

Abstract
The candidate tumor suppressor gene, FHIT, encompasses the common human chromosomal fragile site at 3p14.2, the hereditary renal cancer translocation breakpoint, and cancer cell homozygous deletions. Fhit hydrolyzes dinucleotide 5′,5‴-P1,P3-triphosphate in vitro and mutation of a central histidine abolishes hydrolase activity. To study Fhit function, wild-type and mutant FHIT genes were transfected into cancer cell lines that lacked endogenous Fhit. No consistent effect of exogenous Fhit on growth in culture was observed, but Fhit and hydrolase “dead” Fhit mutant proteins suppressed tumorigenicity in nude mice, indicating that 5′,5‴-P1,P3-triphosphate hydrolysis is not required for tumor suppression.
The structure and expression of the FHIT gene encompassing the FRA3B common fragile site frequently are altered in primary or cultured esophageal, head and neck, lung, gastric, breast, and cervical carcinomas (1–8). Structural alterations tend to be because of deletion within both FHIT alleles, resulting in loss of exons and concomitant absence of full-length FHIT transcript and protein (ref. 6; for review, ref. 9). It has been argued that the FHIT gene may be altered in cancer cells simply because it encompasses the fragile region and is likely to be very susceptible to breakage (7). We agree that the locus is highly susceptible to carcinogen damage, explaining why deletion is much more frequent than point mutation in the gene, but we argue that loss of Fhit function provides a selective advantage for the tumor cell; otherwise, frequent expansion of the deleted FHIT clones in tumors and tumor-derived cell lines would be difficult to explain.
Fhit-related proteins have been found in mammals and yeasts (1, 10, 11) and constitute a branch of the histidine triad (HIT) superfamily (12). The Fhit branch includes the Schizosaccharomyces pombe diadenosine tetraphosphate hydrolase [dinucleoside 5′,5‴-P1,P4-tetraphosphate (Ap4A) hydrolase] (10) to which Fhit is similar. Barnes et al. (13) have shown that Fhit behaves in vitro as a typical dinucleoside 5′,5‴-P1,P3-triphosphate (Ap3A) hydrolase (EC 3.6.1.29); site-directed mutagenesis of FHIT demonstrated that the conserved histidines are required for full activity, and the central histidine of the triad is essential for Ap3A hydrolase activity.
To investigate mechanisms for a selective growth advantage of Fhit negative tumors, we have prepared vectors for expression of Fhit in cancer-derived cells and have examined the phenotypes of the Fhit-expressing clones relative to the Fhit negative parental cells. To determine if the in vitro enzymatic activity was associated with a role in tumor suppression, the hydrolase “dead” mutant gene, FHITH96N, with the central histidine codon of the HIT changed to an asparagine codon, also was expressed in Fhit negative cancer cells.
MATERIALS AND METHODS
Cells.
The MKN74 cell line (kindly provided by Eiichi Tahara, University of Hiroshima, Japan), was derived from a gastric carcinoma (14) and forms tumors rapidly in nude mice. The H460 large cell lung cancer line exhibits homozygous deletion in intron 5, does not express Fhit protein by Western blot or immunohistochemistry, and is highly tumorigenic in nude mice. Human cell lines, AGS, HKI (15), RC48 (16), and 293 (17), were characterized previously for structure and expression of endogenous FHIT alleles (6); 293 cells express Fhit protein, whereas the three cancer cell lines exhibit homozygous deletions within FHIT and do not express Fhit protein.
Plasmids.
Wild-type FHIT cDNA (1) was ligated in-frame to a FLAG octapeptide coding sequence (Eastman Kodak) and cloned into the HindIII and XbaI sites of the pRcCMV vector (Invitrogen) under control of the immediate early human cytomegalovirus (CMV) promoter (13). Codon 96 (CAC, His) was mutated to AAC (Asn) via site-directed mutagenesis to create the plasmid pRcFHITH96N; expression of Fhit wild-type and mutant proteins from these vectors was described (13).
Transfection.
Four Fhit negative cancer-derived cells, and the Fhit positive 293 cells, were transfected with 15 μg of pRcFHIT or control plasmid (pRcCMV) DNA by using the calcium phosphate method (GIBCO/BRL); the AGS, RC48, MKN74, and 293 cell lines also were transfected with the pRcFHITH96N plasmid. Clones were selected in the presence of 200–400 μg/ml G418 (Geneticin; GIBCO/BRL).
For electroporation, exponentially growing H460 cells (1.5 × 107) were resuspended in 1 ml of DMEM supplemented with 50% fetal bovine serum (FBS), mixed with 50 μg of plasmid DNA, and incubated at 4°C for 15 min. Electroporation was performed with a Bio-Rad gene pulser by using a setting of 960 μF and 250 V; cells were plated in DMEM supplemented with 10% FBS and 700 μg/ml of G418 and cultured for 3 weeks.
Western Blot Analysis.
