

Abstract
Female mice lacking the transcription factor C/EBPβ are infertile and display markedly reduced estrogen (E)-induced proliferation of the uterine epithelial lining during the reproductive cycle. The present study showed that E-stimulated luminal epithelial cells of a C/EBPβ-null uterus are able to proceed through the G1 phase of the cell cycle before getting arrested in the S phase. This cell cycle arrest was accompanied by markedly reduced levels of expression of E2F3, an E2F family member, and a lack of nuclear localization of cyclin E, a critical regulator of cdk2. An increased nuclear accumulation of p27, an inhibitor of the cyclin E-cdk2 complex, was also observed for the mutant epithelium. Gene expression profiling of C/EBPβ-null uterine epithelial cells revealed that the blockade of E-induced DNA replication triggers the activation of several well-known components of the DNA damage response pathway, such as ATM, ATR, histone H2AX, checkpoint kinase 1, and tumor suppressor p53. The activation of p53 by ATM/ATR kinase led to increased levels of expression of p21, an inhibitor of G1-S-phase progression, which helps maintain cell cycle arrest. Additionally, p53-dependent mechanisms contributed to an increased apoptosis of replication-defective cells in the C/EBPβ-null epithelium. C/EBPβ, therefore, is an essential mediator of E-induced growth and survival of uterine epithelial cells of cycling mice.
The ovarian steroid hormones estrogen (E) and progesterone (P) play critical roles in the maintenance of the mammalian uterus through cyclical rounds of cell proliferation and differentiation during the reproductive cycle (50). In rodents, ovarian E produced during the proestrus stage stimulates uterine luminal and glandular epithelial cell proliferation, preparing the uterus for potential pregnancy. During this growth phase, E causes distinct physiological changes such as an increased uterine wet weight and structural remodeling of the luminal epithelium (LE) cell layer while also accelerating their entry into the S phase of the cell cycle (17, 18, 29, 50). At the onset of pregnancy, increasing levels of P produced from the newly formed corpora lutea in the ovaries suppresses the E-stimulated proliferation of uterine LE cells. The actions of E and P in these epithelial cells are mediated via their respective nuclear receptors, estrogen receptor alpha (ERα) and progesterone receptor (PR) (23, 26). Acting in concert, these hormones control early events that are essential for providing a suitable environment for blastocyst attachment to LE cells and successful implantation. In the adult female mouse, the administration of exogenous E and P to ovariectomized mice faithfully reproduces the uterine epithelial responses seen during the estrous cycle and early pregnancy. Therefore, mouse uterine LE cells offer an excellent model with which one may explore the molecular mechanisms by which steroid hormones control cell proliferation.
Several previous studies documented the mitogenic effects of E on rodent uterine LE cells and examined the mechanisms underlying this steroid-stimulated proliferation (29, 50). The administration of E to ovariectomized mice led to the transcriptional induction of proto-oncogenes such as c-fos, c-myc, and n-myc, epidermal growth factor (EGF) and its receptor EGFR, transforming growth factor α (TGF-α), and insulin-like growth factor 1 (IGF-1) concomitant with uterine epithelial cell proliferation (29, 33, 37, 50). Based on these findings, it was proposed that proto-oncogene and growth factor pathways mediate the E-induced growth response in the uterus (5, 9, 53, 55). However, later studies using knockout mouse models failed to provide unequivocal evidence in support of a role of certain of these factors in the female reproductive tract (24, 25). Interestingly, the administration of P to ovariectomized mice, which downregulated E-induced uterine LE cell proliferation, did not alter the expression profiles of many of these growth-promoting factors, indicating that these factors may not be participating directly in E-induced cell cycle entry. This observation raised the possibility that additional factors are involved in transmitting the E-induced growth response to the cell cycle machinery (5, 50, 55).
In recent studies, we identified and characterized the transcription factor CCAAT enhancer binding protein beta (C/EBPβ) as a steroid-hormone-regulated gene that is critical for uterine functions (28). C/EBPβ is a member of the bZIP family of leucine zipper proteins and has been implicated as a regulator of proliferation and differentiation in diverse tissues (11, 30, 41, 54, 56). The importance of C/EBPβ in female fertility was revealed when C/EBPβ-null females were found to have severely compromised ovarian and uterine functions. In addition to a failure in ovarian follicle rupture (48), the C/EBPβ-null uterus is refractory to embryo attachment to the uterine LE, as demonstrated by embryo transfer experiments performed with ovariectomized mice supplemented with E and P (28). The expression of C/EBPβ is induced rapidly in the uterine LE during the early phases of the E-induced proliferation of this tissue (28). Its importance in uterine growth was highlighted by the markedly diminished level of expression of the Ki67 antigen, a widely used marker of cell proliferation, in E-treated uterine LE cells of C/EBPβ-null mice (28).
In this study, we noted that simultaneous treatment with P, which reduces E-induced uterine LE cell proliferation, also decreased the level of expression of C/EBPβ, thereby positioning this transcription factor as a biologically relevant mediator of E action in this tissue. We postulated that the E-induced expression of C/EBPβ and its downstream targets controls the production and/or activity of one or more major cell cycle regulatory molecules in uterine LE cells. Using stage-specific cell cycle markers, we determined that the loss of C/EBPβ leads to a significant impairment in DNA replication in uterine LE cells. We also identified alterations in the expression and cellular localization of cyclin E, E2F3, and p27, key molecules that modulate the G1-S-phase transition of the cell cycle. We further showed that the proliferation defect in C/EBPβ-null uterine LE cells is associated with the activation of well-known DNA damage response pathways involving ataxia telengiectasia mutated (ATM), ATM-Rad3 related (ATR), the checkpoint kinases Chk1 and Chk2, and p53. Activated p53 helps to maintain the G1-S-phase blockade of the cell cycle by inducing the synthesis of the inhibitor p21. p53 also promotes the apoptosis of replication-defective uterine LE cells. Collectively, these studies identified the molecular pathways by which C/EBPβ mediates E-induced proliferation and the survival of uterine LE cells during the reproductive cycle and early pregnancy.
MATERIALS AND METHODS
Animals, hormone treatments, and tissue collection.
All experiments involving animals were conducted in accordance with National Institutes of Health standards for the use and care of animals. The animal protocols were approved by the University of Illinois Institutional Animal Care and Use Committee. Heterozygous mice carrying a mutation in the C/EBPβ gene were provided by Peter F. Johnson of the National Cancer Institute. Female mice carrying a null mutation in the C/EBPβ gene were derived from crosses of heterozygous females with nullizygous male mice as described previously (48). Female wild-type (WT) and C/EBPβ-null mice of the 129Sv background were ovariectomized at 10 to 11 weeks of age and rested for 2 weeks. These mice were then treated with a single dose of 17β-estradiol (E) (250 ng in sesame oil) by intraperitoneal (i.p.) injection for various durations. In experiments where progesterone (P) was used, mice were injected with a single dose of P (1 mg in sesame oil) along with E (250 ng). For each treatment group, at least five mice were used at each time point. In some experiments, animals were injected i.p. with bromodeoxyuridine (BrdU) (2 mg/animal; BD Pharmingen) 1 h prior to sacrifice. Uteri were collected and fixed in 10% formalin prior to immunohistochemistry (IHC) analysis. Alternatively, uteri were pooled for the isolation of primary epithelial cells.
Isolation of mouse primary uterine epithelial cells.
Hormone-treated uterine horns were excised, trimmed of fat, and dissected longitudinally to expose uterine lumen. Dissected horns were then cut into 4- to 5-mm-long pieces and washed in Hanks balanced salt solution (HBSS; Gibco). Uterine tissue pieces were then placed into HBSS containing 6 g/liter dispase (Invitrogen) and 25 g/liter pancreatin (Sigma) for 1 h at room temperature (RT), followed by 10 min at 37°C. The tubes were gently agitated to release luminal epithelial cells from the rest of the uterine tissue. The cell suspension was filtered through a 100-μm-pore-size sieve (BD) to remove any tissue debris. The filtrates containing luminal epithelial cells were centrifuged at 2,000 rpm for 5 min to pellet the cells. The cells were washed twice in phosphate-buffered saline (PBS) and repelleted. The pellets were lysed with Trizol for RNA extraction or with radioimmunoprecipitation assay (RIPA) lysis buffer for the preparation of whole-cell protein lysates.
Real-time PCR analysis.
Total RNA was isolated from uterine cells by standard Trizol-based protocols and converted to cDNA as described previously (20). The cDNA was amplified by real-time PCR to quantify gene expression using gene-specific primers and Sybr green (Bio-Rad Laboratories). As a loading control, the expression level of the 36B4 gene, which encodes a ribosomal protein, was determined. For each treatment, the mean threshold cycle (CT) and standard deviation were calculated from CT values obtained individually from 3 to 4 replicates of that sample. Each sample was subjected to three independent real-time PCR trials. The fold change was derived from the mean CT values as described previously (20). Analysis of variance (ANOVA) single-factor analysis was conducted on the grouped means to determine statistical significance at a significance level of a P value of <0.05.
DNA microarray analysis.
WT and C/EBPβ-null mice were ovariectomized and treated with E, and after 18 h, the LE cell layer was isolated and total RNA was prepared. The RNA was hybridized to Affymetrix mouse arrays (GeneChipMouse Genome 430 2.0 array) containing probes that represented ∼14,000 known genes. They were processed and analyzed according to the Affymetrix protocol as described previously (20).
IHC.
