

Abstract
We compared TLR responsiveness in PBMC from HIV-1-infected and uninfected individuals using the TLR agonists: TLR7 (3M-001), TLR8 (3M-002) and TLR7/8 (3M-011). Activation and maturation of plasmacytoid dendritic cells (pDC)were measured by evaluating CD86, CD40 and CD83 expression and myeloid dendritic cell (mDC) activation was measured by evaluating CD40 expression. All agonists tested induced activation and maturation of pDC in PBMC cultures of cells from HIV+ and HIV− individuals. The TLR7 agonist induced significantly less pDC maturation in cells from HIV+ individuals. Quantitative assessment of secreted IFN-α and pro-inflammatory cytokines at the single cell level showed that pDC from HIV+ individuals stimulated with TLR7 and TLR7/8 induced IFN-α. TLR8 and TLR7/8 agonists induced IL-12 and COX-2 expression in mDC from HIV+ and HIV− individuals. Understanding pDC and mDC activation and maturation in HIV-1 infection could lead to more rational development of immunotherapeutic strategies to stimulate the adaptive immune response to HIV-1.
Keywords: Human immunodeficiency virus, Toll-like receptor 7, Toll-like receptor 8, Toll-like receptor agonists, interferon-alpha, COX-2, pro-inflammatory cytokines, dendritic cells
1. Introduction
The innate immune system is at the frontline of the immune response to pathogens and mediates the development of adaptive immune responses (1). Advanced HIV-1 infection is associated with diminished immune responses to pathogens. It is also associated with markedly reduced numbers and function of innate immune cells including plasmacytoid (pDC) and myeloid (mDC) dendritic cells (2–5). The low number of pDC in chronic HIV-1 infection is associated with a diminished innate immune response and decreased levels of IFN-α production, which is also associated with an increased risk of opportunistic infections (3, 6). Given the central role played by dendritic cells in linking innate and adaptive immunity, abnormalities in pDC and mDC number and function may play a role in the diminished pathogen specific responses observed in HIV-1 disease.
Dendritic cells are activated through germ-line encoded pattern recognition receptors, the most important of which are the toll-like receptors (TLR). Peripheral blood dendritic cells have distinct patterns of TLR expression: TLR7 and TLR9 on pDC, and TLR2, TLR3, TLR4 and TLR8 on mDC (7, 8). Activation through these receptors results in co-stimulatory molecule expression, cytokine production and migration of dendritic cells into lymphoid tissues (9, 10). TLR7 and TLR8 are intracellular endosomal receptors that recognize single-stranded RNA (ssRNA) and small molecule imidazoquinolines (11–13). The cellular uptake and endosomal maturation of TLR7 and TLR8 ligands are needed in order to trigger NF-κB and MAP kinase mediated signals through a MyD88-dependent pathway (14).
Synthetic imidazoquinoline-like molecules (8) induce dendritic cell maturation and the production of cytokines which promote direct anti-viral effects and adaptive cellular anti-viral immunity respectively (15–18). Three distinct synthetic imidazoquinolines have been identified based on structural differences; they are designated TLR7-selective, TLR8-selective and TLR7/8-selective agonists. These agonists stimulate dendritic cells to produce type I interferon (IFN-α/β) and cytokines essential in the development and maintenance of immune responses. Production of IFN-α/β by pDC is essential in antiviral innate immunity through direct inhibition of viral replication (19) and induction of an antiviral state in neighboring cells (20). In addition, IFN-α has a strong adjuvant effect on a variety of immune cells including monocytes, mDC, NK cells, and B and T lymphocytes (21).
TLR ligands also regulate innate immunity and the inflammatory response through COX-2, a pivotal enzyme in cellular biosynthetic pathways that leads to prostaglandin and thromboxane synthesis from arachidonic acid. COX-2 expression is regulated through the MyD88 dependent pathway (22, 23) and is induced by pathogens, microbes and synthetic TLR agonists including CpG DNA (TLR9) and lipopolysaccharides (LPS, TLR4) (24). Continuous antigenic stimulation and inflammation observed during chronic infections, including HIV-1, may inhibit effector T cell function through COX-2 expression and the production of prostaglandins (25).
In addition to the physiological immune and pro-inflammatory roles of IFN-α and COX-2, increasing evidence supports a role for these proteins in HIV-1 infection. HIV-1-induced quantitative and qualitative abnormalities in pDC have been identified in both the acute and chronic phases of HIV-1 infection. Specifically, a decrease in IFN-α production was associated with high viral load and development of AIDS (6, 26–28). However, it is unknown if the reduced production of IFN-α in vitro by stimulated mononuclear cells from HIV-1-infected individuals is a consequence of an intrinsic pDC defect or the effect of the reduced pDC frequency.
In vitro incubation with HIV-1 can directly activate pDC to produce IFN-α and upregulate CD80 and CD86 expression in cells that have not been exposed to HIV-1 in vivo (29, 30). No one has previously determined whether the continued exposure to HIV-1 that takes place during the course of HIV-1 infection alters the ability of DC to respond to HIV-1 or to TLR agonists. The goal of these studies is to characterize the effect of synthetic TLR7-selective, TLR8-selective and TLR7/8-selective imidazoquinoline-like molecules on dendritic cells from HIV-1-infected individuals. Both direct and indirect effects were measured since activation was in PBMC cultures.
2. Materials and Methods
2.1 Study population and clinical data
HIV-1 infected (HIV+, n=17) and uninfected individuals (HIV−, n=15) were enrolled in this study to assess pDC and mDC responses to TLR agonists. A subset of 5 HIV+ and 5 HIV− individuals (Group 1) were used to evaluate dendritic cell responses to TLR7, TLR7/8 and TLR8 agonists while a separate subset (Group 2) of 12 HIV+ and 10 HIV− individuals were used for assessment of intracellular IFN-α, IL-12, and COX-2 expression following addition of TLR agonists or inactivated HIV-1 virus. The study participants on HAART had undetectable viral loads (VL <75 copies/ul) and 357-992 CD4 cells/ul. The two HAART naïve individuals used in these studies had CD4 counts of 880 and 134 CD4 cells/μl and a VL of 3190 and <50 copies/ml, respectively. All HIV+ individuals were recruited from the Mark Weiss Memorial Infectious Diseases Clinic of Rush University Medical Center (Chicago, IL). HIV− individuals were recruited from Rush University Medical Center. The demographics of the HIV+ individuals are shown in Table 1. This study was reviewed and approved by the Institutional Review Board of Rush University Medical Center. Informed consent was obtained from all study participants. All viral loads (VL) and CD4 T cell counts were performed by CLIA-certified commercial laboratories at the time the samples were collected for this study.