Preparation of cell lysates and Western blots were described previously (13). Fhit proteins were detected by using anti-FLAG M2 monoclonal IgG or rabbit polyclonal anti-Fhit, and signal was detected by using the SuperSignal chemiluminescent substrate (Pierce). Two rabbit polyclonal antisera were used: the previously described anti-glutathione S-transferase (GST)-Fhit antisera (6, 13) for immunoblot and immunohistochemistry of the H460 clones; and a polyclonal anti-Fhit antiserum commercially prepared against purified Fhit and used at 1:5,000 in other immunoblot experiments.
Immunoprecipitation.
Protein was immunoprecipitated from lysates with the anti-FLAG monoclonal IgG, electrophoresed on a 12% SDS/polyacrylamide gel, transferred to nitrocellulose filters, and protein detected with the indicated antisera. Immunoreactive bands were visualized by using horseradish peroxidase-conjugated secondary antiserum and enhanced chemiluminescence (Amersham). One microgram per milliliter of anti-FLAG M2 mAb (Eastman Kodak) was used both for immunoprecipitation and immunoblot detection, and anti-GST-Fhit polyclonal antibody (6) was used at a 1:500 dilution for detection of precipitated Fhit-FLAG in immunoblots.
Assay of Dinucleoside Polyphosphate Hydrolase Activity.
Transfected cells were assayed for Ap3A hydrolase activity as described (13). Crude supernatants were assayed in duplicate at three different protein masses in an experiment, and the specific activity, expressed as nmol of Ap3A hydrolyzed/min per mg of protein, was calculated from a linear regression analysis of the activity (Ap3A hydrolyzed/min) as a function of protein mass. Each experiment was done two or three times, and mean values are reported.
Growth Kinetics.
For growth rate determination, cells were seeded in 12-well plates in MEM with 10% FBS and 200 μg/ml G418. The medium was replaced with fresh medium every 48 hr. Every 24 hr, cells were trypsinized and counted. For low serum growth cells were seeded in medium containing 10% FBS, and the medium was replaced with 1% FBS 24 hr later.
Soft Agar Growth.
The soft agar culture was composed of an underlay (MEM, 10% FBS, G418, 0.6% agar) and overlay (MEM, 10% FBS, G418, 0.3% agar). The overlay included 1 × 103 or 1.5 × 103 (H460) cells in 2 ml for each 35-mm plate. RC48 and AGS colonies were counted at 8 weeks, MKN74 and H460 colonies at 18 days.
Tumorigenicity Assay.
Transfected clones from tissue culture passages 3–5 were harvested and resuspended in PBS. One (AGS), two (H460), or 5 × 106 (MKN74, RC48, HK1, 293) cells were injected subcutaneously between the shoulder blades or into the right flank of 6-week-old female nude mice, four or five mice per cell line. The animals were monitored twice weekly for tumor formation up to 4 months after inoculation. Tumor growth rate was calculated from tumor volume data, obtained from measurements of tumor dimensions by using linear calipers. At termination of the experiment tumors were excised for histology, assessment of Fhit expression, and, in some cases, growth in tissue culture.
Immunohistochemistry.
The immunoperoxidase assay was carried out with an avidin-biotin peroxidase complex (ABC) kit (Vector Laboratories). Briefly, trypsinized cells were spun onto slides in a cytocentrifuge and fixed in cold acetone for 10 min. The fixed cytospun cells were rehydrated and incubated for 1 hr at room temperature with anti-GST-Fhit polyclonal serum diluted 1:500. After rinsing in PBS, the slides were processed according to the kit staining procedure.
PCR and Reverse Transcription (RT)–PCR Amplification.
Total RNA isolation and RT-PCR amplification was as described (6). For RT 250 pM of dT or SP6 oligonucleotide primers were used and 5 μl of the RT product or 100 ng of genomic DNA served as templates for PCR amplification with 25 pM of primers HITF and HITR from FHIT exons 5 and 9 (13). The amplifications were performed in a Perkin–Elmer Cetus thermal cycler for 30 cycles of 94°C for 45 sec, 58°C for 45 sec, and 72°C for 45 sec.
RESULTS
Characteristics of Parental Cancer Cell Lines for Transfection.
There were several criteria for selection of cancer cell lines for expression of exogenous Fhit. First, it was important that the parental cancer cell lines should have endogenous FHIT alleles inactivated; thus, we chose cell lines exhibiting homozygous deletion of FHIT coding exons (6) and/or no detectable Fhit protein expression. Second, we wanted cell lines derived from cancers originating from various organs, and third, the chosen parental cancer cell lines should be tumorigenic in nude mice. The HKI nasopharyngeal and AGS gastric carcinoma cell lines lack exon 5 and are reportedly tumorigenic (15), and the RC48 renal carcinoma lacks exons 8–10 (6). The MKN74 gastric cancer cell line was tested for deletion within the FHIT gene by PCR amplification of FHIT locus markers encompassing mid intron 4 to distal intron 5, and homozygous deletion of exon 5 was observed (data not shown). The H460 large cell lung cancer line exhibits homozygous deletion of markers in intron 5, but each FHIT exon is represented in H460 DNA. MKN74 and H460 cells are negative for endogenous Fhit protein by Western blot and immunocytochemistry, as will be shown below. The 293 adenovirus 5 transformed human kidney cell line expresses endogenous Fhit and is nontumorigenic in early passage.