Formalin-fixed uterine pieces were processed for paraffin embedding. Cross sections (5-μm thickness) were mounted onto microscope slides (Fisher Scientific). For immunostaining, uterine sections were deparaffinized in xylene (three times for 5 min each), rehydrated through a graded series of treatment with ethanol (100%, 95%, 85%, and 70% for 5 min each), and rinsed in tap water. For all samples, antigen retrieval was performed by boiling the sections in 0.01 M sodium citrate buffer (pH 6.0) for 20 min, followed by incubation at RT for 30 min. A 5% solution of normal donkey serum (Jackson Immunoresearch) in PBS was used as a blocking buffer. Sections were incubated with the following primary antibodies diluted in blocking solution (0.25% bovine serum albumin [BSA], 0.3% Triton X-100, sterile PBS) overnight at 4°C: BrdU and Ki67 (BD Pharmingen), phospho-Ser10 histone H3 (Upstate Biotechnology); cyclin E, cyclin A, and Rad18 (Abcam); cyclin D1 (LabVision NeoMarkers); E2F3 (Santa Cruz Biotechnology); p27 (BD Transduction Laboratories); and phospho-(Ser1981)-ATM, phospho-(Ser139)-H2AX, caspase 3, and cleaved caspase 3 (Cell Signaling Technology). The sections were washed and incubated with biotinylated secondary antibodies (Jackson Immunoresearch Laboratories Inc.) for 60 min, followed by incubation with streptavidin-conjugated horseradish peroxidase (Histostain kit; Zymed Laboratories Inc.) for 45 min. Sections were stained with 3-amino-9-ethyl carbazole (AEC) solution (Zymed Laboratories Inc.) and counterstained with Mayer's hematoxylin (Sigma). Immunofluorescence staining was performed by incubating sections with the Cy3-conjugated streptavidin complex (Jackson Immunoresearch Laboratories) following incubation with secondary antibodies. Counterstaining was done by using 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen Inc.). Stained sections were mounted in fluorescence mounting medium [20% glycerol, 8% (wt/vol) polyvinyl alcohol (PVA), diazabicyclo(2,2,2)octane (DABCO), Tris-Cl (pH 8.5), and sterile double-distilled water (ddH2O)]. Negative controls included incubation with donkey serum and omission of the primary antibody for all samples.
TUNEL staining.
To detect apoptotic cells containing fragmented DNA, in situ labeling of free 3′ OH of nicked DNA was carried out by using the In Situ Cell Death Detection kit (Roche Applied Science) according to the manufacturer's protocol. Briefly, deparaffinized and rehydrated sections were incubated at RT for 15 min with proteinase K (20 μg/ml in 10 mM Tris-HCl [pH 8.0]). Sections were washed and incubated for 1 h at 37°C in a terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) reaction mixture containing TdT enzyme, fluorescein-labeled nucleotide mixture, and diluent buffer (30 mM Tris-HCl [pH 8.0]). For negative controls, TdT enzyme was omitted from the reaction mixture. After rinsing in PBS, sections were counterstained with DAPI and mounted in fluorescence mounting medium.
Image capture and quantitation of immunostaining.
The images of immunohistochemical staining were captured by using a Leica (Nussloch, Germany) DM2500 light microscope fitted with a Qimaging Retiga 2000R camera (Qimaging, British Columbia, Canada). For counting, at least 5 to 6 individual 40× fields from each sample were captured. The numbers of positively stained uterine LE nuclei in each field were averaged and expressed as a percentage of the total number of these cells. The standard deviation was determined for each averaged total. ANOVA single-factor analysis was conducted on the grouped means to determine statistical significance at a significance of a P value of <0.05. Fluorescent images for phospho-histone H3, cyclin E, p27, and E2F3 were captured by using the Leica DM2500 microscope. For all other antibodies, fluorescence microscopy and image capturing were performed by using a Leica DMR microscope and a Retiga EXI monochrome charge-coupled-device (CCD) camera (Qimaging). All images were processed by using Adobe Photoshop CS, version 8.0.
Western blotting.
Whole-cell lysates were prepared from primary LE cells isolated from uteri collected from control (untreated) or E-treated WT and C/EBPβ-null mice. The cells were homogenized using RIPA lysis buffer. Protein concentrations of cell lysates were determined by using the Bradford assay from Bio-Rad. Twenty micrograms of each cell lysate was separated by SDS-PAGE (10%) and transferred onto Hybond-P membranes (Amersham Pharmacia Biotech). The membranes were blocked in Tris-buffered saline with 0.1% (vol/vol) Tween 20 (TBS-T) and 5% (wt/vol) nonfat dry milk for 1 h. The membranes were then incubated overnight at 4°C with primary antibodies diluted 1:1,000 in TBS-T containing 5% (wt/vol) BSA. These antibodies were directed against Chk1, p53, phospho-Thr187-p27, and calnexin (Santa Cruz Biotechnology); p21 (BD Pharmingen); phospho-Ser296-Chk1 and phospho-Ser15-p53 (Cell Signaling Technology); and cdk2 and phospho-Thr160-cdk2 (Abcam). Membranes were then incubated for 1 h at RT with secondary antibodies conjugated to horseradish peroxidase and diluted 1:5,000. After thorough washes with TBS-T, chemiluminescence detection was performed by using SuperSignal Femto and Pico reagents from Pierce (Thermo Scientific).
ChIP.
Chromatin immunoprecipitation (ChIP) analysis was conducted by using an EZ-ChIP kit (Upstate Biotechnology) according to the manufacturer's protocol. Briefly, mouse uterine LE cells were isolated from 11-week-old, ovariectomized WT mice of the 129Sv background that were subjected to E treatment (250 ng) for 12 h. The cells (7.5 × 106 cells) were washed in PBS and cross-linked with 1% formaldehyde at room temperature for 10 min. The cross-linked cells were lysed by using SDS lysis buffer and sonicated 5 times for 10 s each pulse. Lysates were precleared with salmon sperm DNA-protein A, and the DNA-protein complexes were subsequently immunoprecipitated by using antibodies against RNA polymerase II, rabbit IgG, or C/EBPβ (Santa Cruz Biotechnology). The immune complexes were recovered with protein A agarose. The beads were then repeatedly washed, and protein complexes were eluted. The cross-linking was reversed, and proteins were digested by using 0.5 mg/ml proteinase K. Purified DNA were used as templates for real-time PCR using various primer sets to amplify specific regions of the E2F3 promoter.
In silico promoter analysis.
Putative C/EBPβ binding sites at the E2F3 promoter were determined by in silico analysis of the proximal promoter region (bp −1 to −1900) using TESS (45), TFsearch (15), and Consite (44) software. The following primers were designed to amplify specific regions containing putative C/EBPβ binding sites: TTCATACCCCTCCCACAAGA and TTTTATTGTCCTTCTAGCCATGA for bp −329 to −340, CCAGTGATTCAGCATACATTACA and TTATTGCCCTCACCACCTTC for bp −458 to −473, ACAGTCTTGGGTGAGCTGGT and CCCCTACACACTCGGTTCCT for bp −584 to −595, and AGCCTCTTTGACTGGGACTG and TCAGTATTTTGCTGGGGTCTC for bp −1054 to −1066.
Statistical analysis.
Statistical significance was assessed by ANOVA at a significance level of a P value of <0.05 and is indicated by asterisks in the figures. NS indicates nonsignificant changes (P > 0.05).
RESULTS
P suppresses E-induced C/EBPβ expression in uterine epithelial cells.
It is well known that P antagonizes the E-induced proliferation of uterine LE cells (40, 50, 51). Our previous studies described that C/EBPβ expression is markedly induced in the uterine LE of ovariectomized mice in response to an acute administration of E (28). We report here that the simultaneous administration of P inhibited acute E-induced uterine LE cell proliferation, as measured by Ki67 immunostaining (Fig. 1A). We also determined the levels of C/EBPβ mRNA and protein in uterine LE cells of ovariectomized mice after treatment with E or E plus P for 2 and 11 h. We noted that treatment with E alone leads to a robust increase in C/EBPβ mRNA and protein levels within 2 h, followed by a decline in its expression to basal levels by 11 h (Fig. 1B and C). When mice were treated with a combination of E and P, the E-induced increase in C/EBPβ mRNA levels at 2 h was strongly suppressed. P therefore counteracts the transcriptional induction of C/EBPβ by E. These results are consistent with our hypothesis that C/EBPβ is a critical downstream mediator of E-stimulated uterine epithelial cell proliferation and that P exerts its antiproliferative effects, at least in part, by suppressing C/EBPβ expression in these cells.
FIG. 1.

Uterine epithelial cell proliferation and C/EBPβ expression in response to E and P. WT mice were ovariectomized and treated with E or E plus P as described in Materials and Methods. Uteri were collected from these mice at 2 and 11 h after hormone administration. (A) IHC analysis of Ki67 expression in uterine sections. (B) Primary uterine LE cells were isolated from uteri of WT mice treated with E or E plus P. Total RNA was analyzed by real-time PCR using C/EBPβ-specific primers. The fold changes indicate gene expression levels at different times relative to those of E-treated cells at 0 h. Statistically significant differences (P < 0.05) are indicated by asterisks. (C) IHC analysis of C/EBPβ expression in uterine sections. LE, luminal epithelium; S, stroma.
E-dependent S-phase activity is impaired in C/EBPβ-null uterine epithelial cells.