Table 1.
Description of HIV-1infected individuals
| HAART* | No HAART | ||
|---|---|---|---|
| Number | 15 | 2 | |
| Males | 12 | 2 | |
| Females | 3 | 0 | |
| CD4 T cells/mm3 | |||
| Minimum | 69 | 50 | |
| Median | 510 | 465 | |
| Maximum | 992 | 880 | |
| Viral load, copies/ml | |||
| Minimum | <50 | 3190 | |
| Median | <75 | 3595 | |
| Maximum | 94387 | 4000 | |
| Treatment length, months | |||
| Minimum | 1 | NA** | |
| Median | 50 | NA | |
| Maximum | 204 | NA | |
HAART, highly active antiretroviral therapy
NA, not applicable
2.2 Monoclonal antibodies and reagents
The following mouse anti-human monoclonal antibodies were obtained from BD Biosciences (BD; San Jose, CA): Lin1 FITC (CD3, CD14, CD16, CD19, CD20 and CD56 cocktail), CD123 PE, CD11c PE, CD11c APC, HLA-DR PerCP, HLA-DR APC, CD86 APC, CD40 APC, CD83 APC, COX-2 and IL-12 PE. Custom conjugates of mouse anti-human monoclonal antibodies against IFN-α PE (clone 7N4-1) and CD123 PerCP-Cy5.5 were also provided by BD Biosciences. The 7N4-1 antibody reacts with human IFN-α2b (31, 32) and to a lesser extent IFN-α7 (33). It does not react with IFN-α1 or IFN-α4 (33). Matched fluorchrome-conjugated isotype-control antibodies were used in these studies. The BD Cytofix/Cytoperm™ Kit was used for the intracellular staining for IFN-α, IL-12 and COX-2. The FcγR blocking reagent was from Miltenyi Biotec (Auburn, CA).
The synthetic small molecule imidazoquinolines, 3M-001 (TLR7), 3M-002 (TLR8) and 3M-011 (TLR7/8), and the 3M-006 non-reactive control TLR7/8 agonists were provided by 3M Pharmaceuticals (St. Paul, MN). Endotoxin-free A-class CpG ODN 2216 (TLR9 agonist; 5′-ggGGGACGATCGTCgggggG-3′) was provided by Coley Pharmaceutical Group (Wellesley, MA). Purified LPS from Escherichia coli 055:B5 (TLR4 agonist) was purchased from Sigma (St Louis, MO). Noninfectious Aldithriol-2 (AT-2) treated HIV-1Ada (R5-tropic) and HIV-1MN (X4-tropic) viral preparations along with matched control microvesicles, SUPT1-CCR5 (MV-CCR5) and CEMX174 (T1) (MV-X4), isolated from uninfected cell cultures (34) were kindly provided by Dr. Jeff Lifson from the AIDS Vaccine Program, National Cancer Institute (Frederick, MD).
2.3 TLR agonists for stimulation of PBMC
The TLR agonists were utilized at the following concentrations; TLR7 agonist (3M-001; 0.3 μM), TLR8 agonist (3M-002; 3 μM), TLR7/8 agonist (3M-011; 3.0 μM) for in vitro stimulation of PBMC. Media alone or a non-reactive control TLR7/8 imidazoquinoline (3M-006, 3.0 μM) served as negative controls. A TLR9 agonist (A-Class CpG ODN 2216; 4 μ/ml) or a TLR4 agonist (LPS; 5 μg/ml) were added as positive controls in the assays evaluating intracellular cytokines and COX-2 expression in dendritic cells. Optimal concentrations of these agonists were determined previously (8, 26). The concentrations used in these experiments are listed in Table 2.
Table 2.
TLR agonists used in these studies
| TLR Agonist | Designation | Cognate TLR | Test Concentration |
|---|---|---|---|
| Lipopolysaccharide* | LPS | TLR4 | 5.0 μg/ml |
| Imidazoquinoline | 3M-001 | TLR7 | 0.3 μM |
| Imidazoquinoline | 3M-002 | TLR8 | 3.0 μM |
| Imidazoquinoline | 3M-011 | TLR7/8 | 3.0 μM |
| Imidazoquinoline Control | 3M-006 | none | 3.0 μM |
| Class A CpG ODN | CpG-2216 | TLR9 | 4.0 μg/ml |
from E. coli 055:B5
2.4 Cell Culture for activation with TLR agonists
Whole blood was collected by venipuncture into vacutainer tubes containing sodium heparin (BD Vacutainer Systems, Franklin Lakes, NJ). Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation using lymphocyte separation medium (BioWhittaker, Walkersville, MD). Isolated pDC and mDC were not studied because HIV-1 infected individuals have much lower numbers of circulating pDC and mDC than uninfected individuals and too much blood would be required to obtain a sufficient number of purified cells from HIV-1 infected individuals. PBMC (1×106 cells/ml) were incubated in the presence of TLR agonists or controls in RPMI 1640 (BioWhittaker) supplemented with 10% heat-inactivated fetal bovine serum (GemCell, Woodland, CA), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma, St Louis, MO) and 2 mM of L-glutamine (Sigma). For dendritic cell activation/maturation, PBMC were cultured in 6-well (6–8 ml) polystyrene tissue culture plates in a humidified 37°C 5% CO2 incubator. After 24 hours, dendritic cell activation and maturation markers were evaluated by flow cytometry and culture supernatants were frozen at −20°C for cytokine analysis.
For intracellular IFN-α, IL-12 or COX-2, PBMC were incubated with the appropriate TLR agonist (5–7 ml) in 15 ml polypropylene tubes at a 5° slant in a humidified 37°C 5% CO2 incubator. The protein transport inhibitor, Brefeldin A (BFA; 5 μg/ml; BD), was added at the beginning of the culture period (Time = 0 hours) to cultures containing Media, TLR7, TLR8, TLR7/8 and control TLR7/8 agonists or 2 hours following the addition of the TLR9 agonist (CpG ODN). After 20 hours, the cells were harvested and intracellular protein expression was measured using flow cytometry.