Expression of Fhit in Transfected Clones.
The AGS, RC48, MKN74, and 293 cells were transfected with each of the three vectors, pRcCMV control vector, pRcFHIT, and pRcFHITH96N, whereas the HK1 and H460 cells were transfected with the control and wild-type vectors, as reflected in Table 1.
Table 1.
Characterization of FHIT transfected cancer cells
| Cell | Transfection parameters
|
Growth parameters
|
||||||
|---|---|---|---|---|---|---|---|---|
| Total | Number | Number | Soft agar col/plate | Doubling time, hr
|
Tumors, cm3
|
|||
| colonies/plate | analyzed | expressing Fhit | 10% FBS | 1% FBS | Number | Vol | ||
| AGS/CMV | 240 | 2 | 0 | 43 (cl1) | 37.4 | 60.1 | 3/4 | .48 |
| AGS/FHIT | 121 | 86 | 2 (cl25, cl40) | 61 (cl25) | 36.1 | 43.6 | 0/4 | |
| AGS/FHITH96N | 112 | 57 | 0 | NA | NA | NA | ||
| HK1/CMV | 107 | 2 | 0 | 0 | 52.8 | 67.2 | No tumors | |
| HK1/FHIT | 58 | 36 | 1 | 0 | 48.3 | 54.6 | ||
| RC48/CMV | 105 | 2 | 0 | 53 (cl1) | 49.5 | 58.1 | 3/4 | .50 |
| RC48/FHIT | 68 | 67 | 2 (cl15, cl31) | 66 (cl15) | 43.2 | 50.2 | 1/5 | 0.2 |
| RC48/FHITH96N | 4 | 4 | 1 | NT | 46.5 | 50.8 | ||
| H460/CMV | >200 | 2 | 0 | 26 (mean) | 20 | NT | 9/10 | 1.2 |
| H460/FHIT | ≪100 | 34 | 4 (cl1.2, | 6 (mean) | NT | NT | cl1.2, 0/5 | |
| cl2.1, | 40 | NT | cl2.1, 0/5 | |||||
| cl3.2, | NT | NT | cl3.2, 3/5 | 0.1 | ||||
| cl2.3) | NT | NT | cl2.3, 4/4 | 0.6 | ||||
| MKN74/CMV | 259 | 4 | 0 | 37 (mean) | NT | NT | P, 5/5 | .30 |
| E4, 4/4 | .35 | |||||||
| MKN74/FHIT | 141 | 116 | 10 | 35 (mean) | NT | NT | A66, 5/5 | .05 |
| A116, 5/5 | .10 | |||||||
| MKN74/FHITH96N | 223 | 92 | 7 | 10 (mean) | NT | NT | B27, 3/5 | .05 |
| B59, 5/5 | .13 | |||||||
| 293/CMV | 118 | 2 | 0 | 3 | 35.4 | 43.2 | No tumors | |
| 293/FHIT | 114 | 55 | 20 | 5 (cl56) | 31.2 | 36.8 | ||
| 293/FHITH96N | 108 | 18 | 15 | NT | 33.6 (cl24) | 40.1 | ||
Numbers in parentheses are specific clone names; NA, not applicable; NT, not tested; Number analyzed means number of colonies tested for Fhit expression by Western blot analysis with anti-FLAG IgG. For RC48/FHITH96N the low number of colonies observed was probably because of an inefficient transfection.
Two to three weeks after transfection, colonies were counted and colonies from each transfection were isolated, grown, and tested for Fhit expression, as summarized in Table 1. The numbers of colonies obtained after transfection with the FHIT wild-type or mutant vectors were reduced, usually about 2-fold, compared with the empty vector transfections except in the 293 transfections, where each vector yielded about the same number of colonies. Additionally, FHIT and mutant transfectants rarely expressed the Fhit-FLAG or FhitH96N-FLAG protein, as detected by immunoblot analysis of stable transfectant cell lysates by using anti-FLAG mAb (results shown in Fig. 1 Upper, summarized in Table 1). For example, of 86 AGS/FHIT clones, only two expressed detectable Fhit protein, and none of the 57 AGS/FHITH96N clones expressed the Fhit mutant. Conversely, the 293 stable clones were about equal in number after transfection with the three vectors, and many of the FHIT wild-type and mutant clones expressed the exogenous protein. For the MKN74 and H460 cell lines we obtained several Fhit-FLAG expressing clones, as summarized in Table 1 and illustrated in Fig. 1 (Upper), which shows immunoblotting results, and Fig. 1 (Lower) with immunocytochemical detection of Fhit in H460 transfectants.
Figure 1.
