

Abstract
Abstract. Objectives: Intercellular communication in non‐excitable cells is restricted to a limited range close to the signal source. Here, we have examined whether modification of the intracellular microenvironment could prolong the spatial proposition of signal generation and could increase cell proliferation. Material and methods: Mathematical models and experimental studies of endothelial repair after controlled mechanical injury were used. The models predict the diffusion range of injury‐released growth factors and identify important parameters involved in a signalling regenerative mode. Transfected human umbilical vein endothelial cells (HUVECs) were used to validate model results, by examining intercellular calcium signalling range, cell proliferation and wound healing rate. Results: The models predict that growth factors have a limited capacity of extracellular diffusion and that intercellular signals are specially sensitive to cell phospholipase C‐delta (PLCδ) levels. As basal PLCδ levels are increased by transfection, a significantly increased intercellular calcium range, enhanced cell proliferation, and faster wound healing rate were observed. Conclusion: Our in silico and in vitro studies demonstrated that non‐excitable endothelial cells respond to stimuli in a complex manner, in which intercellular communication is controlled by physicochemical properties of the stimulus and by the cell microenvironment. Such findings may have profound implications for our understanding of the tight nature of autocrine cell growth control, compensation to stress states and response to altered microenvironment, under pathological conditions.
INTRODUCTION
Excitable and non‐excitable cells employ different modes of intercellular communication. In the case of the former, neurons, myocytes and smooth muscle cells allow electrical signals, and to a lesser extent, chemical signals to pass successively from an activated cell to its neighbours. The neighbour cells produce new electrical signals of comparable amplitude or strength to that of the activated cell, forming a regenerative signalling mode over long ranges with little attenuation. In contrast, non‐excitable cells, such as endothelial cells, lack regenerative signalling so that intercellular communication rapidly abates and is restricted to a limited range close to the signal source. The restricted nature of non‐excitable cell signalling has been observed experimentally (Drumheller & Hubbell 1991; Demer et al. 1993; Domenighetti et al. 1998), and mathematical models have been developed to clarify roles of different parameters of the second messenger system in the signalling process. Previous mathematical models focused almost exclusively on the diffusion of second messengers, specifically inositol 1,4,5‐trisphosphate (IP3) and calcium (Sneyd et al. 1994, 1995; Wiesner et al. 1996; Hofer et al. 2002). These models confirmed experimental findings of limited calcium signalling ranges and also predicted that calcium signalling ranges depend critically on signal initiation, activation of IP3 production by calcium, and loading of intracellular calcium stores. As intercellular signal range is likely to be influenced by a cell's intrinsic capacity of reproducing IP3, one wonders whether a ‘regenerative’ signalling mode similar to that of excitable cells could be established through modification of the intracellular microenvironment.
Non‐excitable cells interact primarily through extracellular contact with chemical stimuli. Ligand coupling with their cell‐surface receptors leads to a series of activation states that include phosphorylation of second messengers and their subsequent passage to neighbouring cells through gap junctions (Gilula et al. 1972; Newmark 1977; Sibley et al. 1988; Bennett & Verselis 1992). Thus, diffusion of chemical stimuli within the extracellular space and gap junctional transport are two important factors governing intercellular communication. It debates, however, which of the two factors dominate in non‐excitable cells. Experimental studies with pulmonary artery endothelial cells (Hinman et al. 1997; Sammak et al. 1997; Moerenhout et al. 2001) and epithelial cells (Klepeis et al. 2001) suggest that the soluble stimuli, not gap junctions, are responsible for triggering intercellular signalling. Others have demonstrated that gap junctions are closely involved in mediation of epithelial (Sammak et al. 1997) and capillary endothelial cell communication (Pepper et al. 1989; Hinman et al. 1997; Domenighetti et al. 1998), raising the possibility that there is a cell‐specific effect. Most recently, it has been proposed that the two mechanisms may not be mutually exclusive but likely act concurrently and synergistically (Sammak et al. 1997; Gomes et al. 2006).
Extent and nature of the tissue response itself also plays a role in cell signalling; this is particularly true of endothelial cells. Connectivity of endothelial cells in a confluent state induces quiescence and resistance to self‐stimulation and stimulation of adjacent smooth muscle cells (Antonelli & D’Amore 1991; Dodge et al. 1993; Nugent et al. 1993; Ettenson et al. 2000). Potent growth factors such as fibroblast and vascular endothelial growth factors (FGF, VEGF) are relatively inert until endothelium is injured or disrupted (McNeil et al. 1989; Ettenson & Gotlieb 1995; Castilla et al. 1999). Injury‐released growth factors traverse the extracellular space to bind to and activate cell‐surface receptors and subsequently generate second messengers passing through gap junctions. Yet, it remains unclear whether growth factors in the extracellular space or gap junctions dominate intercellular communication in endothelial cells.
In this study, we have investigated intercellular signalling mechanisms in non‐excitable cells using a mathematical model, and empirical examination of endothelial repair after controlled mechanical injury. Our mathematical model incorporates extracellular diffusion of injury‐released growth factors, cell‐surface receptor binding, and passage of second messengers through gap junctions. This model delineates the relative contributions of growth factors and gap junctions to cell communication and identifies the important parameters involved in the regenerative signalling mode. Variations of the model parameters are linked to onset of the regenerative signalling mode in our studies. The model is particularly sensitive to phospholipase C‐delta (PLCδ), one of the three major isoforms of phospholipase C, which mediates release of calcium from the endoplasmic reticulum and activate protein kinase C (Berridge 1993; De Smedt & Parys 1995; Nishizuka 1995). PLCδ is mediated by cytoplasmic calcium levels and serves as a catalyst for formation of IP3, independent of a ligand‐receptor binding. Intriguingly, positive feedback reinforces the calcium signal (Allen et al. 1997; Kim et al. 1999; Rhee 2001; Thore et al. 2005) providing a mechanism for a regenerative intercellular calcium signal. A controlled mechanical injury model with transfected endothelial cells was used to validate the mathematical model results. Experimental observations showed that the regenerative signalling mode increases cell proliferation and wound healing rate in endothelial cells. Integration of the mathematical and biological model systems demonstrated that intercellular communication in non‐excitable cells is governed by physicochemical properties of the stimulus and the cell microenvironment.