2.5 Induction of IFN-α by aldrithiol-2 (AT-2) inactivated HIV-1 virus
PBMC (1×106/ml) from 10 HIV− and 4 HIV+ individuals were cultured with AT-2 HIV-1Ada or AT-2 HIV-1MN at 500 ng/ml p24CA equivalent in RPMI-10% FBS media. Media alone and matched control microvesicles, MV-CCR5 and MV-CEMX, served as negative controls. Cultures (1–2 ml) were incubated at a 5° slant for 20 hours in a humidified 37°C 5% CO2 incubator for 14 hours with 5μg/ml BFA. After incubation, we measured intracellular IFN-α accumulation in pDC by flow cytometry.
2.6 Flow cytometry analysis
PBMC from TLR agonist or AT-2 HIV-1 stimulated cultures were harvested, washed and resuspended to 1×107 cells/ml in Dulbecco’s phosphate buffered saline (DPBS) that contained 0.5% bovine serum albumin and 0.1% sodium azide (FACS buffer). Non-specific antibody binding to Fc receptors was blocked by pre-incubation of the cells with Fcγ-receptor blocking reagent (Miltenyi Biotec) for 20 minutes at 4°C. PBMC for dendritic cell activation/maturation were surface stained with the appropriate antibodies for 20 minutes, washed with FACS buffer, fixed with 2% formaldehyde and stored at 4°C. PBMC cultures for intracellular IFN-α, IL-12 and COX-2 were surface stained for pDC or mDC, washed and incubated with BD Cytofix/Cytoperm™ solution for 20 minutes at 4°C. Cells were washed twice with BD Perm/Wash™ buffer and stained with the intracellular staining monoclonal antibodies for 30 minutes. Finally, the samples were washed twice in Perm/Wash™ buffer, fixed with 1% formaldehyde and stored at 4°C until analysis. All samples were evaluated within 24-hours of staining using either a FACSCalibur™ or LSRII flow cytometer.
Four-color flow cytometry was used to evaluate dendritic cell activation or maturation state and intracellular IFN-α, IL-12 and COX-2 expression after stimulation with TLR agonists or AT-2 inactivated HIV-1 virus. Logical gating was used to identify the pDC (Lin1−/CD123+/HLA DR+) and mDC (Lin1−/CD11c+/HLA DR+) populations. Dendritic cell activation (pDC-CD86, pDC-CD40, mDC-CD40), maturation (pDC-CD83, mDC-CD83), and intracellular protein accumulation (pDC-IFN-α; mDC-IL-12 and mDC-COX-2) were expressed as a percentage of the parent population.
2.7 Measurement of cytokines by ELISA or Cytometric Bead Array (CBA)
Commercial ELISA kits were used to measure the concentration of IFN-α (PBL Biomedical Laboratories, Piscataway, NJ). The BD™ Cytometric Bead Array (CBA) Inflammation Kit was used to measure IL-1β, TNF-α, IL-10, and IL-12p70 in cell culture supernatants following TLR7, TLR8, and TLR7/8 agonist stimulation of PBMC from uninfected and HIV-1 infected individuals. ELISA and CBA assays were performed according to the manufacturer’s guidelines.
2.8 Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM). Groups were compared using a 2-tailed Student’s t test (confidence level of 95 %.) with GraphPad PRISM software v4.03. P values of <0.05 were considered statistically significant.
3. Results
3.1 pDC from HIV infected individuals are activated by TLR agonists
HIV-1 infection is associated with low numbers of circulating pDC in the peripheral blood (6, 26, 27, 35) which may lead to impaired innate immune function and diminished initiation of adaptive immunity. We incubated PBMC with synthetic small molecule imidazoquinolines that are TLR7 and TLR8 agonists and evaluated in vitro activation and maturation of pDC from HIV+ and HIV− individuals. Since pDC have TRL 7 and 9 but do not have TLR 8, activation with TLR 8 is indirect.
Unstimulated pDC from HIV+ individuals had significantly higher CD86 expression than pDC from HIV− individuals. TLR7, TLR8, and TLR7/8 agonist stimulation significantly upregulated the expression of the activation markers, CD86 (B7-2) and CD40, and the maturation marker, CD83, on pDC from HIV+ (p<0.05) and HIV− (p<0.05) individuals compared to their respective media controls (Figure 1a–c). The TLR7 agonist induced significantly less CD83 expression on pDC (1.6 fold; p=0.02) from HIV+ than HIV− individuals, while the degree of CD86 and CD40 expression in cells from infected and uninfected individuals was similar. The TLR8 and TLR7/8 agonists induced the same degree of activation/maturation in pDC from HIV+ and HIV− individuals.
Figure 1. pDC from HIV infected individuals are activated by TLR agonists.

PBMC from 5 HIV+ and 5 HIV− individuals were incubated for 24 hours with TLR7, TLR8, TLR7/8, control cTLR7/8 agonists, or media alone. Flow cytometry was used to determine pDC expression of (a) CD86, (b) CD40 and (c) CD83. Results are expressed as the mean ± SEM of the percentage of pDC that express each cell surface marker. Statistical comparisons were made between the media, control and the corresponding TLR7, TLR8 or TLR7/8 agonist stimulated cultures using a Student’s t-test; values were considered significant if p<0.05. *Significantly different from HIV− control; **Significantly different from HIV+ control; •–• Significant difference between HIV− and HIV+ cultures.
3.2 TLR 8 agonist activates mDC from HIV infected individuals
Activation and maturation of mDC is an essential process in the initiation of an adaptive immune response. We incubated PBMC with TLR7 and TLR8 agonists and evaluated activation of mDC from HIV+ and HIV− individuals. Both mDC and pDC from HIV infected individuals have very high percentages of activated cells after overnight culture with media alone or TLR controls. Activation in controls was more pronounced for mDC than for pDC. Activation of mDC measured by increased percentages of CD86 and CD83 positive cells following overnight culture in the absence of stimulation was so high that it precluded evaluation of activation with TLR agonists. When cells were from individuals who were infected with HIV-1, the background activation seen with CD40 was significantly higher than when cells were from an uninfected individual (Figure 2a). Even though this was the case, we were still able to see significant activation of mDC from an HIV infected individual with the TLR8 agonist. The TLR8 agonist significantly increased (p<0.05) both the percentage and mean fluorescence intensity (MFI) of CD40 on mDC from both HIV+ and HIV− individuals (Figures 2a – b). A similar percentage of cells from infected and uninfected individuals was activated by the TLR 7/8 agonist but since the control values were so much higher, activation with the TLR 7/8 agonist for cells from infected individuals was not significant.
Figure 2. TLR 8 agonist activates mDC from HIV infected individuals.