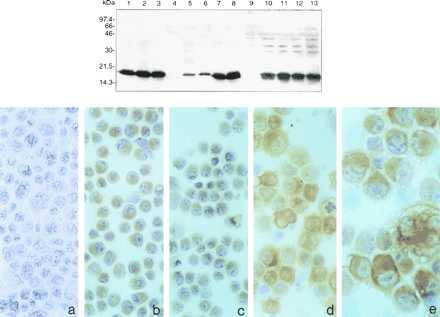
Fhit expression in stably transfected cell lines. (Upper) Detection of Fhit expression of transfectant clones after immunoblotting of 50–100 μg/lane of protein from the following lysates: Lane 1, control Cos cells transiently transfected with pRcFHIT; lane 2, RC48/FHIT cl15; lane 3, RC48/FHITH96N cl1; lane 4, RC48/CMV, lane 5, HKI/FHIT cl26; lane 6, AGS/FHIT cl25; lane 7, 293/FHIT cl56; lane 8, 293/FHITH96N cl24; lane 9, MKN74/CMV; lane 10, MKN74/FHIT clA66; lane 11, MKN74/FHIT clA116; lane 12, MKN74/FHITH96N clB27; and lane 13, MKN74/FHITH96N clB59. Protein in lanes 1–8 was detected by using the monoclonal M2 anti-FLAG antibody; protein in lanes 9–13 was detected by using rabbit polyclonal anti-Fhit antiserum (1:5,000). (Lower) Detection of Fhit expression by immunocytochemistry. Cytospins of H460 cells and pRcFHIT transfected clones were tested for Fhit-FLAG expression by immunocytochemistry by using rabbit polyclonal anti-GST Fhit antiserum (1:500) followed by detection by using immunocoupled horseradish peroxidase: (a) H460 cells; (b) Fhit expressing transfected clone 1.2; (c) clone 3.2; (d) clone 2.I; and (e) clone 2.3.
To determine the status of the transfected FHIT gene in the majority of G418 resistant colonies that did not express detectable Fhit-FLAG, three MKN/FHIT and three MKN/FHITH96N clones that did not express Fhit protein were analyzed further. The three MKN/FHIT clones did not retain an intact FHIT cDNA as determined by attempted amplification of the cDNA ORF from the transfectants. Similarly, two of three MKN/FHITH96N clones did not retain an intact FHITH96N cDNA, an indication that in these five clones, the vector may have integrated into cellular DNA through the FHIT portion of the transfecting plasmid DNA. The remaining MKN/FHITH96N clone, which did retain an intact FHIT cDNA, also expressed FHITH96N mRNA, as determined by RT-PCR, with FHIT exon 5 and 9 primers. For this clone the lack of FhitH96N expression may have been caused by a posttranscriptional block or a very low level of expression.
Ap3A Hydrolase Activity in Stable Fhit-FLAG Transfectants.
Previous studies had shown that transiently expressed Fhit-FLAG fusion proteins were enzymatically active in Ap3A hydrolysis, whereas the FhitH96N-FLAG mutant protein lacked detectable enzymatic activity (13). Similarly, crude supernatants from wild-type and mutant stably transfected cell clones were tested for Ap3A enzymatic activity, and results are summarized in Table 2. The specific activities in the 293/FHIT transfectants are 14–55 times larger than the control. Activities in RC48/FHIT and MKN74/FHIT transfectants are at least 24 times larger than the activities in the corresponding control cells. However, activities in HK1/FHIT and AGS/FHIT transfectants are only two and four times larger, respectively, than activities in corresponding control cells. The level of enzymic activity detected for each cell line roughly paralleled the level of Fhit protein detected by Western blot: 293/FHIT>RC48/FHIT>MKN74/FHIT>AGS/FHIT>HK1/FHIT (Fig. 1). Activities in the FHITH96N transfectants of 293 and MKN74 cells are similar to those of the 293 and MKN74 control cells, respectively. Thus, Ap3A hydrolase activity can be readily detected in stable cancer cell transfectants.
Table 2.
Ap3A hydrolase activity in transfected clones
| Transfected cell lines | Specific activity
|
|---|---|
| nmol Ap3A hydrolyzed/min per mg | |
| 293/CMV | 0.62 |
| 293/FHIT clNK3 | 24.2 |
| cl27 | 28.8 |
| cl44 | 8.45 |
| cl56 | 34.0 |
| 293/FHIT H96N cl3 | 0.62 |
| cl4 | 1.52 |
| cl11 | 0.70 |
| cl24 | 1.66 |
| cl30 | 1.67 |
| RC48/CMV | 0.74 |
| RC48/FHIT cl15 | 23.3 |
| AGS/CMV | 1.40 |
| AGS/FHIT | 6.10 |
| HK1/CMV | 0.99 |
| HK1/FHIT | 1.99 |
| MKN74/CMV | 0.36 |
| MKN74/FHIT A66 | 8.55 |
| MKN74/FHIT A116 | 13.6 |
| MKN74/FHIT H96N B27 | 0.42 |
| MKN74/FHIT H96N B59 | 0.40 |
Phenotype of FHIT Transfectants.