MATHEMATICAL MODEL
A mathematical model was developed (glossary at the end of text) to distinguish the relative contributions to cell signal propagation of direct contact with soluble growth factors and signal propagation through gap junctions. Models enable quantification of the spatial range, and temporal kinetics of growth factor diffusion and calcium levels that cannot be achieved empirically. Our model also evaluated intercellular signalling under multiple conditions and subsequently selected critical parameters involved in the signalling mode. We built on previous work of fibroblast growth factor (FGF) receptor interactions with heparan sulfate proteoglycans (HSPGs) (Filion & Popel 2004) and calcium signalling in astrocytes (Hofer et al. 2002) to include analysis of diffusion ranges of injury‐released growth factors, binding of injury‐released growth factors to cell‐surface receptors, and binding‐induced intercellular calcium propagation through gap junctions. Two major injury‐released growth factors, bFGF and VEGF were considered (McNeil et al. 1989; Ettenson et al. 1995).
The computational model was designed to mimic events occurring in a controlled mechanically induced injury, through a confluent monolayer of human umbilical vein endothelial cells (HUVECs) (Fig. 1a). Growth factors are released from ruptured cells and diffuse towards the surface of the culture medium (y‐axis in Fig. 1a) and perpendicularly to the wound edge (x‐axis in Fig. 1a). The three‐dimensional model can be simplified into two‐dimensional x–y plane assuming that diffusion is uniformly distributed within the plane parallel to the monolayer. Thickness of cells was ignored to simplify calculations.
Figure 1.
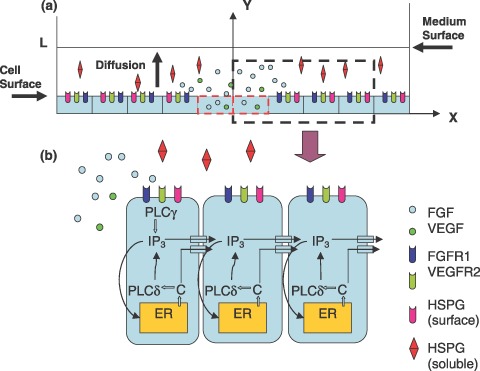
Mathematical model. (a) FGF and VEGF are released from mechanically ruptured endothelial cells (dashed red rectangular) and diffuse in the extracellular medium, to bind to cell‐surface receptors. (b) Growth factor‐receptor binding initiates intra‐ and intercellular calcium signalling. In cells immediately adjacent to a ruptured cell, cell‐surface receptor binding activates PLCγ, which hydrolyzes PIP2 into IP3. IP3 releases calcium from the endoplasmic reticulum (ER) through IP3‐sensitive channels. PLCδ responds to the increased calcium by producing more IP3. Calcium and IP3 pass through gap junctions, releasing calcium from the ER in neighbouring cells, thus, intercellular calcium signalling. FGF, fibroblast growth factor; HSPG, heparan sulfate proteoglycan; PLC, phospholipase C; VEGF, vascular endothelial growth factors.
Diffusion of injury‐released FGF and VEGF in the extracellular space
Injury induces release of FGF monomers and VEGF from ruptured cells, into the culture medium. Released FGF monomers dimerize and soluble HSPGs interact with FGF monomers and dimers (Filion et al. 2004). Concentration change of diffusive components in cell culture medium with time can be described by a set of diffusion equations (Appendix). We estimated that the concentration of FGF released by one ruptured cell was 4.35 × 10−5 ng/mL, based on previous data (McNeil et al. 1989). This was assumed to be constant during calcium signalling time (McNeil et al. 1989) as was that of VEGF. The initial concentration of FGF dimer, FGF–HSPG and the complex of FGF dimer with soluble HSPG was set to zero in the medium layer. Boundary conditions were defined as no diffusion flux across the media surface:
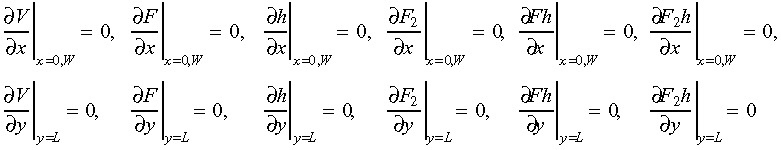 |
(1) |
where L and W are depth and diameter of the cell culture medium layer, which were estimated to be 1500 µm and 17 500 µm, respectively, based on 2 mL medium in a 35‐mm cell culture dish.
Binding of FGF and VEGF to cell‐surface receptors
Growth factors binding to cell‐surface receptors and HSPGs as components and complexes in the media diffuse to neighbouring cells in the model x–y plane. FGF monomers and dimers bind to FGF receptors (FGFR1) and cell‐surface HSPGs, forming various complexes including a relatively stable triad (FGF–HSPG–FGFR1) complex. Binding of VEGF to HSPGs has been found to be weak (Gitay‐Goren et al. 1992; Houck et al. 1992) compared to that between FGF and cell‐surface HSPGs (Nugent & Edelman 1992), thus was assumed to be negligible. Although VEGF binds to two receptors VEGFR1 and VEGFR2 (both are identified and expressed in endothelial cells), VEGFR2 is tyrosine‐phosphorylated more efficiently on binding and seems to mediate all observed endothelial cell responses to VEGF (Waltenberger et al. 1994); thus, our model only considers VEGF binding to VEGFR2. Based on previous studies (Filion et al. 2004), coupling between cell culture medium layer and intracellular space is given by a set of reaction equations describing concentration changes with time, of components and complexes on the cell surface due to binding, association, dissociation and internalization (Appendix). Initial concentrations of FGFR1 and HSPG on the cell surface were estimated based on number of FGFR1 (104) and HSPG (106) per cell (Filion et al. 2004) and the experimentally measured average surface area (400 µm2) of a HUVEC (Table 1). The number of the VEGFR2 on one cell surface was 9000 (MacGabhann & Popel 2004) and initial concentrations of VEGFR2 on a HUVEC was estimated in the same manner as FGFR1 (Table 1). Initial concentrations of other components and complexes on the cell surface were set to zero.
Table 1.