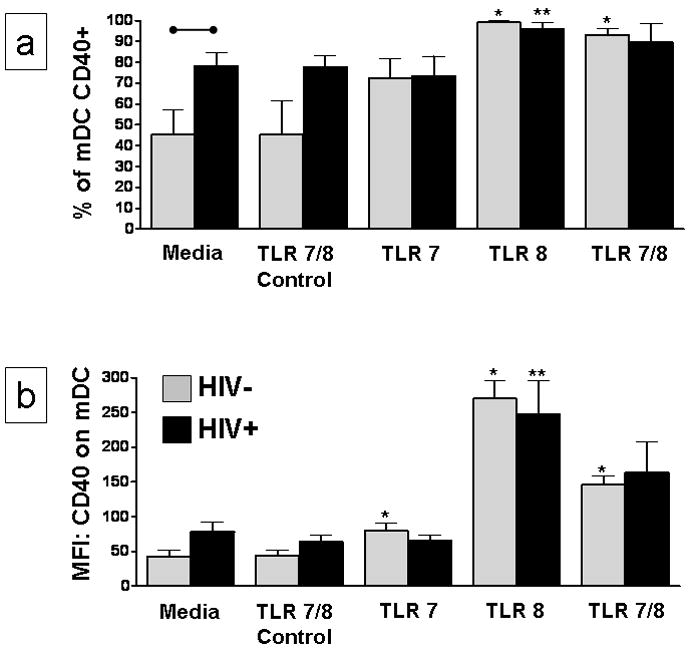
PBMC from 5 HIV+ and 5 HIV− individuals were incubated for 24 hours with TLR7, TLR8, TLR7/8, control cTLR7/8 agonists, or media alone. Flow cytometry was used to determine the (a) percentage of CD40 positive mDC and (b) CD40 mean fluorescence intensity (MFI) on mDC. Results are expressed as the mean ± SEM of the percentage of mDC that express CD40 or mDC-CD40 MFI. Statistical comparisons were made between the media control and the corresponding TLR7, TLR8 or TLR7/8 agonist stimulated cultures using a Student’s t-test; values were considered significant if p<0.05. *Significantly different from HIV− control; **significantly different from HIV+ control; •–• significant difference between HIV− and HIV+ cultures.
3.3 TLR7 and TLR8 agonists induced secretion of cytokines
Supernatants collected from cultures of PBMC from HIV+ and HIV− individuals stimulated with the TLR7, TLR8, and TLR7/8 agonists were evaluated for IFN-α, IL-1β, IL-10, IL-12p70 and TNFα. IFN-α secretion induced by TLR7, TLR8, and TLR7/8 agonist stimulation were respectively 4.8, 2.6, and 2.0 fold higher in the HIV uninfected individuals than in HIV infected individuals. IFN-α concentrations were somewhat lower for all TLR agonists when cells from HIV infected individuals were stimulated (Figure 3a). TLR7 agonist activation of PBMC from HIV infected individuals (mean IFN-α = 152.5 pg/ml) was significantly (p<0.05) lower than IFN-α levels induced in uninfected individuals (mean IFN-α = 730.7 pg/ml)..
Figure 3. TLR7, TLR8 and TLR7/8 agonists induce pro-inflammatory cytokine secretion.

PBMC from 5 HIV+ and 5 HIV− individuals were incubated for 24 hours with a TLR7, TLR8, TLR7/8, control cTLR7/8 agonists or media alone. Culture supernatants were collected and the concentration of (a) IFN-α was measured by ELISA, while (b) IL-1β, TNF-α, IL-10 and IL-12p70 were measured by CBA. Results are expressed as the mean ± SEM concentration (pg/ml). Statistical comparisons were made between the media control and the corresponding TLR7, TLR8 or TLR7/8 agonist stimulated cultures using a Student’s t-test; values were considered significant if p<0.05. *Significantly different from HIV− control; **Significantly different from HIV+ control; •–• Significant difference between HIV− and HIV+ cultures.
The TLR8 and TLR7/8 agonists were potent inducers of IL1-β, TNF-α, IL-10 and IL-12p70 production in HIV− and HIV+ individuals (Figure 3b). These values were all significantly greater (p<0.05) than their corresponding media controls with the exception of TLR7/8 induced IL12p70. Overall, PBMC cultures from HIV+ individuals showed a trend toward higher concentrations of pro-inflammatory cytokines, but these differences were not statistically significant. When incubated with the TLR8 agonist, PBMC from HIV+ individuals secreted more IL-10 and less IL12p70 than PBMC from HIV− individuals. When incubated with the TLR7/8 agonist, PBMC from HIV+ individuals secreted more IL1-β and less IL12p70 than PBMC from HIV− individuals. Less IFN-α was produced in cultures of cells from HIV+ individuals than from uninfected individuals in the presence of the TLR7 agonist. The TLR7 agonist induced concentrations of IL-1β, TNF-α, and IL-10 that were significantly higher in PBMC from HIV+ individuals than in cultures containing PBMC from HIV− individuals: (IL-1β: p=0.014; TNF-α: p=0.032; IL-10: p=0.044). We did not observe any induction of IL-12p70 by the TLR7 agonist.
3.4 HIV-1 and TLR agonists activate IFN-α production in pDC in the absence of costimulatory cytokines
pDC play an important role in innate immunity through the production of IFN-α. PBMC from HIV-1 infected individuals were stimulated with natural or synthetic TLR agonists in the presence of the protein transport inhibitor, Brefeldin A (BFA); we then measured the intracellular accumulation of IFN-α in pDC (Figure 4). The addition of BFA at the beginning of the culture prevented cytokines produced by other cells from being released and influencing IFN-α production by pDC. It also prevented the release of IFN-α and enabled us to measure intracellular accumulation in pDC using phycoerythrin (PE) conjugated anti-human IFN-α monoclonal antibody. Representative results from 1 HIV+ individual are presented in Figure 4a. IFN-α expression was significantly greater when PBMC were stimulated with TLR7, TLR7/8, or TLR9 agonists than in the control cultures (Figure 4b). As expected, no intracellular IFN-α production was stimulated by TLR8 in the absence of indirect effects mediated by mDC. The TLR4 agonist, LPS, was included as a negative control since TLR4 is not present on pDC and TLR9 was included as a positive control since TLR9 agonist activation of cells from HIV infected individuals has been previously reported (26). We observed a significantly lower percentage of IFN-α positive pDC in response to TLR7 and TLR7/8 agonists in PBMC from HIV+ individuals compared with PBMC from uninfected individuals (p=0.015 and p=0.023, respectively) (Figure 4b). These results demonstrate that pDC from HIV+ individuals recognize and respond to the appropriate TLR agonists, but the magnitude of their response to the TLR7 and 7/8 agonists on a per cell basis was significantly less than in uninfected individuals. Infectious and non-infectious AT-2 treated HIV-1Ada (R5-tropic) and AT-2 treated HIV-1MN (X4-tropic) virus are potent stimulators of pDC, inducing activation, maturation and secretion of IFN-α (29, 30, 36). Therefore, in vitro responses to AT-2 inactivated HIV-1 virus were evaluated in PBMC from HIV-1 individuals and compared with responses in uninfected individuals. PBMC from HIV+ or HIV− individuals were cultured with AT-2 HIV-1Ada, AT-2 HIV-1MN, or negative control matched microvesicles and evaluated for intracellular IFN-α expression within the pDC population. Exposure to non-infectious HIV-1 virus significantly increased the number of IFN-α expressing pDC in HIV+ and HIV− individuals when compared to the microvesicle (MV) controls (Figure 4c). However, among HIV+ individuals, the number of IFN-α positive pDC cells was lower in response to AT-2 HIV-1Ada (1.6 fold) or AT-2 HIV-1MN (1.1 fold) virus than from HIV− individuals, but these differences were not statistically significant.