To determine if expression of exogenous wild-type or mutant Fhit affects ability of the stable transfectants to grow, transfected clones were tested for growth in liquid medium supplemented with 10% or 1% FBS, in 0.3% agar and for ability to form tumors in nude mice. Transfectant cell lines expressing Fhit, FhitH96N, and the vector alone were grown continuously in the presence of 10% or 1% FBS. For the stable transfected clones of each of four cell lines, 293, HKI, RC48, and AGS, we found no significant difference in the growth rate among wild-type, mutant, and vector transfected cells in 10% FBS (see Table 1). In 1% serum, cells stably expressing Fhit or Fhit mutant showed a slightly decreased doubling time compared with control vector transfectants grown under the same conditions (see Table 1). In contrast to the calcium phosphate-transfected Fhit expressing clones, an H460/FHIT stable transfectant expressing Fhit showed reduced growth ability with a doubling time of 40 hr compared with the parental H460 cells with a doubling time of 20 hr (see Table 1).
Transfected cell lines then were examined for anchorage-independent growth. The soft agar assay for AGS- and RC48-transfected cell lines showed no significant difference in numbers of colonies formed by the FHIT or empty vector transfectants (Table 1), but AGS and RC48 cells expressing Fhit formed larger colonies compared with control vector clones. HKI and 293 cells, transfected with either FHIT or control vector, demonstrated no colony formation in the soft agar assay. The H460 stably transfected cell clones showed reduction in efficiency and size of colony growth for Fhit expressing H460 transfected clones vs. H460 control vector transfectants (see Table 1).
Because in vitro results do not necessarily predict in vivo behavior, we implanted 1, 2, or 5 × 106 tumor-derived cells, with or without Fhit expression, on the shoulder or flank of 6-week-old female nude mice. The tumorigenicity study of AGS cells showed that three of four mice injected with 1 × 106 cells not expressing Fhit (AGS/CMV) formed tumors within 9 weeks, and the average size of tumors reached ≈0.5 cm3 volume after 13 weeks. In contrast, no tumors were observed after injection of 1 × 106 AGS/FHIT cl25 cells. RC48 cells (5 × 106/mouse) formed tumors in 3 of 4 mice in 2 months, whereas the RC48/FHIT expressing clone produced a tumor in 1 of 5 mice (Table 1). HK1 and 293 parental cells did not form tumors in the time period observed (from 3 to 6 months).
MKN74 gastric cancer transfected clones all formed tumors after injection of 5 × 106 cells but tumors from Fhit wild-type or mutant-expressing clones were smaller and grew more slowly than the MKN74 control vector transfectants (see Fig. 2 a and b). The tumors appeared about 10 days after injection and were observed and measured every 2 days up to 3 weeks after injection. Results in Fig. 2a show that the MKN74 cells not expressing Fhit reached an average size of ≈0.3 cm3 at day 20, whereas Fhit and Fhit mutant expressing clones reached an average size of 0.1 cm3 or less in the same time, as shown photographically in Fig. 2b for excised tumors. The tumors were all similar histologically, with an undifferentiated phenotype.
Figure 2.
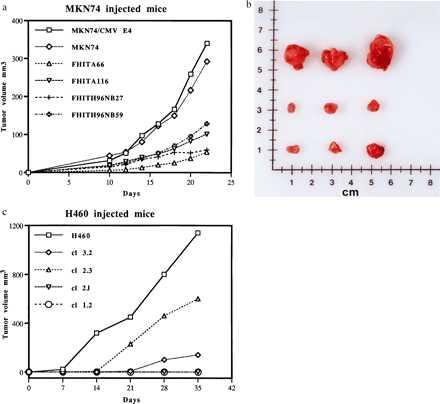
Stable Fhit wild-type and H96N mutant expression in transfected cancer cell clones suppresses tumor growth in nude mice. (a) MKN74 transfectants. Two MKN74 clones stably expressing wild-type or H96N mutant Fhit protein were injected (5 × 106 cells/mouse) into nude mice (5 mice per cell line), and tumor growth was compared with growth in mice injected with 5 × 106 MKN74 parental cells or a MKN/CMV clone. Tumor dimensions were measured for each mouse over a 3-week period, and tumor volumes were determined. Volumes were averaged for each time point for the five mice injected with the individual clones. Although there is clonal variation in size of tumors, all tumors of Fhit wild-type or mutant expressor clones were two to three times smaller than the MKN74 or MKN74/CMV tumors. (b) MKN74 tumors. (Top) MKN74/CMV tumors. (Middle) MKN74/FHIT tumors. (Bottom) MKN74/FHITH96N mutant tumors. (c) H460 transfectants. Nude mice were inoculated in the right flank with 2 × 106 H460 cells (10 mice) or 2 × 106 cells of individual Fhit expressing clones (5 mice each) and mice were observed over a 5-week period. Animals without tumors were not included in calculation of average tumor volumes.
Representative MKN74/FHIT tumors were excised for each clone, and the level of Fhit protein was assessed by immunoblot of equal amounts of protein from the MKN74/FHIT and MKN74/FHITH96N tumors compared with the MKN/FHIT clA66 parental clone. The level of Fhit protein detected in each of the tumors was less than detected in lysate from the MKN74/FHIT clA66 parental cells (not shown), consistent with the idea that growth of these Fhit-expressing clones, including the hydrolase “dead” mutant expressors, selects for cells expressing less Fhit.