Model parameter values
| Parameter | Value | Unit | Source | Default value |
|---|---|---|---|---|
| CER 0 | 50–2000 | µm | Text | 100 |
| DF | 220 | µm2/s | Filion et al. 2004 | |
| D F2 | 220 | µm2/s | Filion et al. 2004 | |
| D F2h | 220 | µm2/s | Filion et al. 2004 | |
| Dh | 220 | µm2/s | Filion et al. 2004 | |
| DFh | 220 | µm2/s | Filion et al. 2004 | |
| DV | 100 | µm2/s | MacGabhann et al. 2004 | |
| DC | 20 | µm2/s | Hofer et al. 2002 | |
| DI | 280 | µm2/s | Hofer et al. 2002 | |
| F 0 | 4.35 × 10−5 | ng/mL | Text | |
| h 0 | 5.56 × 10−6 | µm | Filion et al. 2004 | |
| H 0 | 5.81 × 10−8 | µm min/cm2 | Text | |
| k 1 | 10 | µm/s | Text | |
| k 2 | 0.0–5.0 | µm/s | Text | 0.1 |
| k 3 | 0.0–2.0 | s | Text | 0.08 |
| k 4 | 0.0004 | s | Hofer et al. 2002 | |
| k 5 | 0.08 | s | Hofer et al. 2002 | |
| k 6 | 0.0–5.0 | s | Hofer et al. 2002 | 0.5 |
| k 7 | 0.025 | µm/s | Hofer et al. 2002 | |
| k 8 | 0.0–5.0 | µm/s | Hofer et al. 2002 | 0.2 |
| k 9 | 0.0–5.0 | s | Hofer et al. 2002 | 0.5 |
| k 10 | 0.0–5.0 | s | Hofer et al. 2002 | 4.0 |
| K 1 | 0.0 | µm | Wiesner et al. 1996 | |
| K 2 | 0.0–5.0 | µm | Hofer et al. 2002 | 0.3 |
| K 5 | 0.0–5.0 | µm | Hofer et al. 2002 | 0.2 |
| Ki | 0.0–5.0 | µm | Wiesner et al. 1996 | 1.6 |
| K 8 | 0.0–5.0 | µm | Hofer et al. 2002 | 1.0 |
| K 10 | 0.0–5.0 | µm | Wiesner et al. 1996 | 1.0 |
| kinF | 8.33 × 10−5 | s | Filion et al. 2004 | |
| kinFH | 8.33 × 10−5 | s | Filion et al. 2004 | |
| kinFHR | 71.67 × 10−5 | s | Filion et al. 2004 | |
| kinFFHR | 71.67 × 10−5 | s | Filion et al. 2004 | |
| kinFFhR | 71.67 × 10−5 | s | Filion et al. 2004 | |
| kinFhR | 71.67 × 10−5 | s | Filion et al. 2004 | |
| k inF2R | 130 × 10−5 | s | Filion et al. 2004 | |
| k inF2H | 8.33 × 10−5 | s | Filion et al. 2004 | |
| kinH | 8.33 × 10−5 | s | Filion et al. 2004 | |
| kinV | 10−5 | s | MacGabhann et al. 2004 | |
| kinFR | 1300 × 10−6 | s | Filion et al. 2004 | |
| kinVR | 280 × 10−6 | s | MacGabhann et al. 2004 | |
| koffFR | 1.32 × 10−2 | s | Filion et al. 2004 | |
| k offF2 | 1.32 × 10−2 | s | Filion et al. 2004 | |
| k offF2R | 1.32 × 10−2 | s | Filion et al. 2004 | |
| koffFH | 2.28 × 10−2 | s | Filion et al. 2004 | |
| koffFh | 2.28 × 10−2 | s | Filion et al. 2004 | |
| koffFHR | 6.33 × 10−4 | s | Filion et al. 2004 | |
| koffFhR | 6.33 × 10−4 | s | Filion et al. 2004 | |
| koffVR | 410 × 10−6 | s | MacGabhann et al. 2004 | |
| konFR | 7.0 | µm/s | Filion et al. 2004 | |
| kon F2 | 10−2 | µm/s | Filion et al. 2004 | |
| k onF2R | 7.0 | µm/s | Filion et al. 2004 | |
| k onF2H | 2.0 | µm/s | Filion et al. 2004 | |
| konFH | 2.0 | µm/s | Filion et al. 2004 | |
| konFh | 2.0 | µms | Filion et al. 2004 | |
| k onF2 h | 2.0 | µm/s | Filion et al. 2004 | |
| konFHR | 108 | µm min/cm2 | Filion et al. 2004 | |
| konFFHR | 7.0 | µm/s | Filion et al. 2004 | |
| konFFhR | 7.0 | µm/s | Filion et al. 2004 | |
| konFhR | 5.6 | µm/s | Filion et al. 2004 | |
| konV | 1.2 | µm/s | MacGabhann et al. 2004 | |
| PC | 0.01PI | µm/s | Hofer et al. 2002 | |
| PI | 1.0–5.0 | µm/s | Hofer et al. 2002 | 3.0 |
| r | 0–20 | Dimensionless | Wiesner et al. 1996 | 6.0 |
| R F0 | 0.0095 | µm | Text | |
| R V0 | 0.01 | µm | Text | |
| SF | 48.4 × 10−13 | µm/(cm2 s) | Filion et al. 2004 | |
| SH | 48.4 × 10−19 | µm/(cm2 s) | Filion et al. 2004 | |
| SV | 3.8 × 10−19 | µm/(cm2 s) | MacGabhann et al. 2004 | |
| V 0 | 4.35 × 10−5 | ng/mL | Text |
Intercellular calcium propagation
Binding of a released growth factor to an endothelial cell‐surface receptor activates membrane‐bound enzyme PLC (Fig. 1b). One isoform, PLCγ, participates in calcium signal pathways triggered by binding of receptors containing tyrosine kinase (Margolis et al. 1989). Activated PLCγ hydrolyses phosphatidylinositol 4,5‐bisphosphate to IP3 and diacylglycerol. As a second messenger, IP3 induces release of calcium from the endoplasmic reticulum through IP3 receptor (IP3R) channels and store‐operated calcium channels. In this manner, IP3 increases cytoplasmic calcium concentration, which activates PLCδ, another isoform of PLC. PLCδ is highly sensitive to [Ca2+] compared to other isoforms of PLC and can regenerate IP3 in non‐excitable cells (Allen et al. 1997; Kim et al. 1999; Thore et al. 2005). Other calcium transport events were built into the model as well, including calcium uptake into the endoplasmic reticulum, influx and leak of calcium across the cell plasma membrane. In this model, a positive feedback loop was established to describe the relationships between calcium, IP3 and PLCδ.