Figure 4. HIV-1 and TLR agonists activate IFN-α production in pDC in the absence of costimulatory cytokines.
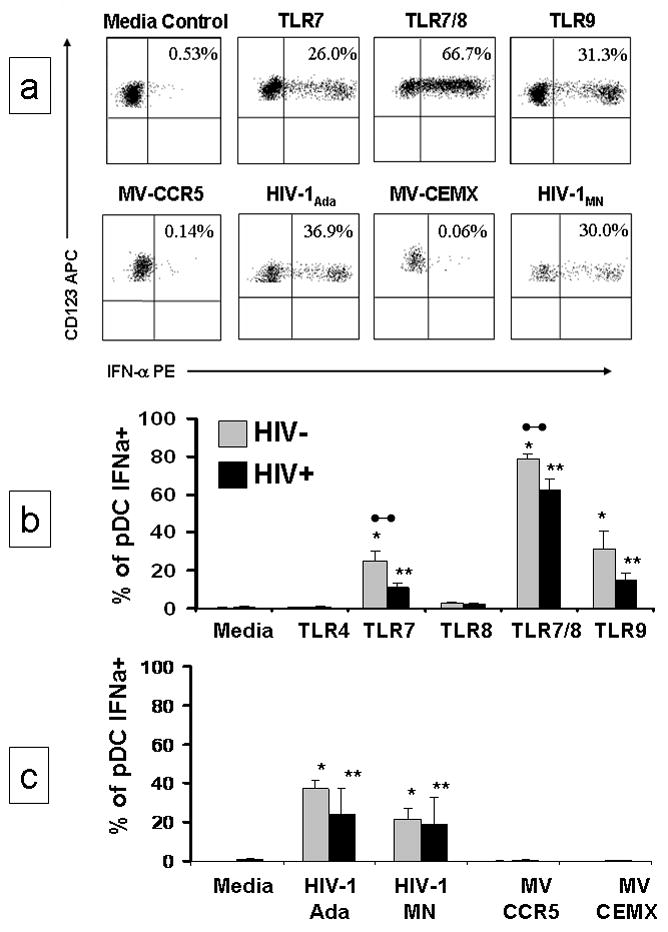
PBMC were incubated for 20 hours with inactivated HIV-1 or synthetic TLR agonists in the presence of 5 ug/ml the protein transport inhibitor Brefeldin A (BFA). Intracellular accumulation of IFN-α was measured. (a) Representative flow cytometric analysis of the selective detection of intracellular IFN-α in pDC [logical gating was used to identify the pDC population from 1 HIV-1 infected individual (Lin1−/HLA DR+/CD123+)] following stimulation. (b) 12 HIV+ and 10 HIV− individuals were evaluated for intracellular accumulation of IFN-α following stimulation with TLR4, TLR7, TLR8, TLR7/8 and TLR9 agonists in the presence BFA. TLR4 was included as a negative control and TLR9 was included as a positive control. (c) 4 HIV+ and 10 HIV− individuals were evaluated for intracellular accumulation of IFN-α following stimulation with AT-2 inactivated HIV-1Ada (R5-tropic), AT-2 inactivated HIV-1MN (X4-tropic) and matched control microvesicles (MV-CCR5, MV-CEMX) in the presence of BFA. Results are expressed as the mean ± SEM of the percentage of pDC that expresses intracellular IFN-α. Statistical comparisons were made between the media control and the corresponding TLR agonist or AT-2 inactivated HIV-1 virus using a Student’s t-test; values were considered significant if p<0.05. *Significantly different from HIV− control; **significantly different from HIV+ control; •–• significant difference between HIV− and HIV+ cultures.
3.5 TLR agonists induce IL-12 and COX-2 in mDC in the absence of costimulatory cytokines
mDC play an active role in regulating innate and adaptive immune responses to foreign pathogens through the production of IL-12. PBMC from HIV-1 infected individuals were stimulated with TLR agonists in the presence of the protein transport inhibitor, Brefeldin A (BFA); we then measured the intracellular accumulation of IL-12 and COX-2 in mDC (Figure 5). Representative results from 1 HIV infected individual are presented in Figure 5a. Unlike the pDC population, the frequency of circulating mDC was similar between HIV+ and HIV− individuals (0.37% versus 0.31%, p>0.05) in this study. PBMC from HIV+ and HIV− individuals stimulated with TLR4, TLR7, TLR8, TLR7/8 and TLR9 agonists were evaluated to determine the percentage of mDC that produce IL-12p40/p70 (Figure 5b). TLR4, TLR8 and TLR7/8 stimulated PBMC from HIV+ and HIV− individuals had significantly more mDC that expressed IL-12 (p<0.05) than their respective media controls. The percentage of IL-12 producing mDC was higher in HIV+ than in HIV− individuals for all of the TLR agonists, but only those responses mediated by TLR4, TLR7, and TLR9 agonists were significantly higher (p = 0.003, p = 0.003 and p= 0.012, respectively).
Figure 5. TLR agonists induce IL-12 and COX-2 in mDC in the absence of costimulatory cytokines.