Previous studies demonstrated that the E-induced proliferation of uterine LE cells is significantly reduced in C/EBPβ-null mice compared to wild-type (WT) mice of the same genetic background (28). The goal of the present study was to determine the precise stage at which cell cycle progression is impaired in mutant uterine LE cells. To assess cell cycle entry and progression through the G1 phase, we monitored the expression of two cell cycle markers, minichromosome maintenance molecule 3 (MCM3) and Ki67, by IHC analysis. MCM3 is a major component of the “prereplicative licensing complex” that is highly expressed during G1 phase in preparation for entry into S phase (7). Ki67 is a nuclear antigen that is expressed at all stages of the active cell cycle (7, 14). At 8 and 12 h after E treatment, which correspond to early and late G1 phase, both MCM3 and Ki67 were expressed at comparatively equal levels in WT and C/EBPβ-null epithelial cells (Fig. 2). This observation suggested that both WT and mutant LE cells are competent to enter G1 from G0 phase upon E stimulation and are able to progress through the G1 phase of the cell cycle.
FIG. 2.
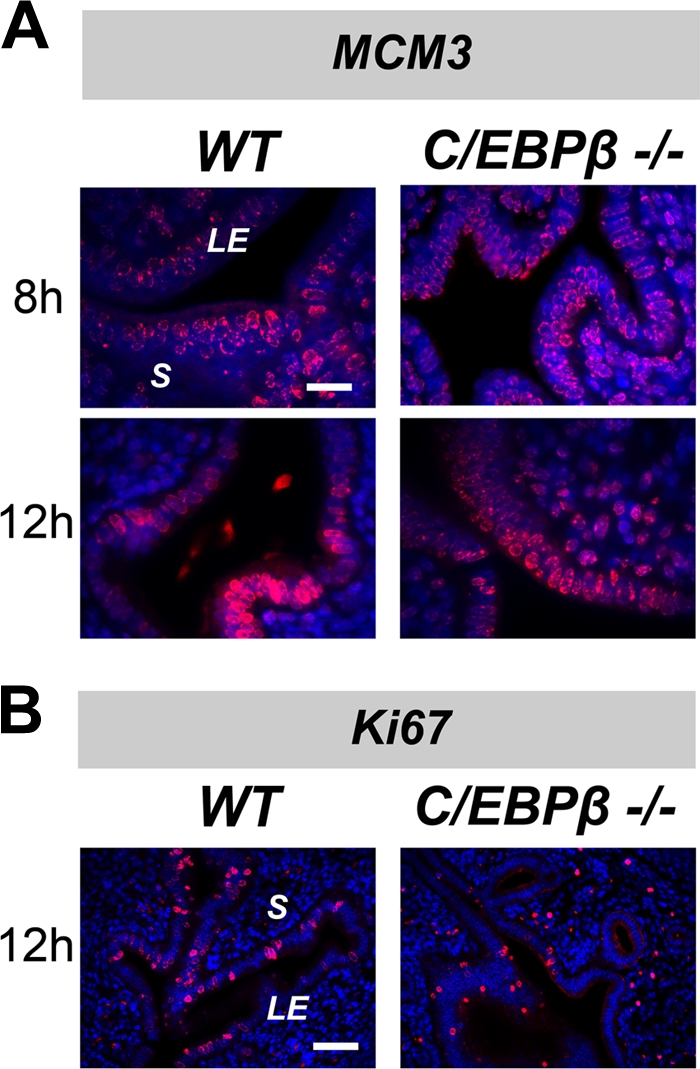
C/EBPβ-null uterine epithelial cells enter into G1 phase in response to E. WT or C/EBPβ−/− mice were ovariectomized and treated with E for 8 or 12 h. Sections of uteri from E-treated mice were subjected to IHC analysis. LE, luminal epithelium; S, stroma. (A) IHC analysis of MCM3 expression. Bar, 20 μm. (B) IHC analysis of Ki67 expression. Staining for MCM3 or Ki67 is indicated in red, and DAPI-stained nuclei are shown in blue. Bar, 50 μm.
To examine the G1-S-phase transition and S-phase activity, ovariectomized WT and C/EBPβ-null mice were treated with E for durations of 8, 12, 15, and 18 h. A 1-h pulse of BrdU, a thymidine analogue, was administered to these mice immediately prior to the collection of uterine tissue. Nuclear immunostaining of BrdU, which is indicative of S-phase activity, was absent in untreated ovariectomized WT and C/EBPβ-null mice (data not shown). Sporadic staining of BrdU was visible for ∼5% of WT LE cells at 8 h following E treatment (Fig. 3Aa and B). This staining was significantly increased by 12 h, indicating the entry of a substantial number (∼16%) of uterine LE cells into S phase (Fig. 3Ac and B). The BrdU incorporation increased further at 15 h (Fig. 3Ae). By 18 h of E treatment, almost all uterine LE cells are actively undergoing DNA replication in WT mice (Fig. 3Ag). In comparison, in the uterine LE of C/EBPβ-null mice, the level of BrdU incorporation was drastically reduced at all time points tested following E treatment (Fig. 3Ab, d, f, and h and B). While more than 90% of WT LE cells showed BrdU-positive staining at 18 h following E treatment, only 10% were BrdU positive in mutant epithelial cells (Fig. 3). These results suggested that in the absence of C/EBPβ, E-stimulated uterine LE cells are severely impaired in their ability to undergo DNA replication.
FIG. 3.
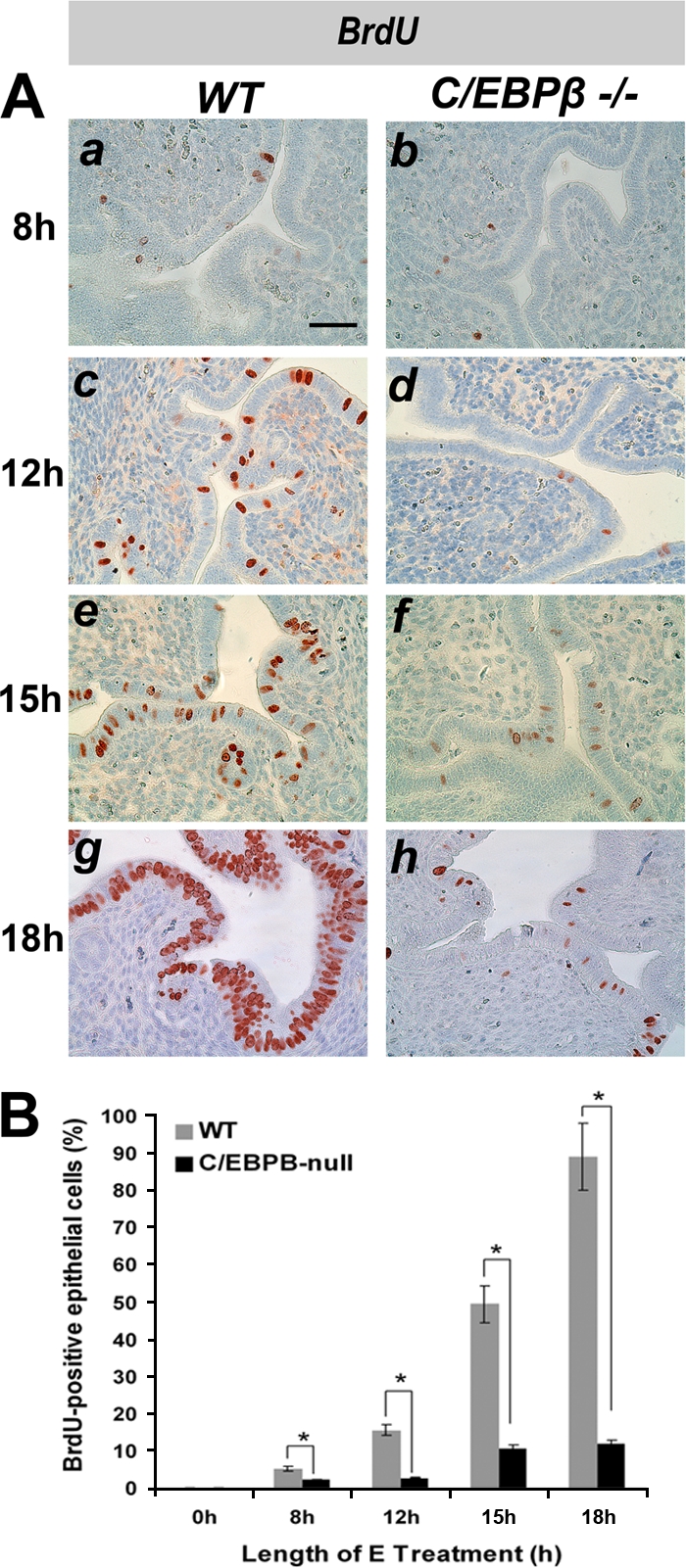
E-induced S-phase activity is impaired in C/EBPβ-null uterine epithelial cells. WT or C/EBPβ−/− mice were ovariectomized and treated with E. Uteri were collected at 8, 12, 15, and 18 h following E administration. Mice were injected with BrdU 1 h prior to sacrifice. (A) IHC of uterine sections using an anti-BrdU antibody. Bar, 50 μm. (B) For quantitation of BrdU incorporation, positively stained nuclei of LE cells were counted as described in Materials and Methods. Statistically significant differences (P < 0.05) are indicated by asterisks.