The dynamic nature of intracellular calcium signalling in endothelial cells is represented by a set of kinetic equations describing IP3 generation and degradation, [Ca2+] release from endoplasmic reticulum, and the normalized activated IP3 receptor levels with time (Appendix). The intercellular boundary conditions of concentration change at time t for calcium and IP3 are:
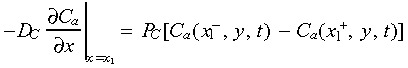 |
(2) |
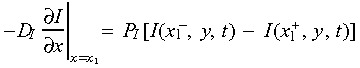 |
(3) |
where x 1 is the contact location of two adjacent cell membranes. Rate of IP3 generated by PLCγ induced by binding, was assumed to be proportional to the sum of signalling receptor complexes on the cell surface, due to lack of experimental data. The parameter value of IP3 generation k 1 was estimated so that cytoplasmic [Ca2+] increase was in the range of documented experimental data (peak magnitude 0.4–1.3 µm) (Drumheller et al. 1991; Demer et al. 1993; Tran et al. 1999). Sensitivity analysis was used to select the parameters involved in calcium signalling (Table 1).
Computational simulation
Model results were obtained using the finite difference scheme with spatial interval of 5 µm in both x and y directions and a temporal interval of 1 ms. The computational results of FGF and VEGF diffusion ranges were observed 60 s after injury, long enough compared to the time required (usually within 5 s) to trigger injury‐induced calcium signalling. Calcium concentration for a cell to be considered active was set to 0.5 µm. The calcium signal range is defined as number of cell rows from the wound edge.
MATHEMATICAL MODEL RESULTS
Injury‐released FGF and VEGF diffusion in the extracellular space
The diffusion of injury‐released FGF and VEGF was predicted to be limited to those areas close to the wound edge (Fig. 2). Concentrations of injury‐released growth factors are maximal (0.068 µm) at a wound edge, and gradually decreased to zero with cell locations further away. Sixty seconds after injury, VEGF diffusion should go no further than the first three rows of cells. FGF concentration decreased faster than that of VEGF. Diffusion range of FGF is inversely related to the initial concentration of cell‐surface HSPG (Fig. 3a), for example, the higher the initial concentration of cell‐surface HSPG, the lower the diffusion range of FGF. Diffusion range of FGF decreased from six to one row from the wound edge, when the initial concentration of cell‐surface HSPG increased from 102 to 106 receptors per cell. Interestingly, FGF diffusion was not significantly affected by initial concentration of soluble HSPG in cell culture medium (data not shown).
Figure 2.
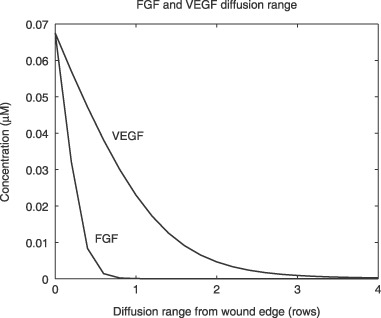
Diffusion ranges of injury‐released VEGF and FGF. Diffusion Ranges are predicted by the mathematical model and are plotted as number of cell rows from the wound edge at 60 s after injury. FGF, fibroblast growth factor; VEGF, vascular endothelial growth factors.
Figure 3.
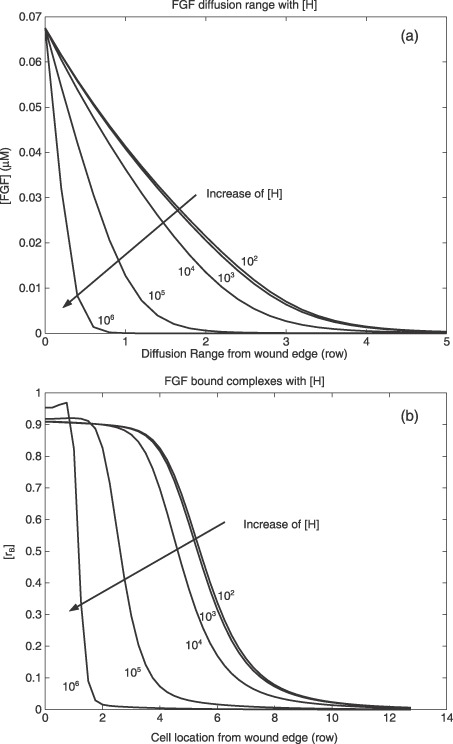
Diffusion range of (a) FGF and (b) normalized FGF‐bound complexes on cell surfaces versus initial concentration of cell‐surface HSPG (receptor/cell). Diffusion ranges are predicted by the mathematical model and are plotted as number of cell rows from wound edge at 60 s after injury. Normalized FGF‐bound complexes are plotted versus cell location starting from wound edge. FGF, fibroblast growth factor; HSPG, heparan sulfate proteoglycan; VEGF, vascular endothelial growth factors.
Cell‐surface binding
Concentrations of FGF‐bound complexes on the cell surface changed with cell location and initial concentration of cell‐surface HSPG ([H]) (Fig. 3b). Concentration of FGF‐bound complexes was normalized by concentration of initial FGF receptors on the cell surface to create the ratio ([rB]). Sixty seconds after injury, when [H] was 102 receptors per cell, maximal [rB] was ~0.91 for cells located in the first row from the wound edge. This value of rB suggests that 91% of FGF receptors were bound to FGF monomers, dimers or FGF complexes. As the model results show, [rB] decreased significantly from 0.91 to 0.03 in cell locations progressively farther from the wound edge. With increase of [H], the ratio of FGF‐bound complexes decreased much faster for a given cell location. When [H] increased from 102 to 106 receptors per cell, normalized concentration of FGF‐bound complexes in cells of the second row from the wound edge decreased from 0.91 to approximately 0.005.