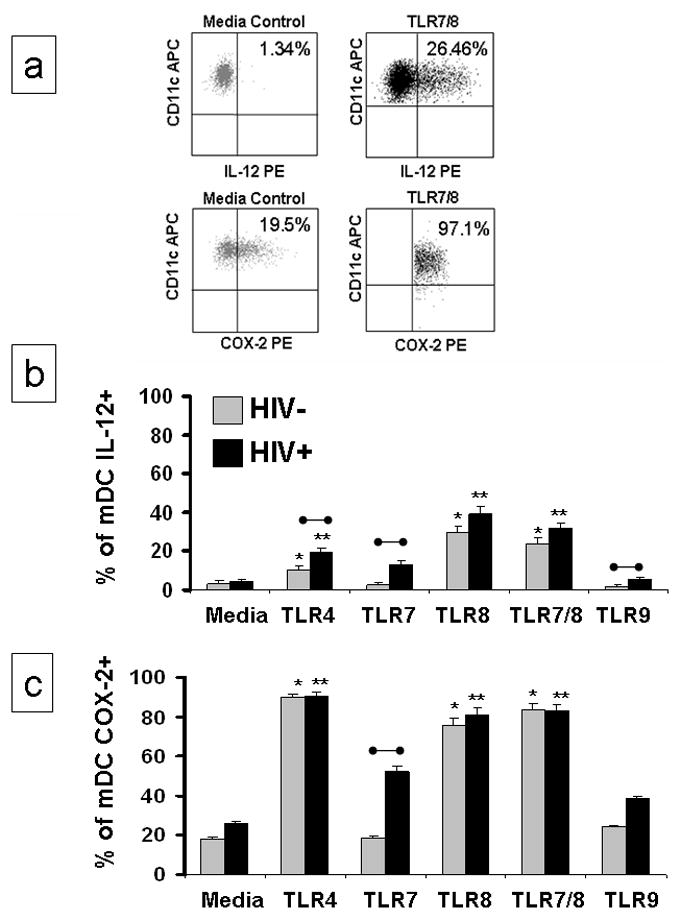
PBMC were incubated for 20 hours with inactivated HIV-1 or synthetic TLR agonists in the presence of 5 ug/ml the protein transport inhibitor Brefeldin A (BFA). Intracellular accumulation of IFN-α was measured. (a) Representative flow cytometric analysis of the selective detection of intracellular IL-12p40/p70 or COX-2 in mDC; [logical gating was used to identify the mDC population from 1 HIV infected individual (Lin1-/HLA DR+/CD11c+)] following stimulation. (b) 12 HIV+ and 10 uninfected individuals were evaluated for intracellular accumulation of IL-12 in mDC following stimulation with TLR4, TLR7, TLR8, TLR7/8 and TLR9 agonists in the presence BFA. TLR4 was included as a negative control and TLR9 was included as a positive control. (c) The same individuals were evaluated for intracellular accumulation of COX-2 in mDC. Results in b and c are expressed as the mean ± SEM percentage of mDC expressing the intracellular marker. Statistical comparisons were made between the media control and the corresponding TLR agonist using a Student’s t-test; values were considered significant if p<0.05. *Significantly different from HIV− control; **significantly different from HIV+ control; •–• significant difference between HIV− and HIV+ cultures.
COX-2, a pivotal enzyme in prostaglandin biosynthetic pathways, was evaluated in mDC following TLR agonist stimulation (Figure 5a and 5c). TLR4, TLR8 and TLR7/8 agonist stimulation significantly increased the percentage of COX-2 positive mDC in both the HIV− and HIV+ individuals; the percentages of mDC-COX-2 expression was similar between the HIV− and HIV+ individuals. Constitutive mDC COX-2 expression (media controls) was 1.5 fold higher in HIV+ than in HIV− individuals but this difference was not statistically significant (p = 0.640). In Figure 5 since cells are incubated with agonists in the presence of BFA, we would not expect to see mDC activation with TLR7 or TLR9 since mDC do not have these TLR receptors. TLR7 and TLR9 responses in cultures of cells from HIV infected individuals are not significantly greater than their matched control. Higher cytokine production is seen in cells from HIV-1 infected individuals presumably because of in vivo activation of these cells. These results suggest that TLR agonist stimulation induces both positive (IL-12) and negative (COX-2) regulatory proteins to be expressed by mDC.
4. Discussion
In this study, we compared the ability of synthetic small molecule imidazoquinolines to activate pDC and mDC from HIV+ and HIV− individuals. Since a mixed cell culture more accurately reflects what might happen in vivo, activation was done in PBMC cultures. As a result of this, activation of DC could be either direct or indirect. The TLR7, TLR8 and TLR7/8 agonists were equally able to mediate dendritic cell activation as measured by CD86 and CD40 expression on pDC and CD40 expression on mDC. The imidiazoquinolines were also effective at inducing pDC maturation (pDC-CD83). However, pDC maturation was significantly reduced among HIV+ individuals stimulated with the TLR7 agonist. Similarly, all three types of imidazoquinolines induced IFN-α and pro-inflammatory cytokine secretion in the PBMC cultures. The TLR8 and TLR7/8 agonists were the strongest inducers of IL-1β, TNF-α, IL-10 and IL-12p70 in HIV+ individuals. These observations are consistent with previous reports of increased dendritic cell activation/maturation and expression of pro-inflammatory cytokines in uninfected individuals (8, 37) following TLR7 and TLR8 agonist stimulation and points to the potential of achieving a similar response in dendritic cells from HIV-1-infected individuals using these agonists.
The innate immune response to pathogens plays a crucial role in mediating direct antiviral effects and in the development of the adaptive immune response. Early in HIV-1 disease, innate immune cells such as pDC, mDC, NK, and iNKT are either diminished due to the direct cytopathic effects of HIV-1 or are dysfunctional due to a dysregulation of the cytokine network (38–43). TLR agonists are promising molecules for initiating and/or maintaining immune cell function. They have been shown to directly trigger antigen presentation by up-regulating activation (CD86, CD40) and maturation (CD83) markers on dendritic cells and by inducing the synthesis of large amounts of cytokines like IFN-α, TNF-α and IL-12. These events can lead to a protective host immune response to HIV-1 infection through the development of CD8+ T-cell antiviral activity (37).
Distinct patterns of cytokine production were observed in TLR7 and TLR8 agonist stimulated cultures. The TLR7/8 agonist induced a broader range of cytokines including IFN-α, TNF-α, and IL-12p70. The TLR7 agonist preferentially induced IFN-α while the TLR8 agonist induced TNF-α, IL-12p70 and interestingly low levels of IFN-α. The TLR8 and TLR7/8 agonists were more effective at producing high levels of pro-inflammatory cytokines including IL-1β and IL-10. These results are consistent with previous reports on TLR7 and TLR8 agonist induced cytokine production in PBMC from healthy uninfected individuals (8).