For the H460 transfectants, four wild-type Fhit-expressing clones (2 × 106/mouse) were injected into four or five mice each, and growth of tumors in these mice was compared with growth of tumors after injection of Fhit negative H460 cells. Two Fhit-expressing clones, clones 1.2 and 2.I did not form tumors in mice by 5 weeks after injection. Clone 3.2 formed very small tumors (average size, ≈0.1 cm3) in 3 of 5 mice at 5 weeks, and clone 2.3 formed tumors of an average volume of 0.6 cm3 in 4 of 4 mice compared with the 1.2 cm3 tumors formed in 9 of 10 mice injected with Fhit negative H460 cells. These results are presented in graphic form in Fig. 2c.
Several of the H460 tumors were excised, and the expression of exogenous Fhit was evaluated by immunocytochemistry, by using anti-GST-Fhit polyclonal antibody, and by immunoprecipitation with anti-FLAG IgG, either on primary tumors or cell lines established from individual tumors. Tumor-derived cell lines from clone 3.2 showed absence of the Fhit protein in both assays (not shown), whereas those from clone 2.3 expressed less Fhit protein (Fig. 3). Fhit protein expression was barely detectable with anti-Flag and anti-GST-Fhit antibodies after immunoprecipitation of cell lysates prepared from the cell lines established from tumors (not shown). Immunohistochemical analysis revealed that cells established from clone 2.3 tumors were only 10–20% Fhit-positive compared with 50–60% positive cells observed in the corresponding 2.3 parental clone (Fig. 3). Conversely, parental clone 2.I, which was completely suppressed for in vivo tumorigenicity, showed more than 90% positive cells (Fig. 3). These results suggest that selective pressure against Fhit expression had occurred during in vitro and in vivo passage of clone 2.3 cells. Thus, the degree of tumor growth inhibition by transfectants may be dependent on the level of expression of the Fhit protein.
Figure 3.
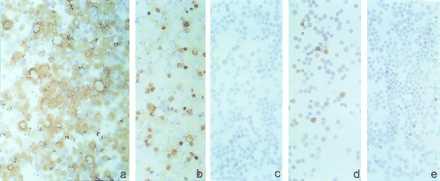
Immunocytochemical detection of Fhit in tumor-derived cells. Fhit protein expression, evaluated by immunocytochemistry, of clone 2.I (a), clone 2.3 after several tissue culture passages (b), and cultured cells from tumors of mice injected with clone 2.3 (c–e).
DISCUSSION
In 1996 the FHIT gene, at human chromosome region 3p14.2, was cloned and shown to be a large gene that encompassed the FRA3B common fragile site, numerous cancer cell-specific homozygous and hemizygous deletions, and a familial renal cancer chromosome translocation break (1, 18). The gene is ≈1 Mb in size and encodes a 1.1-kb cDNA with 10 small exons; exon 5 is the first protein coding exon and is flanked in intron 4 and intron 5 by the specific FRA3B fragile sites thus far described (19–22). Early studies of a number of important human tumor types showed that FHIT RNA expression frequently was altered (1–3, 5, 23), and the alterations to RNA expression were shown to correlate with deletions within the FHIT gene (6). Furthermore, lack of detectable Fhit protein in cancer-derived cell lines was correlated with FHIT gene deletions and altered RNA expression; that is, cell lines derived from gastric, cervical, nasopharyngeal, head and neck, and breast cancers did not express Fhit protein (6). Cancer cell-specific homozygous deletions within a gene and lack of expression of the protein product are hallmarks of tumor suppressor genes; so from many angles the FHIT gene was a strong candidate tumor suppressor gene that could play an important role in major human cancers. Because many tumor deletion endpoints were near positions of fragile sites in introns 4 and 5 and the deletions occurred frequently in tumors that are known to be carcinogen-induced, such as lung cancer (24), we proposed that the FRA3B was particularly susceptible to chromosome breaks caused by environmental factors.
There were, however, aspects of the FHIT locus alterations in cancer cells that were puzzling when viewed in the framework of knowledge generated by classical tumor suppressor genes thus far studied. The major puzzles were: occurrence of small homozygous deletions within large FHIT introns, apparently not affecting FHIT exons; occurrence of apparent full-length FHIT transcripts accompanied by aberrant transcripts in cancer-derived cell lines and tumors; lack of point mutations in remaining FHIT alleles in cancer-derived cell lines; multiple noncontiguous homozygous deletions within the FHIT locus in some cancer-derived cell lines; and oligoclonality of FHIT genomic alterations in some tumor-derived cell lines.