Intercellular calcium signalling range
Sensitivity analysis showed that the range of intercellular calcium signalling was dictated by several model parameters, including rate of IP3 generation by PLCδ, k 2, initial [Ca2+] in the endoplasmic reticulum, C ER0, IP3 degradation rate, k 3, and half‐saturation constant for IP3 activation of IP3R, Ki (Fig. 4). Given fixed k 3, C ER0, and Ki, the range of intercellular calcium signalling increased approximately linearly with increase of k 2, the rate of IP3 generation by PLCδ. When k 2 increased from 0 to 5.0 µm/s, the range of intercellular calcium signalling was extended from 5 rows to ~40 rows from the wound edge. The range of intercellular calcium signalling was inversely exponentially related to IP3 degradation rate k 3, given a fixed rate of k 2, C ER0 and Ki. The slower the IP3 degradation rate, the larger the calcium signalling range. When k 3 decreased from 2.0 to 0.0 s, the calcium signalling range increased from 0 to ~9 rows from the wound edge. The range of intercellular calcium signalling increased with increase of initial calcium concentration of the endoplasmic reticulum, C ER0, given a fixed rate of k 2, k 3, and Ki. Model results show that when C ER0 increased from 50 to 2000 µm, the calcium signalling range increased from 3 to ~13 rows from the wound edge. Intercellular calcium signalling was inversely exponentially related to half‐saturation constant for IP3 activation of IP3R, Ki, given a fixed rate of k 2, k 3 and C ER0. When Ki increased from 0 to 5.0 µm, the calcium signalling range was virtually eliminated, falling from 25 to ~1 row from the wound edge.
Figure 4.
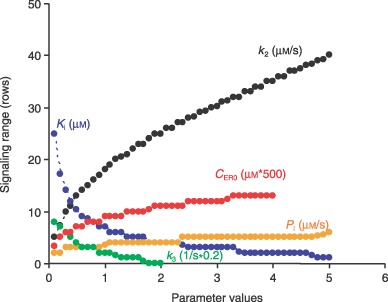
Sensitivity analysis of calcium signalling range. Signalling range is estimated by the mathematical model and plotted as the number of cell rows away from the wound edge. Ki: half‐saturation constant for IP3 activation of IP3R; k 2: rate of IP3 generation by PLCδ; C ER0: the initial [Ca2+] in ER; k 3: IP3 degradation rate; and PI: permeability of IP3.
Counterintuitively, increasing gap junction permeability did not significantly increase the range of intercellular calcium signalling. Increasing it to IP3, PI from 0 to 5.0 µm/s only extended the signalling range by ~3–4 rows (Fig. 4), gap junction permeability controlled the signalling range to a very limited extent. Sensitivity analysis did not show significant influence of calcium signalling range by other parameters, including the rate constant of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump, k 6, ratio of volume between cytoplasmic and endoplasmic reticulum, r, and rate constant of IP3R inactivation, k 10 (data not shown).
EXPERIMENTAL MATERIALS AND METHODS
Our mathematical model predicts that intercellular calcium signals, induced by growth factor diffusion, is limited to an area near a wound edge and that range of the intercellular calcium signal is governed by several parameters, including initial calcium concentration in the endoplasmic reticulum, IP3 degradation rate, and rate of IP3 generation by PLCδ. We have validated the model's prediction and the effect of PLCδ level on range of the intercellular calcium signal, by examining endothelial cell repair kinetics after controlled injury. We chose the PLCδ level as the focus of our experimental studies as this protein is easier to manipulate than the other parameters.
Basal levels of PLCδ in HUVECs were increased by cell transfection and confirmed by Western blot analysis. Effects of increased PLCδ level were investigated by comparing the range of intercellular calcium signal, cell proliferation and wound closure rates in PLCδ‐transfected and control cells although quantification of basal and changing calcium level requires sensitive equipment and is beyond the scope of the present study. In addition, quantification of calcium level change by mechanical injury has already been well documented as a stepwise function with time in endothelial cells (Drumheller et al. 1991; Laskey et al. 1992; Demer et al. 1993; Domenighetti et al. 1998; Tran et al. 1999). All experimental measurements were performed blind to which cultures were control or transfected.
Cell culture and transfection
Human umbilical vein endothelial cells obtained from Cambrex (Walkersville, MD, USA), were grown routinely in endothelial basal medium (EBM) supplemented with EBM‐2 Bullet Kit (Cambrex) and 10% foetal bovine serum (Hyclone, Logan, UT, USA) at 37 °C, humidified in 5% CO2; medium was routinely changed every 2 days. The HUVECs were transiently transfected with either empty (control) plasmid pNeoSRαII or plasmid containing the PLCδ gene (both gifts from Dr. Grant Kelley, SUNY, Syracuse, NY, USA). Plasmid DNA for cell transfection was amplified in competent Escherichia coli and was purified using PureYield Plasmid Midiprep System (Promega, Madison, WI, USA) as per manufacturer's instructions. One day prior to transfection, HUVECs were plated into 35 mm dishes so that 80–90% of cell confluence was achieved at the time of transfection. The liposome‐mediated gene transfer method was employed combined with Magnetofection method. Briefly, 2 µg plasmid DNA, 6 µL of Fugene 6 (Roche, Indianapolis, IN, USA) and 2 µL of CombiMag reagent (OriGene, Rockville, MD, USA) were used to form a transfection complex following manufacturer's instructions. Experiments were performed 48 h after transfection.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis
For immunoblot analysis, control and transfected HUVECs were washed with phosphate‐buffered saline (PBS) and lysed in lysis buffer containing 20 mm Tris pH 8.0, 150 mm NaCl, 1% Triton X‐100, 1% sodium dodecyl sulfate, 2 mm phenylmethylsulphonyl fluoride, and complete protease inhibitor pellet (Roche). Cell protein content was measured using the bicinchoninic acid protein method (Pierce, Rockford, IL, USA). Equal amounts of cell protein (20 ng) were electrophoresed in 4–15% pre‐cast gel (Bio‐Rad, Hercules, CA, USA) and then transferred to PVDF membranes (Millipore, Billerica, LA, USA). Western blot analysis was performed using anti‐PLCδ antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by incubation with horseradish peroxidase‐conjugated secondary antibody (Santa Cruz Biotechnology), the final reaction product of which was detected using a chemiluminescent substrate kit (Perkin‐Elmer, Wellesley, MA, USA). Blots were visualized with a Fluorochem SP imager (Alpha Innotech, San Leandro, CA, USA) and quantified with AlphaEase (Alpha Innotech) image analysis software.