One would predict that the blockade in S-phase activity in C/EBPβ-null uterine LE cells would result in a loss of E-induced mitotic activity in these cells. To test this prediction, we employed an antibody that specifically recognizes phosphorylated serine 10 of histone H3. This marker typically localizes to distinct regions of chromosome condensation during mitosis, appearing as punctuate loci in the nucleus (16). As expected, the levels of phospho-histone H3 staining steadily increased from 18 to 22 h in E-treated WT uterine LE cells (Fig. 4A). However, in the absence of C/EBPβ, phospho-histone H3 staining was absent or drastically reduced in LE cells compared to WT uteri (Fig. 4A and B). Consistent with the S-phase impairment that we observed, the C/EBPβ-null mice displayed virtually no evidence of mitotic activity throughout the LE cell layer. The mitotic block was further confirmed by examining the mRNA and protein levels of cyclin B, a well-defined regulator of mitosis. As expected, the levels of expression of cyclin B2 mRNA (Fig. 4Ca) and protein (Fig. 4Cb) were greatly reduced in C/EBPβ-null LE cells compared to WT tissue (Fig. 4C).
FIG. 4.

Mitotic activity is absent in E-treated C/EBPβ-null uterine epithelial cells. WT or C/EBPβ−/− mice were ovariectomized and treated with E. Uteri were collected at 18, 19, 20, 22, and 24 h following E administration, and sections were subjected to IHC analysis. (A) Analysis of mitotic activity in uterine LE cells using an anti-phospho-histone H3 (Ser10) antibody. Specific staining is indicated in red, and DAPI-stained nuclei are shown in blue. Bar, 50 μm. (B) Quantitation of nuclei stained positively for phospho-histone H3. (C) Expression of cyclin B2 mRNA and protein in uterine LE cells upon E treatment. (a) Primary uterine LE cells were isolated from uteri obtained from mice following 19 h of E treatment. Total RNA was prepared, and the expression level of cyclin B2 mRNA was determined by real-time PCR. The fold changes indicate cyclin B2 mRNA expression levels relative to those of E-treated WT cells at 0 h. (b) Uterine sections of mice treated with E for 19 h were subjected to IHC using an anti-cyclin B2 antibody. Positive staining for cyclin B2 is indicated in red, and DAPI-stained nuclei are shown in green. Bar, 50 μm. Statistically significant differences (P < 0.05) are indicated by asterisks.
Altered expression and activities of multiple G1-S-phase regulatory factors in C/EBPβ-null uterine epithelial cells.
The progression of the cell cycle from G1 to S phase is orchestrated by stage-specific cyclins, cyclin-dependent kinases, and other cell cycle regulatory factors (14). Cyclin D1 is typically expressed early in the G1 phase and associates with cdk4 to promote S-phase entry. Cyclins E and A then associate with cdk2 and directly regulate S-phase progression. cdk2 achieves full functionality through the Cdk-activating kinase (CAK)-dependent phosphorylation of a threonine residue (Thr160) (13, 32). To analyze the basis of the lack of E-induced proliferation in C/EBPβ-null epithelial cells, we therefore examined the expression levels of each of these S-phase-regulatory cyclins and cdk's in WT and C/EBPβ-null LE cells at 15 h following E treatment.
When the expression of cyclin D1 and cyclin A mRNAs in WT and C/EBPβ-null uterine LE cells was examined, no significant difference in their levels in WT and mutant epithelial cells was seen (Fig. 5Aa and c). IHC of uterine sections at 15 h following E treatment revealed that the levels of the cyclin D1 protein and its nuclear localization were unaltered (Fig. 5Ab). Interestingly, we noted a modest decrease in the levels of the cyclin A protein in the nuclei of mutant LE cells (Fig. 5Ad). We did not observe any significant difference in the expression levels of cdk2 and cdk4 mRNAs in WT and C/EBPβ-null uterine epithelial cells at 15 h following E treatment (Fig. 5Ae and data not shown). In contrast, we noted an approximately 50% reduction in the expression of cyclin E mRNA in C/EBPβ-null uterine epithelial cells compared to WT cells (Fig. 5B, top). This decrease in cyclin E levels in mutant epithelial cells was further confirmed by IHC analysis of tissue sections collected at 15 h following E treatment (Fig. 5B, bottom). Most notably, we failed to detect a nuclear localization of cyclin E in mutant epithelial cells. While the presence of this cyclin was prominent in the nuclei of WT epithelial cells (Fig. 5Ba and c), its immunostaining was markedly reduced and predominantly cytoplasmic in C/EBPβ-null epithelial cells (Fig. 5Bb and d). Coincident with the reduced nuclear presence of cyclin E, we observed that the activation of cdk2 via the phosphorylation of its Thr160 residue was diminished in C/EBPβ-null epithelial cells (Fig. 5Af). The reduced level of expression of cyclin E, combined with a lack of its nuclear localization in epithelial cells, therefore contributes to the impaired activation of cdk2, leading to the defect in E-induced S-phase activity in C/EBPβ-null uterine LE cells.
FIG. 5.

Expression and localization of G1-S-phase cyclins and cdk's in E-treated C/EBPβ-null uterine epithelial cells. (A) LE cells were isolated from uteri of ovariectomized WT or C/EBPβ−/− mice treated with E for 0 and 15 h. (a, c, and e) Expression levels of cyclin D1 (a), cyclin A (c), and cdk2 (e) mRNAs were determined by real-time PCR. NS indicates differences that are not statistically significant (P > 0.05). (b and d) Sections of uteri collected at 15 h following E treatment were subjected to IHC using antibodies against cyclin D1 (b) and cyclin A (d). Positive staining for cyclin D1 is indicated in red, and DAPI-stained nuclei are shown in blue. (f) Lysates of LE cells were prepared at 0 and 15 h of E treatment and analyzed by Western blotting using antibodies against cdk2 and phosphorylated cdk2 (Thr160). (B) LE cells were isolated from uteri of mice treated with E for 0, 12, and 15 h. The level of expression of cyclin E mRNA (top) was determined by real-time PCR. The fold changes indicate cyclin E mRNA expression levels relative to those of E-treated WT cells at 0 h. Statistically significant differences (P < 0.05) are indicated by asterisks. (Bottom) Sections of uteri obtained from mice treated with E for 15 h were subjected to IHC using anti-cyclin E antibody. Panels c and d are displayed at a magnification of 100 μm. White arrows indicate nuclear staining for cyclin E, while yellow arrowheads indicate cytosolic localization. Positive staining for cyclin E is indicated in red, and DAPI-stained nuclei are shown in blue. LE, luminal epithelium; S, stroma.
E2Fs 1, 2, and 3 belong to a family of six known E2Fs and have been implicated in several studies as being the most important regulators of the G1-S-phase checkpoint (35). Upon mitogenic stimulation, activated E2F factors transactivate downstream genes, including cyclin E, promoting the progression of the cell cycle into S phase. The mRNA levels of these three E2Fs in WT and C/EBPβ-null LE cells were investigated. While the expression levels of E2F1 and E2F2 displayed no statistically significant difference between WT and mutant epithelial cells, the E2F3 mRNA level was diminished by approximately 50% in LE cells lacking C/EBPβ (Fig. 6Aa to c). This observation was further confirmed by IHC analysis showing reduced nuclear expression of the E2F3 protein in LE cells from WT and C/EBPβ-null tissue sections following 15 h of E treatment (Fig. 6B). This finding is consistent with previous reports that E2F3 plays an essential role in promoting S-phase activity (19, 22). It is of interest that the steady-state expression level of E2F3 mRNA is considerably higher than that of E2F1 or E2F2 mRNA in E-treated uterine LE cells (Fig. 6Ad). Therefore, the reduced levels of E2F3 may have a strong impact, resulting in a diminished level of expression of cyclin E mRNA and the consequent impaired ability of C/EBPβ-null uterine epithelial cells to progress through the S phase of the cell cycle.
FIG. 6.

C/EBPβ regulates the expression of E2F family genes in E-treated uterine epithelial cells. (A) LE cells were isolated from uteri of ovariectomized WT or C/EBPβ−/− mice following treatment with E for 0, 12, and 15 h. (a to c) Expression levels of E2F1 (a), E2F2 (b), and E2F3 (c) were determined by real-time PCR. The fold changes indicate mRNA expression levels of the E2F genes relative to those of E-treated WT cells at 0 h. (d) Relative steady-state levels of mRNAs for E2Fs 1, 2, and 3 in primary LE cells obtained from uteri treated with E for 15 h are shown following normalization with the 36B4 mRNA level. Statistically significant differences (P < 0.05) are indicated by asterisks. (B) Uteri from WT and C/EBPβ-null mice treated with E for 15 h were collected, and sections were subjected to IHC analysis using anti-E2F3 antibody. Positive staining for E2F3 is indicated in red, and DAPI-stained nuclei are shown in blue. (C) ChIP analysis. LE cells were isolated from uteri of ovariectomized WT mice following treatment with E for 12 h. ChIP was performed as described in Materials and Methods using antibodies against C/EBPβ, RNA polymerase II (RNAP II), and rabbit IgG. Relative levels of recruitment at various sites on the E2F3 promoter were determined by real-time PCR and normalized to input DNA and RNA polymerase II values.
C/EBPβ is recruited to the E2F3 promoter in response to E.
