Google Scholar]
- 46.Lu, Z., E. Altermann, F. Breidt, P. Predki, H. P. Fleming, and T. R. Klaenhammer. 2005. Sequence analysis of the Lactobacillus plantarum bacteriophage ΦJL-1. Gene 348:45-54. [DOI] [PubMed] [Google Scholar]
- 47.Lu, Z., F. Breidt, H. P. Fleming, E. Altermann, and T. R. Klaenhammer. 2003. Isolation and Characterization of a Lactobacillus plantarum bacteriophage ΦJL-1 from a cucumber fermentation. Int. J. Food Microbiol. 84:225-235. [DOI] [PubMed] [Google Scholar]
- 48.Lu, Z., F. Breidt, V. Plengvidhya, and H. P. Fleming. 2003. Bacteriophage ecology in commercial sauerkraut fermentations. Appl. Environ. Microbiol. 69:3192-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lubbers, M. W., N. R. Waterfield, T. P. Beresford, R. W. Le Page, and A. W. Jarvis. 1995. Sequencing and analysis of the prolate-headed lactococcal bacteriophage c2 genome and identification of the structural genes. Appl. Environ. Microbiol. 61:4348-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahanivong, C., J. D. Boyce, B. E. Davidson, and A. J. Hillier. 2001. Sequence analysis and molecular characterization of the Lactococcus lactis temperate bacteriophage BK5-T. Appl. Environ. Microbiol. 67:3564-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahdi, A., G. Sharples, T. Mandal, and R. Lloyd. 1996. Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J. Mol. Biol. 257:561-573. [DOI] [PubMed] [Google Scholar]
- 52.Mahony, J., H. Deveau, S. McGrath, M. Ventura, C. Canchaya, S. Moineau, G. F. Fitzgerald, and D. van Sinderen. 2006. Sequence and comparative genomic analysis of lactococcal bacteriophages jj50, 712 and P008: evolutionary insights into the 936 phage species. FEMS Microbiol. Lett. 261:253-261. [DOI] [PubMed] [Google Scholar]
- 53.Makarova, K., A. Slesarev, Y. Wolf, A. Sorokin, B. Mirkin, E. Koonin, A. Pavlov, N. Pavlova, V. Karamychev, N. Polouchine, V. Shakhova, I. Grigoriev, Y. Lou, D. Rohksar, S. Lucas, K. Huang, D. M. Goodstein, T. Hawkins, V. Plengvidhya, D. Welker, J. Hughes, Y. Goh, A. Benson, K. Baldwin, J. H. Lee, I. Díaz-Muñiz, B. Dosti, V. Smeianov, W. Wechter, R. Barabote, G. Lorca, E. Altermann, R. Barrangou, B. Ganesan, Y. Xie, H. Rawsthorne, D. Tamir, C. Parker, F. Breidt, J. Broadbent, R. Hutkins, D. O'Sullivan, J. Steele, G. Unlu, M. Saier, T. Klaenhammer, P. Richardson, S. Kozyavkin, B. Weimer, and D. Mills. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:15611-15616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonald, L. C., H. P. Fleming, and H. M. Hassan. 1990. Acid tolerance of Leuconostoc mesenteroides and Lactobacillus plantarum. Appl. Environ. Microbiol. 56:2120-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minakhin, L., M. Goel, Z. Berdygulova, E. Ramanculov, L. Florens, G. Glazko, V. N. Karamychev, A. I. Slesarev, S. A. Kozyavkin, I. Khromov, H. W. Ackermann, M. Washburn, A. Mushegian, and K. Severinov. 2008. Genome comparison and proteomic characterization of Thermus thermophilus bacteriophages P23-45 and P74-26: siphoviruses with triplex-forming sequences and the longest known tails. J. Mol. Biol. 378:468-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mudgal, P., F. Breidt, S. R. Lubkin, and K. P. Sandeep. 2006. Quantifying the significance of phage attack on starter cultures: a mechanistic model for population dynamics of phage and their hosts isolated from fermenting sauerkraut. Appl. Environ. Microbiol. 72:3908-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muller, P. Y., E. Studer, and A. R. Miserez. 2001. Molecular BioComputing Suite: a word processor add-in for the analysis and manipulation of nucleic acid and protein sequence data. BioTechniques 31:1306-1313. [DOI] [PubMed] [Google Scholar]
- 58.Mundt, J. O. 1970. Lactic acid bacteria associated with raw plant food material. J. Milk Food Technol. 33:550-553. [Google Scholar]
- 59.Mundt, J. O., W. F. Graham, and I. E. McCarty. 1967. Spherical lactic acid-producing bacteria of southern-grown raw and processed vegetables. Appl. Microbiol. 15:1303-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mundt, J. O., and J. L. Hammer. 1968. Lactobacilli on plants. Appl. Microbiol. 16:1326-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60a.Pedersen, A. G., L. J. Jensen, S. Brunak, H. H. Staerfeldt, and D. W. Ussery. 2000. A DNA structural atlas for Escherichia coli. J. Mol. Biol. 299:907-930. [DOI] [PubMed] [Google Scholar]
- 61.Pederson, C. S., and M. N. Albury. 1969. Technical bulletin 824: the sauerkraut fermentation. New York State Agricultural Experiment Station, Geneva.