Injury model and intercellular calcium signalling imaging
Forty‐eight hours after transfection, confluent HUVEC monolayers were washed twice with PBS; cells were then loaded with a dye solution of Fluo‐4 No Wash Assay Kit (Invitrogen‐Molecular Probes, Carlsbad, CA, USA) and were incubated for 30 min at 37 °C, and for an additional 30 min at room temperature. A double‐sided wound (~1000 µm) was made in the middle of the monolayer by scraping cells with a spatula. Immediately after injury, cells that increased their cytosolic calcium became fluorescent and were observed under a Leica DMRA2 Epi‐fluorescence microscope, equipped with a digital camera (Leica Microsystems, Bannockburn, IL, USA). Images were captured on a computer running Metamorph software (Molecular Devices Corp., Downingtown, PA, USA). In parallel studies, recovery of the denuded area was measured as a function of closure distance, by an observer blind to sample category. Repair was quantified by selecting a field of view, which covered approximately 2% of the total cultured area; for each culture, three fields of view, under a 100× objective were chosen. Wound closure rate was measured as distance difference over 24 h. These experiments were performed on cells grown on glass coverslip and with cells seeded on gelatin‐coated and non‐coated tissue culture plastic dishes.
For proliferation studies, injured control or transfected cultures grown on glass coverslips were washed three times with medium, and were re‐incubated for another 24 h with medium containing 5% foetal bovine serum. During the last 3 h of this incubation, a labelling reagent (Amersham Biosciences, Piscataway, NJ, USA; 1 : 1000), containing bromodeoxyuridine (BrdU) and fluorodeoxyuridine (FdU), was added to the cultures. Cells were then fixed with acid‐alcohol (90% ethanol : 5% acetic acid : 5% deionized water) for 30 min at room temperature. Cells were incubated for 1 h with monoclonal anti‐BrdU antibody containing nuclease (Amersham Biosciences; 1 : 100), and then with antimouse IgG‐Texas Red (Santa Cruz Biotechnology). Coverslips were mounted in glycerol/PBS (1 : 1) and were examined blind to category using an epi‐fluorescence microscope.
Statistical analysis
All statistical analyses were performed using Student's paired t‐test with P < 0.01 as level of significance.
EXPERIMENTAL RESULTS
Transfection and intercellular calcium signalling range
Phospholipase C‐delta expression was confirmed by Western blot analysis. PLCδ levels were not detectable in wild‐type HUVEC nor those cells transfected with empty plasmid. In contrast, significant expression of PLCδ was revealed in HUVECs transfected with PLCδ‐containing plasmid (Fig. 5d), demonstrating success of PLCδ overexpression in HUVECs.
Figure 5.
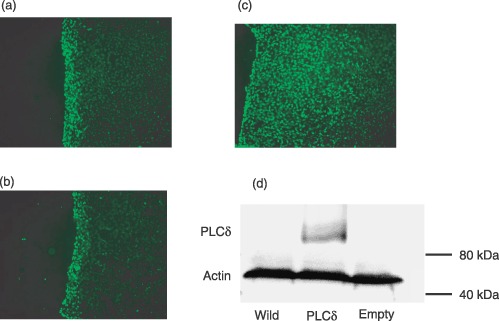
Calcium signalling using Fluo‐4 in HUVECs. (a) Wild‐type HUVECs (b) HUVECs transfected with empty plasmid. (c) HUVECs transfected with PLCδ‐containing plasmid. (d) Western blots of PLCδ (85 kDa) and corresponding staining for actin (45 kDa; to confirm equal loading of protein) in wild‐type, PLCδ‐transfected HUVECs and HUVECs transfected with empty plasmid. PLCδ is only detectable in PLCδ‐transfected HUVECs. Position of molecular weight standards are indicated on the right. HUVEC, human umbilical vein endothelial cells; PLC, phospholipase C.
Cytoplasmic calcium levels were followed after controlled mechanical injury of HUVEC monolayers to examine the range of intercellular calcium signalling and regulation of this range by PLCδ. Calcium levels were followed with Fluo‐4 and range of intercellular calcium signal was measured as the number of cell rows away from a wound edge. Seconds after injury, calcium signals were discernible at wound edges, and they propagated to neighbouring cells at a speed of approximately one cell per second. In wild‐type, non‐transfected HUVECs, injury‐induced intercellular calcium signals were limited to the wound edge (5.2 ± 0.3 rows, n = 5) (Fig. 5a). The transfection procedure per se had no affect on range of intercellular calcium signal as demonstrated by HUVECs transfected with empty plasmid (6.4 ± 0.4 rows, n = 5) (Fig. 5b). In contrast, the intercellular calcium signal propagated five times further from the wound edge in HUVECs transfected with PLCδ plasmid (25 ± 1.2 rows, P < 0.01, n = 5) (Fig. 5c).
Cell proliferation
The population and location of cells involved in injury‐induced proliferation were examined. Proliferating cells were labelled by BrdU staining. Overexpression of PLCδ increased both the number of proliferating cells and the rows of cells involved from the wound edge. In HUVECs transfected with empty plasmid, 24 h after injury, proliferating cells were located within a limited area close to the wound edge (5.5 ± 0.7 rows, n = 3) (Fig. 6a). Wounding of HUVECs overexpressing PLCδ not only increased the number of proliferating cells (by 147.3 ± 0.4%, P < 0.01) but also induced proliferation of cells further away from the wound edge (13.6 ± 1.5 rows, n = 7, P < 0.01) (Fig. 6b).
Figure 6.

Proliferation assay using BrdU staining of HUVECs transfected with (a) empty plasmid and (b) PLCδ‐containing plasmid. Proliferating HUVECs are stained bright red. Arrows indicate the location of the wound edge on the left side. HUVEC, human umbilical vein endothelial cells.