C/EBPβ manifests its gene regulatory role by binding to the promoters of its target genes. To explore the possibility that C/EBPβ directly mediates the E-induced expression of E2F3 and/or cyclin E, we examined the promoter regions of these genes by in silico analysis. We identified four putative binding sites for C/EBPβ in the E2F3 promoter and two such sites in the cyclin E promoter, as indicated in Materials and Methods. We then performed a ChIP analysis to test for C/EBPβ occupancy at these sites. While we did not detect C/EBPβ recruitment to any of the putative binding sites in the cyclin E promoter sites (data not shown), we found a robust recruitment of this transcription factor to the −584 region of the E2F3 promoter following E treatment (Fig. 6C).
Alterations in the G1-S-phase-inhibitory factors p27 and p21 in C/EBPβ-null uterine epithelial cells.
The cyclin-dependent kinase inhibitors (CKIs) block cell cycle progression by negatively regulating the actions of cyclin-cdk complexes (14, 47). The predominant CKIs belong to the INK4 and Cip/Kip families of inhibitors, the latter of which includes p21 and p27, two well-characterized G1-S-phase regulators (4). Both p21 and p27 directly inhibit the cyclin E-cdk2 and cyclin A-cdk2 complexes, thereby controlling S-phase activity. We found that the levels of p27 mRNA were only modestly elevated in C/EBPβ-null LE cells compared to WT cells at 0, 12, and 15 h after E treatment (Fig. 7A). Strikingly, however, a prominent nuclear localization of the p27 protein in C/EBPβ-null epithelial cells was observed at 15 and 18 h of E treatment (Fig. 7Bb and d). This nuclear accumulation of the p27 protein coincided with the block in S-phase progression and the lack of a G2-M-phase transition of these cells. The phosphorylation of p27 at threonine 187 by cdk2 serves as a signal for the ubiquitin-mediated degradation of this inhibitor (46). Consistent with the lack of cdk2 activity in the C/EBPβ-null LE cells, we observed that the level of phosphorylation of p27 at Thr187 was reduced in these cells compared to WT cells, as indicated by IHC and Western blotting (Fig. 7C and D).
FIG. 7.

Expression of the cdk inhibitors p27 and p21 in C/EBPβ-null uterine epithelial cells in response to E. (A) LE cells were isolated from uteri of ovariectomized WT or C/EBPβ−/− mice following treatment with E for 0, 12, 15, and 18 h. Expression levels of p27 mRNA were determined by real-time PCR. The fold changes indicate expression levels of p27 and p21 mRNAs relative to those of E-treated WT cells at 0 h. Statistically significant differences (P < 0.05) are indicated by asterisks, and nonsignificant differences are indicated by NS. (B) Uterine sections of WT and C/EBPβ-null mice treated with E for 15 h (a and b) and 18 h (c and d) were subjected to IHC analysis using an anti-p27 antibody. Positive staining for p27 is indicated in red, and DAPI-stained nuclei are shown in blue. The nuclear presence of p27 is indicated by white arrowheads in b and d. Bar, 50 μm. (C) IHC analysis of phosphorylated p27 (Thr187). Positive staining for phospho-p27 is indicated by red deposits. (D) Lysates of LE cells isolated from uteri of mice treated with E for 15 h were analyzed by Western blotting using the anti-phospho-p27 (p-p27) (Thr187) antibody. Immunostaining of calnexin served as a loading control. (E) Expression levels of p21 mRNA were determined by real-time PCR. (F) LE cells were isolated from uteri of mice treated with E for 0 and 18 h. Cell lysates were analyzed by Western blotting using anti-p21 antibodies. KO, knockout.
We also detected an increased level of p21 mRNA in C/EBPβ-null LE cells relative to WT cells at all times following E treatment (Fig. 7E). Of particular note was the more-than-2-fold-higher level of expression of p21 mRNA (Fig. 7E) and a marked enhancement in the p21 protein level in the mutant epithelium compared to WT tissue at 18 h after E treatment (Fig. 7F). This increased level of expression of p21 in C/EBPβ-null epithelial cells is likely to contribute to the maintenance of the G1-S-phase arrest.
Activation of DNA damage checkpoint machinery and expression of repair proteins in C/EBPβ-null uterine epithelial cells following the G1-S-phase arrest.
To further investigate the gene networks underlying C/EBPβ function during the E-induced proliferation of uterine LE cells, we compared the gene expression profiles of WT and C/EBPβ-null uterine LE cells. Briefly, ovariectomized mice were treated with E, and after 18 h, the LE cell layer was isolated and total RNA was subjected to DNA microarray analysis using Affymetrix murine arrays as described in Materials and Methods. The results indicated that the expressions of 620 genes were upregulated and that the expressions of 116 genes were downregulated greater than 2-fold in C/EBPβ-null uterine LE cells compared to WT cells (C. R. Ramathal and M. K. Bagchi, unpublished results). Using gene ontology analysis, we identified a subset of genes with well-established roles in the DNA damage response among those markedly upregulated in LE cells lacking C/EBPβ. These genes encoded factors regulating cell cycle checkpoints (such as Atm, Atr, Birk5, Skp2, and Cdc20), DNA repair (such as Rad18, Rad51, Rad54B, Rpa1, Mre11A, Exo1, and Xrcc5), and apoptosis (such as Casp3, Cdh2, and Tp73l).
A potential consequence of compromised DNA replication is the collapse of replication forks or the creation of aberrant structures at the replication forks, making the DNA prone to damage, such as single-strand or double-stand break formation (38, 57). These DNA lesions are detected by the ATM and ATR proteins, which are primary sensors and transducers of the DNA damage response (38, 57). These proteins signal downstream of the checkpoint kinases Chk1 and Chk2 and the tumor suppressor p53 in an attempt to correct the defect and remove damaged cells by triggering apoptosis (Fig. 8A). An additional mechanism involves the recruitment of the E3 ubiquitin ligase Rad18 to stalled replication forks. Rad18, in partnership with Rad6, is thought to assist in the disassembly of the aberrant fork (36). Consistent with the results of our microarray analysis, real-time PCR confirmed the upregulation of mRNAs corresponding to Atm and Atr in C/EBPβ-null uterine LE cells (Fig. 8B). The activation of the ATM and ATR kinases via unique phosphorylation events signals to the cell cycle checkpoint molecules that govern the G1-S-phase transition (38, 57). As shown in Fig. 8C, in E-treated WT uterine LE cells, ATM exists in an inactive nonphosphorylated form, indicating the absence of any DNA damage response. In contrast, prominent nuclear staining for the active phosphorylated (Ser1981) form of ATM was detectable throughout C/EBPβ-null uterine LE cells at 18 h following E treatment (Fig. 8C). In addition, an accumulation of phosphorylated (Ser139) histone H2AX, a well-established marker of the ATM-dependent DNA damage response (8, 43), was observed at 18 to 20 h in C/EBPβ-null uterine epithelial cells (Fig. 8Ea and b). As further evidence of replication fork lesions, we observed an increased nuclear expression of Rad18 in C/EBPβ-null epithelial cells compared to WT cells (Fig. 8Ec and d).
FIG. 8.

Activation of the DNA damage checkpoint pathway in E-treated C/EBPβ-null uterine epithelial cells. (A) Schematic of the major DNA damage checkpoint pathways leading to p53-dependent apoptosis or p21-mediated cell cycle arrest. (B) LE cells were isolated from uteri of WT or C/EBPβ−/− mice treated with E for 0 and 18 h. Expression levels of ATM (a) and ATR (b) mRNAs were determined by real-time PCR. Statistically significant differences (P < 0.05) are indicated by asterisks. (C) Uterine sections of WT and C/EBPβ-null mice treated with E for 0 and 18 h were subjected to IHC analysis using an antibody specific for phosphorylated Ser1981 of ATM. Positive staining for phospho-ATM is indicated in red, and DAPI-stained nuclei are shown in blue. LE, luminal epithelium; S, stroma. Bar, 50 μm. (D) Lysates of LE cells, isolated from uteri of mice treated with E for 0 and 18 h, were analyzed by Western blotting using antibodies specific for Chk1, phosphorylated Chk1 (Ser296), p53, and phosphorylated p53 (Ser15). Immunoblotting of calnexin indicated equal loading in the lanes (data not shown). (E) Sections of uteri collected from WT and C/EBPβ-null mice treated with E for 20 h were subjected to IHC analysis using antibodies specific for phosphorylated histone H2A variant X (γ-H2AX) (a and b) and Rad18 (c and d). Positive staining is indicated in red, and DAPI-stained nuclei are shown in green. Bar, 20 μm.
We next assessed the occurrence of the DNA damage response downstream of ATM/ATR activation by monitoring other markers such as checkpoint kinase 1 (Chk1) and p53 in C/EBPβ-null epithelial cells. The phosphorylation of Chk1 at Ser296 by ATM indicates that the DNA damage response and checkpoint activation are initiated (38). The phosphorylation of p53 at the Ser15 residue by the activated ATM-Chk1 pathway causes p53 transcriptional activation (38). Western blotting experiments were performed by using soluble lysates prepared from LE cells obtained from uteri collected from WT and C/EBPβ-null mice following E treatment for 18 h. The data revealed a marked elevation in the levels of phosphorylated forms of Chk1 (Ser296) and p53 (Ser15) (Fig. 8D). There was also a significant increase in the level of total p53 protein in the C/EBPβ-null LE cells relative to WT cells at 18 h of E treatment (Fig. 8D). It was previously reported that a consequence of the upregulation and activation of p53 is an elevated level of expression of its transcriptional target, p21 (3, 38). We indeed saw a significant rise in the level of the p21 protein (Fig. 7E), which presumably helps maintain cell cycle arrest in mutant epithelial cells. Collectively, our results established that the lack of C/EBPβ, which leads to a block in the E-mediated proliferation of uterine epithelial cells, triggers the ATM/ATR-dependent DNA damage response pathway and activates p53-mediated signaling that helps maintain the cell cycle block.