We do not have complete answers to each of these puzzles, but partial explanations follow from the presence of the most inducible of the common fragile regions, FRA3B, within the FHIT gene. The fragility of the FHIT gene, we believe, accounts for the fact that deletions in both alleles of the gene occur far more frequently than loss of one allele and mutation of the other. We already have shown that apparent homozygous deletions in introns are because of overlapping deletions on the two alleles, with each allele carrying hemizygous deletions of exons (6). The nature of damages to the locus, which has not been completely elucidated but presumably involves double-strand breakage and incorrect repair, also may account for the multiple, noncontiguous deletions seen in some cells; that is, maybe two or three regions of one allele can be deleted simultaneously. The oligoclonality, vis-a-vis the FHIT locus, observed in some long-term, cultured cancer-derived cell lines (2, 6) may suggest that loss of Fhit function does not impart a very strong selective pressure in vitro for overgrowth by Fhit minus cells; it also suggests that some primary tumors, from which the oligoclonal cell lines were derived, were not monoclonal for alterations in the FHIT locus. Nevertheless, there was clonal expansion of Fhit minus cells within many tumors, as reflected in the ability to define FHIT alterations, if monoclonal, biclonal, or oligoclonal, in many primary and cultured tumors. Thus, Fhit inactivation provides a selective advantage for clonal expansion in vivo. Others have argued that, because the FHIT introns are very large (>200 kb), the FHIT gene may not be the target of the frequent deletions within the gene, although we and others (7) have looked for other genes without success.
As part of our study of Fhit function, especially as a tumor suppressor, we have expressed exogenous wild-type and mutant Fhit proteins in tumorigenic Fhit minus lung, kidney, and gastric cancer-derived cells and have studied the affect of Fhit expression on growth of the tumor cell clones. For each of four carcinoma-derived cell lines, Fhit expression eliminated or reduced the tumorigenicity of the cancer cells in nude mice. We conclude that Fhit is indeed a tumor suppressor.
The AGS gastric cancer cell clones were the first transfectants analyzed. Only two Fhit expressing clones were isolated, and one of these was assessed for growth and tumorigenicity. The AGS/FHIT cl25 cells grew at least as well, and in 1% FBS better, than the AGS/CMV control clone in tissue culture; nevertheless on injection of 1 × 106 cells per nude mouse, the AGS/FHIT clone produced no tumors, whereas the control vector transfected clone produced sizable tumors in 3 of 4 mice. Similarly, the HK1 and RC48 Fhit expressing clones grew as well as the nonexpressing parental clones in liquid and semisolid medium, but the RC48/FHIT clone was less tumorigenic than the RC48 parental Fhit negative cells. It is likely that a high level of Fhit expression does affect cell survival or growth, as indicated by the very low yield of cancer cell transfectants expressing wild-type or mutant Fhit. It could be that we have, by selecting for constitutive Fhit expression, selected for the fittest Fhit expressors, or it could be that a low level of Fhit expression is compatible with vigorous growth of some cell types in tissue culture. Some of the questions raised as a result of these experiments may be answered by selection of conditionally expressing Fhit transfectants.
For our second round of tumorigenicity experiments we selected two cell lines, H460 and MKN74, which we knew to be highly tumorigenic in nude mice. We also selected and tested many colonies from these transfections to have a panel of Fhit-expressing clones to test for tumorigenicity. For both of these faster-growing tumor cell lines, Fhit expression either suppressed tumor growth completely (H460/FHIT clones 1.2 and 2.I) or slowed the tumor growth significantly. One of the surprising conclusions of the experiments with the MKN74 cells was that the FHITH96N mutant, which has no detectable Ap3A hydrolase activity, suppresses tumorigenicity as well as wild-type Fhit, suggesting that perhaps substrate binding or binding of Fhit to an interacting protein is more important in tumor suppression than hydrolytic activity.
That for 293 cells exogenous Fhit did not affect growth of the cells, as reflected in the high yield of colonies and Fhit expressors among the colonies, is comparable to the report that transfection with wild-type p53 has no effect on growth of benign colorectal tumor cells expressing endogenous wild-type p53 (25) and demonstrates that Fhit overexpression does not result in generalized toxicity.
The H460 transfectants behaved somewhat differently than the other transfectants, in that there was a significant decrease in growth in liquid medium and in efficiency of soft agar growth for H460/FHIT clones. This decrease may be related to the specific cell line, a specific histotype or the transfection method used, electroporation, which may result in higher copy number of transfecting plasmids and higher Fhit protein expression.
Acknowledgments
This work was supported by U.S. Public Health Service Grants CA56336, CA51083 and CA21124, a gift from R.R.M. Carpenter, III and Mrs. Mary K. Carpenter, National Science Foundation Grant MCB-9604124 (to L.D.B.), the Italian Association for Cancer Research (AIRC), and the National Research Council (CNR) of Italy.