Wound closure rate
The calcium signal is one of the earliest signalling pathway events to be activated in endothelial cells following injury. Therefore, It is important to investigate how PLCδ level regulates the signal events and the subsequent biological response in injury repair. Wound closure rate was measured for control and transfected HUVECs seeded on three different types of surface: namely glass coverslips, gelatin‐coated dishes and non‐coated tissue culture plastic dishes. Wound closure rates were significantly increased in a monolayer of HUVECs transfected with PLCδ‐containing plasmid (Fig. 7). For cells seeded on glass coverslips, wound closure rate was 713.4 ± 168.1 µm/day (n = 8) for PLCδ‐overexpressing cells, compared to 560.5 ± 145.8 µm/day (n = 8) for cells transfected with empty plasmid. Wound closure rate of PLCδ overexpressing cells was 27 ± 0.4% (P < 0.01) faster than that of cells transfected with empty plasmid. Similarly, PLCδ‐transfected HUVECs closed faster than cells transfected with empty plasmid, on gelatin‐coated (21 ± 0.3%, P < 0.01, n = 8) and non‐coated dishes (30 ± 0.4%, P < 0.01, n = 8).
Figure 7.
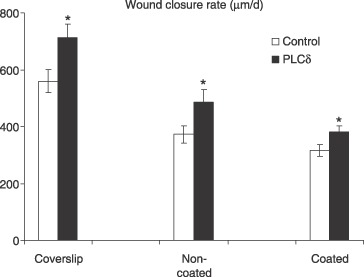
Wound closure rate of HUVECs transfected with PLCδ‐containing plasmid and empty plasmid. Cells were seeded on glass coverslip, non‐coated, and gelatin‐coated tissue culture dishes. Wound closure rate is significantly higher in PLCδ‐transfected cells grown on all three different surfaces (*P < 0.01, n = 8). HUVEC, human umbilical vein endothelial cells; PLC, phospholipase C.
DISCUSSION
Signal propagation is as important in non‐excitable cell biology as it is for excitable cells. While excitable cells may be built for speed, non‐excitable cells are designed for efficiency. We investigated the contribution of gap junctions in intercellular communication and the possibility of establishing a regenerative signalling mode in non‐excitable cells. Mathematical modelling allowed us to explore the sensitivity of the two signalling mechanisms to a range of governing parameters. Our model incorporates extracellular diffusion of injury‐released growth factors, cell‐surface receptor binding and second messenger passing through gap junctions. Model predictions were validated with empirical data. Injury‐released growth factors were predicted to have limited diffusion through layers of endothelial cells. Locally released growth factors did not diffuse more than one to three cell layers (Fig. 2), and are likely absorbed by binding elements like heparan sulfate proteoglycans (Fig. 3). A large accumulation of FGF complexing with HSPGs alone or with FGFR1 (Fig. 3b) on cells close to the injured site suggests that HSPGs might serve as a surface‐binding reservoir in endothelial cells. The predicted limited diffusion range of growth factors is supported by experimental results, that transport of FGF was enhanced dramatically when heparan sulfate binding was inhibited (Dowd et al. 1999). Hence, a powerful paradox arises. In spite of the limited diffusive range of locally active growth factors, there is a demonstrable growth factor effect in more distant cells (5–7 rows). We propose that signal propagation beyond the diffusion range of growth factors is attributable to signal relay through intercellular gap junctions. This hypothesis is based on our computational results and the unique cell connectivity‐related signalling in endothelial cells.
Connectivity of endothelial cells
Under physiological conditions, blood vessels are lined by an intact confluent monolayer of endothelial cells. These regulate local vascular biology, as both a selectively permeable barrier between circulating blood and underlying tissue, and as a cell density state‐responsive bioreactor. Confluent endothelial cells possess the highest degree of connectivity and remain relatively insensitive to stimulatory agents. If connectivity is disrupted, as by injury, endothelial cells switch to a subconfluent phenotype and cells become responsive to growth stimuli. This is particularly the case in the endothelial response to vascular growth factors. Compounds like FGF and VEGF, which are potent drivers of endothelial cell proliferation and protein synthesis in the subconfluent state, have minimal effects on confluent endothelial cells (Antonelli et al. 1991; Dodge et al. 1993; Nugent et al. 1993; Ettenson et al. 2000). Theoretically, cells starting from the second row from the wound edge have intact connections with the neighbouring cells; thus, would not be responsive to growth factors through extraneous transport. The connectivity‐based responsive nature makes gap junctional intercellular signalling an essential pathway for the control of endothelial cell biology irrespective of extracellular transport of regulatory compounds.
Model‐predicted intercellular signalling range
Our mathematical model delineated the relative impact of four important influences on endothelial calcium metabolism. The model predicted that cytoplasmic PLCδ level and the initial [Ca2+] in the endoplasmic reticulum should drive signal propagation, while IP3 degradation and half‐saturation constant for IP3 activation of IP3R should limit signalling. Counterintuitively, increasing concentrations of injury‐released growth factors (data not shown) or gap junction permeability to IP3 did not significantly increase signal propagation range. It can be inferred from our model results (Fig. 4) that endothelial cells maintain PLCδ level under precise and careful control to regulate cell response to external stimuli. Under physiological conditions, PLCδ levels in endothelial cells (Fu et al. 1994) are far lower than in other cells, such as astrocytes, smooth muscle cells and neurons (Mizuguchi et al. 1991; Yamada et al. 1991; Crljen et al. 2004). This may explain why intercellular signal propagation is restricted to a far greater degree in endothelial cells than other cell types.
Positive feedback between PLCδ, IP3 and calcium provides a mechanism for a regenerative intercellular calcium signal. PLCδ is highly sensitive to cytoplasmic calcium levels. An increase in calcium concentration within a physiological range (0.1–10 µm) is sufficient to stimulate PLCδ, but not other PLC isoforms such as PLCγ and PLCβ (Allen et al. 1997). IP3 is generated by PLCγ in endothelial cells that are directly exposed to external stimuli, and by PLCδ in cells that have elevated cytoplasmic calcium concentration following stimulation. As intercellular signals pass to neighbouring cells via gap junctions, PLCδ can be activated to produce more IP3 and release more calcium from the endoplasmic reticulum of the neighbouring cells. Hence, in confluent monolayers, the propagation range can be extended when IP3 serves as a local ‘signal amplifier’ in endothelial cells that are not directly exposed to the external stimuli.