Increased apoptosis in the C/EBPβ-null uterine epithelium following G1-S-phase arrest.
The phosphorylation of p53 at Ser15 by the ATM-Chk1/Chk2 pathway promotes its stabilization by preventing ubiquitination and degradation (1, 38). p53 is known to promote the expression of several proapoptotic genes, which in turn stimulate the mitochondrial pathways leading to caspase activation (43). We therefore investigated the possibility that the activation of an ATM/ATR/Chk1/Chk2 DNA damage response mechanism in C/EBPβ-null uterine LE cells, which results in the activation and accumulation of p53, commits these cells to enter programmed cell death. In order to assess the initiation of apoptosis in E-treated uterine LE cells of WT and C/EBPβ-null mice, we first examined the activation of caspase 3 (Casp3), a well-known marker of this process (27, 43). We performed an IHC analysis to determine the levels of Casp3 and its proteolytically cleaved active form (cl-Casp3) in these cells. Examination of the levels of full-length Casp3 at 48 h following E treatment showed a higher level of expression of this protein in the cytosolic compartment of C/EBPβ-null epithelial cells than in WT epithelial cells (data not shown). When we assayed for the presence of cl-Casp3 at the 48-h time point, an increased presence of this apoptosis marker was evident in C/EBPβ-null epithelial cells (Fig. 9A).
FIG. 9.

Evidence for increased apoptosis in E-treated C/EBPβ-null uterine epithelial cells. (A) Uteri were collected from ovariectomized WT and C/EBPβ-null mice treated with E for 48 h. Uterine sections were subjected to IHC analysis by using an antibody specific for the cleaved form of caspase 3. Positive staining for cleaved caspase 3 is indicated in red, and DAPI-stained nuclei are shown in blue. Arrowheads indicate cells that stained positive for cleaved caspase 3 in uterine LE cells. (B) Uteri collected from WT and C/EBPβ-null mice treated with E for 48 h were sectioned and subjected to a TUNEL assay. Positive staining for apoptotic cells is indicated in green, and DAPI-stained nuclei are shown in blue. Arrows indicate apoptotic cells in uterine LE cells. (C) Quantitation of epithelial nuclei stained positively in the TUNEL assay was performed. The data were plotted as the percentage of total epithelial cells present in the fields. Statistically significant differences (P < 0.05) are indicated by asterisks.
We next performed TUNEL staining to assess the extent of DNA degradation in apoptotic cells of uterine epithelial cells of WT and C/EBPβ-null mice. We detected a notable increase in TUNEL staining at discrete foci in LE cells of mutant mice compared to cells of WT mice at 48 h of E treatment (Fig. 9B and C). These data suggested that the arrest of E-stimulated C/EBPβ-null LE cells at the S phase of the cell cycle triggers the activation of p53-dependent mechanisms that increase their susceptibility to programmed cell death and affect their survival.
DISCUSSION
C/EBPβ is an essential mediator of E-induced S-phase entry of uterine epithelial cells.
Our previous studies revealed that C/EBPβ is a major downstream target of E regulation in the uterus (28). The discovery that the uterine epithelium in C/EBPβ-null mice is nonreceptive to embryo implantation (28) prompted us to utilize this animal model to investigate the functional role of C/EBPβ and its downstream pathways in uterine epithelial biology. The studies presented here demonstrated that the absence of C/EBPβ results in a significant defect in the ability of uterine LE cells to proliferate in response to E stimulation. This is evidenced by a major impairment in DNA replication activity and multiple dysregulated cell cycle components in C/EBPβ-null uterine LE cells. We determined that C/EBPβ-null uterine LE cells are able to enter the cell cycle and progress through the G1 phase before being arrested in the S phase (Fig. 2 and 3). The ablation of the C/EBPβ gene therefore led to a complete loss of E-induced DNA replication and subsequent mitotic activity in uterine LE cells. We therefore uncovered a role for C/EBPβ as an essential mediator of E-induced DNA replication in uterine epithelial cells.
Previous reports documented that E administered to ovariectomized mice promotes the G1-to-S-phase transition of uterine LE cells (29, 50, 51). Previous studies by Tong and Pollard indicated that the E-induced nuclear localization of cyclin D1 is an essential event that permits the S-phase entry of uterine LE cells (50, 51). It was shown that in the presence of E, the activation and nuclear translocation of cyclin D1 occur via the inhibition of glycogen synthase kinase 3β (GSK-3β) activity. It was further proposed that P opposes the proliferative actions of E by allowing the phosphorylation of cyclin D1 by GSK-3β and the subsequent nuclear export of this cyclin. In the present study, we noted that the nuclear presence of cyclin D1 remained unaltered in S-phase-arrested LE cells of C/EBPβ-null uteri (Fig. 5A). These results suggested that the C/EBPβ regulation of uterine LE cell proliferation occurs via a mechanism that is unrelated to the nuclear translocation of cyclin D1. Our findings are corroborated by data from previous reports showing that the nuclear entry of cyclin D1 occurs in the absence of C/EBPβ in the liver and mammary epithelium (11, 12).
A major finding of our study is that, upon E treatment, the level of cyclin E mRNA is notably lower in C/EBPβ-null LE cells than in WT LE cells and that the cyclin E protein failed to localize to the nucleus (Fig. 5B). It is likely that the C/EBPβ-mediated entry of LE cells into the S phase of the cell cycle involves primarily the regulation of cyclin E-cdk2 function. During the S phase of the cell cycle, the active cyclin E-cdk2 complex is localized in the nucleus of WT LE cells and coordinates the assembly of DNA replication factors into the origins of replication (14, 47). In contrast, in C/EBPβ-null uteri, the combination of a reduced level of expression of cyclin E and its absence from the nucleus of LE cells would prevent its partnership with cdk2 and its S-phase-promoting activities. Consequently, inadequate cdk2 activation in C/EBPβ-null LE cells would also contribute to an S-phase blockade (Fig. 5A). Tong and Pollard previously reported that P abolishes E-induced cyclin E-cdk2 activity in uterine epithelial cells (51). Data from that report are consistent with our observation that the simultaneous administration of P suppresses E-induced C/EBPβ expression, which in turn controls the expression and nuclear presence of cyclin E. Our finding is also consistent with data from a previously reported microarray study conducted using whole uteri of mice (17). It was reported that the acute administration of E stimulated the expression of cyclin E and cdk2 in this tissue, although the uterine cell type(s) in which these molecules are expressed remained unclear.
Previous reports indicated that the loss of C/EBPβ results in reduced cyclin E expression levels, reduced cdk2 functionality, and decreased proliferative activity in mammary epithelial cells and in hepatocytes during liver regeneration (11, 12). The precise mechanism by which C/EBPβ regulates the expression and nuclear localization of cyclin E is presently unknown. Several studies proposed that cyclin E is a target of transcriptional and posttranslational regulation by the E2F transcription factors, which are important regulators of the G1-S-phase transition and DNA replication (19, 21, 22, 34, 35, 39). Interestingly, among the E2F family members that we have examined, E2F3 is the most highly expressed in WT LE cells (Fig. 6Ad). It displayed reduced expression levels in C/EBPβ-null uterine epithelial cells (Fig. 6A), while the levels of E2F1 and E2F2 remained unaltered. ChIP analysis demonstrated the recruitment of C/EBPβ to the proximal promoter region of E2F3, strongly suggesting that E2F3 is a primary target of C/EBPβ in uterine LE cells. We postulate that E2F3 regulates cyclin E expression, which in turn controls the entry of uterine LE cells into the S phase of the cell cycle.
We found that the CDK inhibitors p21 and p27 also play important roles in blocking E-induced cell cycle progression in C/EBPβ-null uterine LE cells. In G0 and early G1 phases, a high level of p27 is present in hormone-withdrawn uterine LE cells. p27 binds to the cyclin E-cdk2 complex and inhibits its activity. Upon E stimulation, p27 protein levels decline via ubiquitin-mediated degradation. In late G1 phase, p27 becomes a substrate of phosphorylation by the cyclin E-cdk2 complex. Phosphorylated p27 is released from the cyclin E-cdk2 complex, exits its nuclear location, and undergoes degradation, resulting in cyclin E-cdk2 activation (14, 42, 46, 47). Consistent with this scenario, we observed that E-stimulated WT uterine LE cells efficiently targeted p27 for ubiquitin-mediated degradation by phosphorylation, thereby depleting the nuclear p27 protein upon S-phase entry (Fig. 7C and D). In contrast, a robust nuclear accumulation of p27 was detected in C/EBPβ-null epithelial cells, coincident with the drastically reduced cyclin E level in the nuclear compartment and the cell cycle blockade in S phase (Fig. 7B). Our results supported the view that the accumulation of the p27 protein arises from an inefficient phosphorylation of p27 by the cdk2-cyclin E complex and the consequent lack of a ubiquitin-mediated degradation of p27 in mutant LE cells (Fig. 7C and D). The nuclear retention of p27 suggests that it may interact with and inactivate any residual cyclin E-cdk2 or cyclin A-cdk2 complex that might form in the nuclei. We also noted a remarkable rise in the level of the p21 inhibitor at later stages of cell cycle arrest. The sustained presence of p27 and p21 inhibitors during early and late S phase maintains the blockade of cyclin-cdk2 activities, preventing uterine LE cells from completing DNA replication and entering mitosis.