ABBREVIATIONS
- Ap3A
5′,5‴-P1,P3-triphosphate
- HIT
histidine triad
- CMV
cytomegalovirus
- FBS
fetal bovine serum
- RT
reverse transcription
- GST
glutathione S-transferase
References
- 1.Ohta M, Inoue H, Cotticelli M G, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce C M, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 2.Virgilio L, Shuster M, Gollin S M, Veronese M L, Ohta M, Huebner K, Croce C M. Proc Natl Acad Sci USA. 1996;93:9770–9775. doi: 10.1073/pnas.93.18.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sozzi G, Veronese M L, Negrini M, Baffa R, Cotticelli M G, Inoue H, Tornielli S, Pilotti S, DeGregorio L, Pastorino V, Pierotti M A, Ohta M, Huebner K, Croce C M. Cell. 1996;85:17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- 4.Fong K M, Biesterveld E J, Virmani A, Wistuba I, Sekido Y, Bader S A, Ahmadian M, Ong S T, Rassool F V, Zimmerman P V, Ciaccone G, Gazdar A F, Minna J A. Cancer Res. 1997;57:2256–2267. [PubMed] [Google Scholar]
- 5.Negrini M, Monaco C, Vorechovsky I, Ohta M, Druck T, Baffa R, Huebner K, Croce C M. Cancer Res. 1996;56:3173–3179. [PubMed] [Google Scholar]
- 6.Druck T, Hadaczek P, Fu T-B, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese M L, Rosen D, Rothstein J, McCue P, Cotticelli M G, Inoue H, Croce C M, Huebner K. Cancer Res. 1997;57:504–512. [PubMed] [Google Scholar]
- 7.Boldog F, Gemmill R M, West J, Robinson M, Robinson L, Li E, Roche J, Todd S, Waggoner B, Lundstrom R, Jacobson J, Mullokandov M R, Klinger H, Drabkin H A. Hum Mol Genet. 1997;6:193–203. doi: 10.1093/hmg/6.2.193. [DOI] [PubMed] [Google Scholar]
- 8.Hendricks D T, Taylor R, Reed M, Birrer M J. Cancer Res. 1997;57:2112–2115. [PubMed] [Google Scholar]
- 9.Huebner K, Hadaczek P, Siprashvili Z, Druck T, Croce C M. Biochim Biophys Acta. 1997;1332:M65–M70. doi: 10.1016/s0304-419x(97)00009-7. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Garrison P N, Barnes L D. Biochem J. 1995;312:925–932. doi: 10.1042/bj3120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner C, Garrison P, Gilmour J, Peisach D, Ringe D, Petsko G A, Lowenstein J M. Nat Struct Biol. 1997;4:231–238. doi: 10.1038/nsb0397-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seraphin B. DNA Sequence. 1992;3:177–179. doi: 10.3109/10425179209034013. [DOI] [PubMed] [Google Scholar]
- 13.Barnes L D, Garrison P N, Siprashvili Z, Guranowski A, Robinson A K, Ingram S W, Croce C M, Ohta M, Huebner K. Biochemistry. 1996;35:11529–11535. doi: 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- 14.Matoyama T, Hojo M, Watanabe H. Acta Pathol Japanica. 1986;36:65–83. doi: 10.1111/j.1440-1827.1986.tb01461.x. [DOI] [PubMed] [Google Scholar]
- 15.Huang D P, Ho J H C, Poon Y F, Chew E C, Saw D, Lui M, Li C L, Mak L S, Lai S H, Lau W H. Int J Cancer. 1980;26:127–132. doi: 10.1002/ijc.2910260202. [DOI] [PubMed] [Google Scholar]
- 16.Ueda R, Shiku H, Pfreundschuh M, Takahashi T, Li L T C, Whitmore W F, Oettgen H F, Old L J. J Exp Med. 1979;150:564–579. doi: 10.1084/jem.150.3.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham F L, Smiley J, Russell W C, Naizn R. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 18.Kastury K, Baffa R, Druck T, Ohta M, Cotticelli M G, Inoue H, Negrini M, Rugge M, Huang D, Croce C M, Palazzo J, Huebner K. Cancer Res. 1996;56:978–983. [PubMed] [Google Scholar]
- 19.Wilke M C, Hall B K, Hoge A, Paradee W, Smith D I, Glover T W. Hum Mol Genet. 1996;5:187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- 20.Paradee W, Wilke C M, Wang L, Shridhar R, Mullins C M, Hoge A, Glover T W, Smith D I. Genomics. 1996;35:87–93. doi: 10.1006/geno.1996.0326. [DOI] [PubMed] [Google Scholar]
- 21.Rassool F V, LeBeau M M, Shen M-L, Neilly M E, Espinosa R, III, Ong S T, Boldog F, Drabkin H, McCarroll R, McKeithan T W. Genomics. 1996;35:109–117. doi: 10.1006/geno.1996.0329. [DOI] [PubMed] [Google Scholar]
- 22.Zimonjic D B, Druck T, Ohta M, Kastury K, Popescu N C, Huebner K. Cancer Res. 1997;57:1166–1170. [PubMed] [Google Scholar]
- 23.Mao L, Fan Y-H, Lotan R, Hong W K. Cancer Res. 1996;56:5128–5131. [PubMed] [Google Scholar]
- 24.Sozzi G, Sard L, DeGregorio L, Marchetti A, Musso K, Buttitta F, Tornielli S, Pellegrini S, Veronese M L, Manenti G, Incarbone M, Chella A, Angeletti C S, Pastorino U, Huebner K, Bevilaqua G, Pilotti S, Croce C M, Pierotti M A. Cancer Res. 1997;57:2121–2123. [PubMed] [Google Scholar]
- 25.Baker S J, Markowitz S, Fearon E R, Willson J K V, Vogelstein B. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]