- 62.Pedulla, M. L., M. E. Ford, J. M. Houtz, T. Karthikeyan, C. Wadsworth, J. A. Lewis, D. Jacobs-Sera, J. Falbo, J. Gross, N. R. Pannunzio, W. Brucker, V. Kumar, J. Kandasamy, L. Keenan, S. Bardarov, J. Kriakov, J. G. Lawrence, W. R. Jacobs, Jr., R. W. Hendrix, and G. F. Hatfull. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171-182. [DOI] [PubMed] [Google Scholar]
- 63.Polushin, N., A. Malykh, A. M. Morocho, A. Slesarev, and S. Kozyavkin. 2005. High-throughput production of optimized primers (fimers) for whole-genome direct sequencing. Methods Mol. Biol. 288:291-304. [DOI] [PubMed] [Google Scholar]
- 64.Pouwels, P. H., and R. J. Leer. 1993. Genetics of lactobacilli: plasmids and gene expression. Antonie Van Leeuwenhoek 64:85-107. [DOI] [PubMed] [Google Scholar]
- 65.Schouler, C., S. D. Ehrlich, and M. C. Chopin. 1994. Sequence and organization of the lactococcal prolate-headed bIL67 phage genome. Microbiology 140:3061-3069. [DOI] [PubMed] [Google Scholar]
- 66.Shub, D. A., H. Goodrich-Blair, and S. R. Eddy. 1994. Amino acid sequence motif of group I intron endonucleases is conserved in open reading frames of group II introns. Trends Biochem. Sci. 19:402-404. [DOI] [PubMed] [Google Scholar]
- 67.Slesarev, A. I., K. V. Mezhevaya, K. S. Makarova, N. N. Polushin, O. V. Shcherbinina, V. V. Shakhova, G. I. Belova, L. Aravind, D. A. Natale, I. B. Rogozin, R. L. Tatusov, Y. I. Wolf, K. O. Stetter, A. G. Malykh, E. V. Koonin, and S. A. Kozyavkin. 2002. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. U. S. A. 99:4644-4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sneath, P. H. A., and R. R. Sokal. 1973. Numerical taxonomy. W. H. Freeman, San Francisco, CA.
- 69.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 70.Tatusov, R., N. Fedorova, J. Jackson, A. Jacobs, B. Kiryutin, E. Koonin, D. Krylov, R. Mazumder, S. Mekhedov, A. Nikolskaya, B. S. Rao, S. Smirnov, A. Sverdlov, S. Vasudevan, Y. Wolf, J. Yin, and D. Natale. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tettelin, H., V. Masignani, M. J. Cieslewicz, J. A. Eisen, S. Peterson, M. R. Wessels, I. T. Paulsen, K. E. Nelson, I. Margarit, T. D. Read, L. C. Madoff, A. M. Wolf, M. J. Beanan, L. M. Brinkac, S. C. Daugherty, R. T. DeBoy, A. S. Durkin, J. F. Kolonay, R. Madupu, M. R. Lewis, D. Radune, N. B. Fedorova, D. Scanlan, H. Khouri, S. Mulligan, H. A. Carty, R. T. Cline, S. E. Van Aken, J. Gill, M. Scarselli, M. Mora, E. T. Iacobini, C. Brettoni, G. Galli, M. Mariani, F. Vegni, D. Maione, D. Rinaudo, R. Rappuoli, J. L. Telford, D. L. Kasper, G. Grandi, and C. M. Fraser. 2002. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc. Natl. Acad. Sci. U. S. A. 99:12391-12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van de Guchte, M., S. Penaud, C. Grimaldi, V. Barbe, K. Bryson, P. Nicolas, C. Robert, S. Oztas, S. Mangenot, A. Couloux, V. Loux, R. Dervyn, R. Bossy, A. Bolotin, J. M. Batto, T. Walunas, J. F. Gibrat, P. Bessières, J. Weissenbach, S. D. Ehrlich, and E. Maguin. 2006. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc. Natl. Acad. Sci. U. S. A. 103:9274-9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Sinderen, D., H. Karsens, J. Kok, P. Terpstra, M. H. Ruiters, G. Venema, and A. Nauta. 1996. Sequence analysis and molecular characterization of the temperate lactococcal bacteriophage r1t. Mol. Microbiol. 19:1343-1355. [DOI] [PubMed] [Google Scholar]
- 74.Yoon, S.-S., R. Barrangou-Poueys, F. Breidt, and H. P. Fleming. 2007. Detection and characterization of a lytic Pediococcus bacteriophage from the fermenting cucumber brine. J. Microbiol. Biotechnol. 17:262-270. [PubMed] [Google Scholar]
- 75.Yoon, S.-S., R. Barrangou-Poueys, F. Breidt, T. R. Klaenhammer, and H. P. Fleming. 2002. Isolation and characterization of bacteriophages from fermenting sauerkraut. Appl. Environ. Microbiol. 68:973-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Young, R., I.-N. Wang, and W. D. Roof. 2000. Phages will out: strategies of host cell lysis. Trends Microbiol. 8:120-128. [DOI] [PubMed] [Google Scholar]