E stimulation in the absence of C/EBPβ activates the DNA damage checkpoint pathway in uterine epithelial cells.
The dysregulation of cyclin E was previously reported to cause the destabilization of prereplication complexes necessary for DNA synthesis (6). We postulate that a defect in cyclin E-cdk2-controlled replication fork assembly is encountered when E-stimulated C/EBPβ-null LE cells traverse through G1 phase and attempt to progress through S phase. We favor the hypothesis that the onset of a replication defect in E-stimulated C/EBPβ-null uterine LE cells triggers DNA damage checkpoint activation. In mammalian cells, stalled replication forks or replication-blocking lesions are typically recognized by the ATM/ATR kinase checkpoint pathways as DNA damage signals, triggering a series of well-defined molecular events culminating in cell cycle arrest and DNA repair (1, 10, 38, 43, 57). Stalled replication forks also recruit the ubiquitin ligase Rad18, a key component of the replication repair pathway (2, 36). In our studies, the ATM-dependent checkpoint pathway is activated in C/EBPβ-null uterine LE cells, as evidenced by the enhanced accumulation of active, phosphorylated forms of ATM kinase and its downstream targets, histone variant H2AX, Chk1, and p53 (Fig. 8). The increased presence of Rad18 in the nuclei of replication-arrested C/EBPβ-null epithelial cells further confirms the replication fork lesions in mutant LE cells. The activation of Chk1 via phosphorylation at Ser296 is known to affect S-phase progression (1, 10). Furthermore, the ATM/ATR-dependent phosphorylation of p53 at Ser15 leads to its activation and stabilization. Transcriptionally active p53 in turn promotes the synthesis of p21, which directly inhibits the cdk2-cyclin E complex (4, 14, 38, 47). In this manner, the activation of p53-dependent mechanisms downstream of the DNA damage response helps maintain the DNA replication arrest in hormone-stimulated C/EBPβ-null epithelial cells.
Loss of C/EBPβ expression in uterine epithelial cells triggers apoptosis: implications for E-dependent cell survival.
In addition to its mitogenic effects, E is also known to promote cell survival by acting as an antiapoptotic agent (31, 52). A block in E-induced uterine LE cell proliferation in the absence of C/EBPβ may trigger apoptosis, which is the culmination of a cascade of molecular steps, including cell cycle withdrawal and DNA fragmentation (27). We also considered apoptosis as a potential biological end point for S-phase-arrested C/EBPβ-null uterine LE cells since there is a strong connection between the ATM/p53-dependent checkpoint response and the removal of damaged cells via programmed cell death (38, 43). One of the essential players in apoptosis is the serine-protease Casp3, which plays a central role in coordinating a variety of downstream events leading to cell death (27, 43). Our studies revealed that a subpopulation of E-treated C/EBPβ-null LE cells blocked in S phase entered the apoptotic pathway, as evidenced by the increased presence of the cleaved, active form of Casp3 in these cells compared to WT LE cells (Fig. 9A). Additionally, we noted a significant increase in cell death due to DNA fragmentation in C/EBPβ-null epithelial cells compared to WT tissue (Fig. 9B and C). The genetic deletion of C/EBPβ was previously linked to increased apoptosis and reduced tumorigenesis in skin keratinocytes (49, 58). It is likely that the majority of the C/EBPβ-null uterine LE cells undergo DNA repair and recovery, thereby preventing widespread apoptosis in the epithelium. In support of this concept, the levels of expression of several proteins associated with the DNA repair pathway such as Rad18, Rad51, Rad54B, Rpa1, Mre11A, Exo1, and Xrcc5 were elevated in uterine epithelial cells lacking C/EBPβ.
In conclusion, C/EBPβ is an early and critical mediator of the E-controlled proliferative response in uterine LE cells. The functional link between E and C/EBPβ provides a novel mechanism by which this hormone controls the expression, localization, and activity of specific cell cycle regulatory molecules, such as E2F3, cyclin E, p27, and p21, to influence DNA synthesis in LE cells (Fig. 10). The downstream actions of this key transcription factor are also indispensable for DNA repair and cell survival, allowing the preparation of a functional uterine epithelium for the establishment of pregnancy. Furthermore, the role of C/EBPβ as a mediator of proliferative as well as antiapoptotic effects of E in uterine epithelial cells presents it as a potential target for anticancer therapeutics for preventing E-dependent endometrial cancers.
FIG. 10.

Molecular pathways regulated by C/EBPβ during E-induced proliferation of uterine epithelial cells. In mouse uterine epithelial cells, the expression of C/EBPβ is stimulated by E and opposed by simultaneous treatment with P. C/EBPβ controls the entry of E-stimulated epithelial cells into the S phase of the cell cycle by upregulating the expression of E2F3 and cyclin E. It also promotes the nuclear localization of cyclin E, which enables the formation of an active cyclin E-cdk2 complex critical for DNA replication. The lack of C/EBPβ allows the nuclear accumulation of p27, which contributes to the cell cycle arrest. In E-stimulated C/EBPβ-null uterine epithelial cells, stalled DNA replication activates a DNA damage response pathway involving ATM/ATR, Chk1/Chk2, Rad18, and p53. Activated p53 maintains the block in DNA replication by enhancing the synthesis of the cell cycle inhibitor p21 and also promotes the removal of damaged cells via caspase-dependent apoptosis.
Acknowledgments
We acknowledge Elizabeth Hunt, Katya Dribinsky, and Jarrad Marcell for genotyping.
This work was supported by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement U54 HD055787 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research.
Footnotes
Published ahead of print on 19 January 2010.
REFERENCES
- 1.Bartek, J., and J. Lukas. 2001. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 490:117-122. [DOI] [PubMed] [Google Scholar]
- 2.Bi, X., L. R. Barkley, D. M. Slater, S. Tateishi, M. Yamaizumi, H. Ohmori, and C. Vaziri. 2006. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 26:3527-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunz, F., A. Dutriaux, C. Lengauer, T. Waldman, S. Zhou, J. P. Brown, J. M. Sedivy, K. W. Kinzler, and B. Vogelstein. 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282:1497-1501. [DOI] [PubMed] [Google Scholar]
- 4.Cheng, M., P. Olivier, J. A. Diehl, M. Fero, M. F. Roussel, J. M. Roberts, and C. J. Sherr. 1999. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 18:1571-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cullingford, T. E., and J. W. Pollard. 1994. Growth factors as mediators of sex-steroid hormone action in the uterus during the pre-implantation period, p. 13-30. In S. A. Khan and G. M. Stancel (ed.), Protooncogenes and growth factors in steroid hormone-induced growth differentiation. CRC Press Inc., Boca Raton, FL.
- 6.Ekholm-Reed, S., J. Mendez, D. Tedesco, A. Zetterberg, B. Stillman, and S. I. Reed. 2004. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 165:789-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endl, I. K. E., M. Baack, R. Knippers, J. Gerdes, and T. Scholzen. 2001. The expression of Ki-67, MCM3, and p27 defines distinct subsets of proliferating, resting, and differentiated cells. J. Pathol. 195:457-462. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Capetillo, O., H. T. Chen, A. Celeste, I. Ward, P. J. Romanienko, J. C. Morales, K. Naka, Z. Xia, R. D. Camerini-Otero, N. Motoyama, P. B. Carpenter, W. M. Bonner, J. Chen, and A. Nussenzweig. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993-997. [DOI] [PubMed] [Google Scholar]
- 9.Ghahary, A., and L. J. Murphy. 1989. Uterine insulin-like growth factor-I receptors: regulation by estrogen and variation throughout the estrous cycle. Endocrinology 125:597-604. [DOI] [PubMed] [Google Scholar]
- 10.Gorgoulis, V. G., L.-V. F. Vassiliou, P. Karakaidos, P. Zacharatos, A. Kotsinas, T. Liloglou, M. Venere, R. A. DiTullio, N. G. Kastrinakis, B. Levy, D. Kletsas, A. Yoneta, M. Herlyn, C. Kittas, and T. D. Halazonetis. 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907-913. [DOI] [PubMed] [Google Scholar]
- 11.Greenbaum, L. E., W. Li, D. E. Cressman, Y. Peng, G. Ciliberto, V. Poli, and R. Taub. 1998. CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. Clin. Invest. 102:996-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimm, S. L., A. Contreras, M.-H. Barcellos-Hoff, and J. M. Rosen. 2005. Cell cycle defects contribute to a block in hormone-induced mammary gland proliferation in CCAAT/enhancer-binding protein (C/EBP beta)-null mice. J. Biol. Chem. 280:36301-36309. [DOI] [PubMed] [Google Scholar]
- 13.Gu, Y., J. Rosenblatt, and D. O. Morgan. 1992. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 11:3995-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper, J. V., and G. Brooks. 2005. The mammalian cell cycle: an overview. In H. T. Brooks (ed.), Cell cycle control: mechanisms and protocols. Humana Press Inc., Totowa, NJ.