In this study, the mean percentage of peripheral blood pDC was lower in HIV+ individuals (HIV+ 0.16% pDC versus 0.34%; p<0.05) which is consistent with previous reports of low pDC numbers in association with chronic HIV-1 infection. Previous studies have shown impaired IFN-α secretion by pDC and defective monocyte maturation post-TLR9 (CpG ODN) stimulation in HIV-1-infected individuals (26, 35, 44). To expand on these studies we demonstrated a dysregulation in total IFN-α (pg/ml) secretion in PBMC cultures from HIV+ individuals (Fig. 3A) stimulated with the TLR7 and TLR7/8 agonists. A reduced frequency of pDC in HIV+ individuals which is consistent with previously published reports (6, 26, 27, 35), may be responsible for the lower concentrations of IFN-α detected in HIV-1 infected individuals. To discriminate between qualitative and quantitative abnormalities in pDC function, we measured the intracellular expression of IFN-α at the single cell level using a broad range of synthetic and natural TLR ligands. The percentage of pDC that express IFN-α was reduced in HIV+ individuals stimulated with the synthetic TLR7, TLR7/8 and TLR9 agonists. PBMC stimulated with AT-2 inactivated HIV-1 virus induced pDC IFN-α expression, with no significant differences between HIV+ and HIV− individuals. Taken together, these results demonstrate a dysregulation in IFN-α production in HIV-1-infected individuals which may impact the anti-viral effect of IFN-α and the induction of HIV-1 specific adaptive immune responses. The relative contribution of TLR agonists to the control of HIV replication and enhancement of HIV-specific immunity in an in vitro setting are currently being studied. However, in contrast to the previously reported protective anti-HIV-1 properties of IFN-α (45), recent studies suggest that this cytokine contributes to an increase in CD4+ T cell death mediated by apoptosis in chronically HIV-1-infected individuals (46). The potentially detrimental effects of IFN-α should be considered when assessing the risk/benefit balance of using therapeutic tools that increase IFN-α production in HIV-1 infection.
Another factor in HIV-1 disease progression and immune system dysfunction is related to excessive immune activation (47–49). In HIV-1 infection, dysregulation of inflammatory responses points to the importance of studying critical molecules such as COX-2, which contributes to the pathogenesis of diseases with uncontrolled or chronic inflammation (50, 51). We also observed higher constitutive levels of COX-2 in mDC from HIV-1 infected individuals. COX-2 is up-regulated by TLR agonists such as CpG ODN and LPS in a MyD88-dependent signaling pathway (22–24). In these studies we demonstrate direct modulation of COX-2 expression in mDC using TLR4, TLR7/8, and TLR8 agonists. Interestingly, the TLR9 and TLR7 agonists also induced COX-2 in mDC from HIV+ individuals.
In conclusion, the synthetic small molecule imidazoquinolines displayed functionally distinct cytokine patterns linked to the TLR expressed by innate immune cells. Based on our results, we propose that a TLR7/8 (3M-001) agonist may be the most effective at modulating HIV-1 immunity through increased dendritic cell activation/maturation and the induction of critical cytokines including IFN-α from pDC and IL-12 from mDC. In addition, we must be aware that COX-2 and IL-10 that are induced by TLR7 and TLR8 agonists may dampen or alter the development of antigen-specific T-cell responses. Therefore, strategies to induce a protective host immune response to HIV-1 infection using TLR agonists may need to be combined with strategies that inhibit COX-2 and IL-10 mediated pathways.
Acknowledgments
This work was supported in part by NIH Research Grant AI055793; the Colombian Agency for the Development of Science and Technology-Colciencias and by the Committee for the Development of Research-CODI from the University of Antioquia. Mark Tomai is an employee of 3M Corporation which provided the synthetic TLR7 and TLR8 imidazoquinolines used in this study. Arthur M. Krieg is an employee of Coley Pharmaceutical Group which provided the CpG-ODN used in this study. Smita Ghanekar is an employee of BD Biosciences which provided anti-IFN-α and anti-COX-2 monoclonal antibodies. We thank Dr. Jeff Lifson from the AIDS Vaccine Program, National Cancer Institute (Frederick, MD) for kindly providing the AT-2 inactivated HIV-1 virus and matching microvesicles that were used as controls in these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janeway CA, Jr, Medzhitov R. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Almeida M, Cordero M, Almeida J, Orfao A. Aids. 2005;19:261–71. [PubMed] [Google Scholar]
- 3.Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, Sun Y, Megjugorac N, Fitzgerald-Bocarsly P. Clin Immunol. 2001;101:201–10. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- 4.Sandberg JK, Fast NM, Palacios EH, Fennelly G, Dobroszycki J, Palumbo P, Wiznia A, Grant RM, Bhardwaj N, Rosenberg MG, Nixon DF. J Virol. 2002;76:7528–34. doi: 10.1128/JVI.76.15.7528-7534.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finke JS, Shodell M, Shah K, Siegal FP, Steinman RM. J Clin Immunol. 2004;24:647–52. doi: 10.1007/s10875-004-6250-5. [DOI] [PubMed] [Google Scholar]
- 6.Soumelis V, Scott I, Gheyas F, Bouhour D, Cozon G, Cotte L, Huang L, Levy JA, Liu YJ. Blood. 2001;98:906–12. doi: 10.1182/blood.v98.4.906. [DOI] [PubMed] [Google Scholar]