- 15.Heinemeyer, T., E. Wingender, I. Reuter, H. Hermjakob, A. E. Kel, O. V. Kel, E. V. Ignatieva, E. A. Ananko, O. A. Podkolodnaya, F. A. Kolpakov, N. L. Podkolodny, and N. A. Kolchanov. 1998. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 26:362-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendzel, M. J., Y. Wei, M. A. Mancini, A. Van Hooser, T. Ranalli, B. R. Brinkley, D. P. Bazett-Jones, and D. C. Allis. 1997. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106:348-360. [DOI] [PubMed] [Google Scholar]
- 17.Hewitt, S. C., B. J. Deroo, K. Hansen, J. Collins, S. Grissom, C. A. Afshari, and K. S. Korach. 2003. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol. Endocrinol. 17:2070-2083. [DOI] [PubMed] [Google Scholar]
- 18.Hewitt, S. C., J. C. Harrell, and K. S. Korach. 2005. Lessons in estrogen biology from knockout and transgenic animals. Annu. Rev. Physiol. 67:285-308. [DOI] [PubMed] [Google Scholar]
- 19.Humbert, P. O., R. Verona, J. M. Trimarchi, C. Rogers, S. Dandapani, and J. A. Lees. 2000. E2F3 is critical for normal cellular proliferation. Genes Dev. 14:690-703. [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, J., M. Sato, Q. Li, J. P. Lydon, F. J. DeMayo, I. C. Bagchi, and M. K. Bagchi. 2008. Peroxisome proliferator-activated receptor gamma is a target of progesterone regulation in the preovulatory follicles and controls ovulation in mice. Mol. Cell. Biol. 28:1770-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong, L.-J., J. T. Chang, A. H. Bild, and J. R. Nevins. 2006. Compensation and specificity of function within the E2F family. Oncogene 26:321-327. [DOI] [PubMed] [Google Scholar]
- 22.Leone, G., J. DeGregori, Z. Yan, L. Jakoi, S. Ishida, R. S. Williams, and J. R. Nevins. 1998. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 12:2120-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lubahn, D., J. Moyer, T. Golding, J. Couse, K. Korach, and O. Smithies. 1993. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. U. S. A. 90:11162-11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luetteke, N., T. Qiu, S. Fenton, K. Troyer, R. Riedel, A. Chang, and D. Lee. 1999. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126:2739-2750. [DOI] [PubMed] [Google Scholar]
- 25.Luetteke, N. C., T. H. Qiu, R. L. Peiffer, P. Oliver, O. Smithies, and D. C. Lee. 1993. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 73:263-278. [DOI] [PubMed] [Google Scholar]
- 26.Lydon, J. P., F. J. DeMayo, C. R. Funk, S. K. Mani, A. R. Hughes, C. A. Montgomery, Jr., G. Shyamala, O. M. Conneely, and B. W. O'Malley. 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9:2266-2278. [DOI] [PubMed] [Google Scholar]
- 27.Maddika, S., S. R. Ande, S. Panigrahi, T. Paranjothy, K. Weglarczyk, A. Zuse, M. Eshraghi, K. D. Manda, E. Wiechec, and M. Los. 2007. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist. Updat. 10:13-29. [DOI] [PubMed] [Google Scholar]
- 28.Mantena, S. R., A. Kannan, Y.-P. Cheon, Q. Li, P. F. Johnson, I. C. Bagchi, and M. K. Bagchi. 2006. C/EBPbeta is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the uterine epithelium and stroma. Proc. Natl. Acad. Sci. U. S. A. 103:1870-1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin, L., J. W. Pollard, and B. Fagg. 1976. Oestriol, oestradiol-17β and the proliferation and death of uterine cells. Endocrinology 69:103-115. [DOI] [PubMed] [Google Scholar]
- 30.McKnight, S. L. 2001. McBindall—a better name for CCAAT/enhancer binding proteins? Cell. 107:259-261. [DOI] [PubMed] [Google Scholar]
- 31.Monroe, D. G., R. R. Berger, and M. M. Sanders. 2002. Tissue-protective effects of estrogen involve regulation of caspase gene expression. Mol. Endocrinol. 16:1322-1331. [DOI] [PubMed] [Google Scholar]
- 32.Morgan, D. O. 1995. Principles of CDK regulation. Nature 374:131-134. [DOI] [PubMed] [Google Scholar]
- 33.Mukku, V., and G. Stancel. 1985. Regulation of epidermal growth factor receptor by estrogen. J. Biol. Chem. 260:9820-9824. [PubMed] [Google Scholar]
- 34.Muller, H., A. P. Bracken, R. Vernell, M. C. Moroni, F. Christians, E. Grassilli, E. Prosperini, E. Vigo, J. D. Oliner, and K. Helin. 2001. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15:267-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller, H., and K. Helin. 2000. The E2F transcription factors: key regulators of cell proliferation. Biochim. Biophys. Acta 1470:M1-M12. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima, S., L. Lan, S. Kanno, N. Usami, K. Kobayashi, M. Mori, T. Shiomi, and A. Yasui. 2006. Replication-dependent and -independent responses of RAD18 to DNA damage in human cells. J. Biol. Chem. 281:34687-34695. [DOI] [PubMed] [Google Scholar]
- 37.Nelson, K. G., T. Takahashi, N. L. Bossert, D. K. Walmer, and J. A. McLachlan. 1991. Epidermal growth factor replaces estrogen in the stimulation of female genital-tract growth and differentiation. Proc. Natl. Acad. Sci. U. S. A. 88:21-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nyberg, K. A., R. J. Michelson, C. W. Putnam, and T. A. Weinert. 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36:617-656. [DOI] [PubMed] [Google Scholar]
- 39.Pajalunga, D., and M. Crescenzi. 2004. Regulation of cyclin E protein levels through E2F-mediated inhibition of degradation. Cell Cycle 3:1572-1578. [DOI] [PubMed] [Google Scholar]
- 40.Pan, H., Y. Deng, and J. W. Pollard. 2006. Progesterone blocks estrogen-induced DNA synthesis through the inhibition of replication licensing. Proc. Natl. Acad. Sci. U. S. A. 103:14021-14026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramji, D. P., and P. Foka. 2002. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365:561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed, S. I. 2002. Cell cycling? Check your brakes. Nat. Cell Biol. 4:E199-E201. [DOI] [PubMed] [Google Scholar]
- 43.Roos, W. A., and B. Kaina. 2006. DNA-damage induced cell death by apoptosis. Trends Mol. Med. 12:440-450. [DOI] [PubMed] [Google Scholar]
- 44.Sandelin, A., W. W. Wasserman, and B. Lenhard. 2004. ConSite: Web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 32:W249-W252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schug, J., and G. C. Overton. 1997. TESS: transcription element search software on the WWW. Computational Biology and Informatics Laboratory, School of Medicine, University of Pennsylvania, Philadelphia, PA.
- 46.Sheaff, R. J., M. Groudine, M. Gordon, J. M. Roberts, and B. E. Clurman. 1997. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11:1464-1478. [DOI] [PubMed] [Google Scholar]
- 47.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13:1501-1512. [DOI] [PubMed] [Google Scholar]
- 48.Sterneck, E., L. Tessarollo, and P. F. Johnson. 1997. An essential role for C/EBPbeta in female reproduction. Genes Dev. 11:2153-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sterneck, E., S. Zhu, A. Ramirez, J. L. Jorcano, and R. C. Smart. 2005. Conditional ablation of C/EBP beta demonstrates its keratinocyte-specific requirement for cell survival and mouse skin tumorigenesis. Oncogene 25:1272-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tong, W., and J. W. Pollard. 2002. Female sex steroid hormone regulation of cell proliferation in the endometrium, p. 94-109. In S. R. Glasser, J. D. Alpin, L. C. Giudice, and S. Tabibzadeh (ed.), Endometrium. Taylor & Francis, London, United Kingdom.
- 51.Tong, W., and J. W. Pollard. 1999. Progesterone inhibits estrogen-induced cyclin D1 and cdk4 nuclear translocation, cyclin E- and cyclin A-cdk2 kinase activation, and cell proliferation in uterine epithelial cells in mice. Mol. Cell. Biol. 19:2251-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, Q., X. Li, L. Wang, Y.-H. Feng, R. Zeng, and G. Gorodeski. 2004. Antiapoptotic effects of estrogen in normal and cancer human cervical epithelial cells. Endocrinology 145:5568-5579. [DOI] [PubMed] [Google Scholar]
- 53.Webb, D., B. Moulton, and S. Khan. 1993. Estrogen induces expression of c-jun and jun-B protooncogenes in specific rat uterine cells. Endocrinology 133:20-28. [DOI] [PubMed] [Google Scholar]
- 54.Wedel, A., and H. A. Ziegler-Heitbrock. 1995. The C/EBP family of transcription factors. Immunobiology 193:171-185. [DOI] [PubMed] [Google Scholar]
- 55.Yamashita, S., A. Takayanagi, and N. Shimizu. 1996. Temporal and cell-type specific expression of c-fos and c-jun protooncogenes in the mouse uterus after estrogen stimulation. Endocrinology 137:5468-5475. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, J.-W., Q.-Q. Tang, C. Vinson, and M. D. Lane. 2004. Dominant-negative C/EBP disrupts mitotic clonal expansion and differentiation of 3T3-L1 preadipocytes. Proc. Natl. Acad. Sci. U. S. A. 101:43-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou, B.-B. S., and S. J. Elledge. 2000. The DNA damage response: putting checkpoints in perspective. Nature 408:433-439. [DOI] [PubMed] [Google Scholar]
- 58.Zhu, S., K. Yoon, E. Sterneck, P. F. Johnson, and R. C. Smart. 2002. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. U. S. A. 99:207-212. [DOI] [PMC free article] [PubMed] [Google Scholar]