- 7.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. J Exp Med. 2001;194:863–9. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. J Immunol. 2005;174:1259–68. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 9.Krieg AM. Annual Review of Immunology. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 10.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 11.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 12.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 13.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 14.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 15.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Science. 2004;303:1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 16.Gibson SJ, Lindh JM, Riter TR, Gleason RM, Rogers LM, Fuller AE, Oesterich JL, Gorden KB, Qiu X, McKane SW, Noelle RJ, Miller RL, Kedl RM, Fitzgerald-Bocarsly P, Tomai MA, Vasilakos JP. Cell Immunol. 2002;218:74–86. doi: 10.1016/s0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Amakawa R, Kaisho T, Hemmi H, Tajima K, Uehira K, Ozaki Y, Tomizawa H, Akira S, Fukuhara S. J Exp Med. 2002;195:1507–12. doi: 10.1084/jem.20020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 19.Lapenta C, Santini SM, Proietti E, Rizza P, Logozzi M, Spada M, Parlato S, Fais S, Pitha PM, Belardelli F. Virology. 1999;263:78–88. doi: 10.1006/viro.1999.9869. [DOI] [PubMed] [Google Scholar]
- 20.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 21.Bogdan C. Curr Opin Immunol. 2000;12:419–24. doi: 10.1016/s0952-7915(00)00111-4. [DOI] [PubMed] [Google Scholar]
- 22.Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Int J Biochem Cell Biol. 2006;38:1654–61. doi: 10.1016/j.biocel.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 23.Yeo SJ, Yoon JG, Yi AK. J Biol Chem. 2003;278:40590–600. doi: 10.1074/jbc.M306280200. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Zhang J, Moore SA, Ballas ZK, Portanova JP, Krieg AM, Berg DJ. Int Immunol. 2001;13:1013–20. doi: 10.1093/intimm/13.8.1013. [DOI] [PubMed] [Google Scholar]
- 25.Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. J Immunol. 2006;177:246–54. doi: 10.4049/jimmunol.177.1.246. [DOI] [PubMed] [Google Scholar]
- 26.Martinson JA, Tenorio AR, Montoya CJ, Al-Harthi L, Gichinga CN, Krieg AM, Baum LL, Landay AL. Immunology. 2007;120:526–35. doi: 10.1111/j.1365-2567.2007.02530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montoya CJ, Rugeles MT, Landay AL. Expert Rev Anti Infect Ther. 2006;4:767–80. doi: 10.1586/14787210.4.5.767. [DOI] [PubMed] [Google Scholar]
- 28.Soumelis V, Scott I, Liu YJ, Levy J. Hum Immunol. 2002;63:1206–12. doi: 10.1016/s0198-8859(02)00760-7. [DOI] [PubMed] [Google Scholar]
- 29.Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, Liu YJ, Lifson JD, Littman DR, Bhardwaj N. J Virol. 2004;78:5223–32. doi: 10.1128/JVI.78.10.5223-5232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yonezawa A, Morita R, Takaori-Kondo A, Kadowaki N, Kitawaki T, Hori T, Uchiyama T. J Virol. 2003;77:3777–84. doi: 10.1128/JVI.77.6.3777-3784.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao MJ, Lee N, Dipaola M, Hussain M, Brissette R, Ni D, Smith T, Desai M, Ferencz-Biro K, Testa D. J Interferon Res. 1994;14:183–5. doi: 10.1089/jir.1994.14.183. [DOI] [PubMed] [Google Scholar]
- 32.Dipaola M, Smith T, Ferencz-Biro K, Liao MJ, Testa D. J Interferon Res. 1994;14:325–32. doi: 10.1089/jir.1994.14.325. [DOI] [PubMed] [Google Scholar]
- 33.Lydon NB, Favre C, Bove S, Neyret O, Benureau S, Levine AM, Seelig GF, Nagabhushan TL, Trotta PP. Biochemistry. 1985;24:4131–41. doi: 10.1021/bi00336a048. [DOI] [PubMed] [Google Scholar]
- 34.Bess JW, Jr, Gorelick RJ, Bosche WJ, Henderson LE, Arthur LO. Virology. 1997;230:134–44. doi: 10.1006/viro.1997.8499. [DOI] [PubMed] [Google Scholar]
- 35.Chehimi J, Campbell DE, Azzoni L, Bacheller D, Papasavvas E, Jerandi G, Mounzer K, Kostman J, Trinchieri G, Montaner LJ. J Immunol. 2002;168:4796–801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 36.Del Corno M, Gauzzi MC, Penna G, Belardelli F, Adorini L, Gessani S. J Virol. 2005;79:12597–601. doi: 10.1128/JVI.79.19.12597-12601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, Roederer M, Seder RA, Koup RA. J Immunol. 2003;171:4320–8. doi: 10.4049/jimmunol.171.8.4320. [DOI] [PubMed] [Google Scholar]
- 38.Sloand E. AIDS Rev. 2005;7:187–96. [PubMed] [Google Scholar]
- 39.Sirianni MC, Tagliaferri F, Aiuti F. Immunol Today. 1990;11:81–2. doi: 10.1016/0167-5699(90)90032-5. [DOI] [PubMed] [Google Scholar]
- 40.Mansour I, Doinel C, Rouger P. AIDS Res Hum Retroviruses. 1990;6:1451–7. doi: 10.1089/aid.1990.6.1451. [DOI] [PubMed] [Google Scholar]
- 41.Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. Eur J Immunol. 2001;31:2154–63. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 42.Hu PF, Hultin LE, Hultin P, Hausner MA, Hirji K, Jewett A, Bonavida B, Detels R, Giorgi JV. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:331–40. [PubMed] [Google Scholar]
- 43.Douek DC, Betts MR, Hill BJ, Little SJ, Lempicki R, Metcalf JA, Casazza J, Yoder C, Adelsberger JW, Stevens RA, Baseler MW, Keiser P, Richman DD, Davey RT, Koup RA. J Immunol. 2001;167:6663–8. doi: 10.4049/jimmunol.167.11.6663. [DOI] [PubMed] [Google Scholar]
- 44.Jiang W, Lederman MM, Salkowitz JR, Rodriguez B, Harding CV, Sieg SF. J Virol. 2005;79:4109–19. doi: 10.1128/JVI.79.7.4109-4119.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brassard DL, Grace MJ, Bordens RW. J Leukoc Biol. 2002;71:565–81. [PubMed] [Google Scholar]
- 46.Herbeuval JP, Boasso A, Grivel JC, Hardy AW, Anderson SA, Dolan MJ, Chougnet C, Lifson JD, Shearer GM. Blood. 2005;105:2458–64. doi: 10.1182/blood-2004-08-3058. [DOI] [PubMed] [Google Scholar]
- 47.Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, Hartmann G. Eur J Immunol. 2001;31:3026–37. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 48.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. Immunity. 2004;21:107–19. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, Trinchieri G. J Immunol. 2005;174:727–34. doi: 10.4049/jimmunol.174.2.727. [DOI] [PubMed] [Google Scholar]
- 50.Weber AA, Zimmermann KC, Meyer-Kirchrath J, Schror K. Lancet. 1999;353:900. doi: 10.1016/S0140-6736(99)00498-5. [DOI] [PubMed] [Google Scholar]
- 51.Hoozemans JJ, O’Banion MK. Curr Drug Targets CNS Neurol Disord. 2005;4:307–15. doi: 10.2174/1568007054038201. [DOI] [PubMed] [Google Scholar]