Excitable and non‐excitable cell signalling differs not only in propagation speed, but range and amplitude as well. Excitable cells use action potentials, which can be fully regenerated from an activated cell to its downstream counterpart; thus, signal propagation should have no innate range limit. Non‐excitable cells have a limited signal propagation range because of the damping effect of IP3; the amount of IP3 diffusing into a downstream cell is always lower than that generated in the upstream cell. This ultimately leads to signal amplitude decline and limited propagation range. Our study, however, demonstrated that reduction in signal amplitude can be compensated for if PLCδ level is sufficiently high in the downstream cells; thus, forming a regenerative signal mode for non‐excitable cells. The cell microenvironment rather than the nature of the signal, allows non‐excitable cells to enact a signalling mechanism analogous to that of excitable cells. Cell signal propagation is therefore a balance between signal promotion governed by PLCδ activity and gap junction connectivity, and signal limitation by IP3 depletion and restricted growth factor diffusion.
Experimental validation of the model predictions
Our mathematical predictions were empirically validated in endothelial cells undergoing controlled mechanical injury. Increasing basal expression of PLCδ overcame inhibition of cell connectivity and provided unimpeded signal propagation. Spatial extension of calcium signalling in these cells increased approximately 4‐fold above that seen in control cells (Fig. 5). Transfection did not alter diffusion of Lucifer yellow through gap junctions as measured by the scrape‐load dye transfer assay (6.3 ± 0.2 rows for wild‐type cells, 5.8 ± 0.4 rows for cells transfected with empty plasmid, and 5.4 ± 0.6 rows for cells overexpressing PLCδ, n = 5), validating that signal extension is not from increased diffusivity of the second messengers. It is unknown whether higher PLCδ level affects local potential changes and elicits possible electrical signalling in endothelial cells. Endothelial cells have long been defined as electrically non‐excitable cells, yet electrical coupling between smooth muscle cells and endothelial cells has been proposed in vasomotor response studies (Emerson & Segal 2000). The effect of endothelial membrane potential on calcium signal is still debated. Endothelial cell membrane potential is proposed to impact on the resting and plateau of cytoplasmic calcium level (Luckhoff & Busse 1990; Kamouchi et al. 1999). However, more recent papers have shown that calcium levels in endothelial cells were independent of membrane potential (Cohen & Jackson 2005; McSherry et al. 2005), and voltage‐activated channels have not been found in cultured endothelial cells (Colden‐Stanfield et al. 1987). The calcium speed predicted by our model of ~20 µm/s was in agreement with other studies and remained constant in our experimental observation (data not shown). Based on the above evidence, we assumed that a chemical signal, instead of an electrical signal, played an important role in endothelial cell signalling after injury.
The extension of pharmacological effect of injury‐released growth factors was evidenced by extended proliferation of confluent cells located away from the wound edge, and by an increased wound closure rate (6, 7). The mechanism of enhanced calcium signalling range increasing cell proliferation and healing rate is complex and not clearly understood. Various studies have shown that an increase in cytoplasmic calcium is required for progression through the cell cycle (Whitaker & Patel 1990). In addition, calcium is involved in actin polymerization (Europe‐Finner & Newell 1986; Elsner et al. 1996) and subsequent cell migration (Naito et al. 1989; Liu et al. 1999; Etienne‐Manneville et al. 2000). Our experiments indicated that the extended signalling range achieved with increased PLCδ levels allows for more cells to be activated. Hence, there are a greater number of cells taking part in the repair process, thereby increasing wound healing rate.
Clinical impact
Endothelial integrity and recovery are critical to a range of pathophysiologic processes, including the full spectrum of vascular diseases, malignant tumour growth and metastasis, and modulation of systemic inflammatory and immune diseases. Mechanical relief of vascular disease unavoidably induces endothelial injury, disrupting normal homeostasis and structural integrity of the vessel wall. Subsequently, underlying smooth muscle cell proliferation and migration is induced into the intimal space, forming lesions that can progress until repair of endothelial monolayer is complete. Restoration of intact endothelium is critical to the control of intimal hyperplasia, restenosis and clinical response to vascular intervention. Several therapeutic modalities are proposed to limit hyperplasic response, including controlled drug release from stent; however, their effects are still not ideal. The regulatory role of PLCδ on cell response provides a novel means of modulating wound healing and recovery of endothelial layer integrity. The ability of endothelial cells to respond to different PLCδ levels is evident in our injury model with PLCδ‐transfected cells. To date, responsive PLCδ expression of vascular endothelial cells in pathological states is not clear.
Alteration in PLCδ level has been observed in a number of diseases; increased levels or abnormal accumulation of PLCδ has been documented in coronary spastic angina (Nakano et al. 2002), Pick's disease (Shimohama et al. 1993), progressive supranuclear palsy (Shimohama et al. 1993), diffuse Lewy body disease (Albasanz et al. 2005) and in some carcinomas (Nomoto et al. 1995). Abnormally, high levels of PLCδ in neuronal tissue and low levels of PLCδ in non‐neural brain tissue are evident in Alzheimer's disease (Shimohama et al. 1991, 1995; Matsushima et al. 1995). Increased expression and activity of PLCδ has been observed in skin fibroblasts (Kosugi et al. 2003), aortae (Kato et al. 1992), and renal arterioles (Peng et al. 2007) obtained from patients with hypertension. Elevated blood pressure can disrupt the endothelium. It is possible that an early increase in endothelial PLCδ levels could enhance cell and tissue repair and restoration of normal haemodynamic function. As more is learned about the role of PLCδ in these and other diseases, potential therapeutics can be developed to regulate PLCδ to more appropriate levels.
Limitations
Although our model matches current data and predicts empirical data, it provides a framework for evaluating perturbations to biological systems and can be updated and refined; as more endothelial‐specific values become available, there is still room for improvement; for example, injury‐induced FGF and VEGF release were assumed to occur simultaneously, and in the same concentration. This may not be true in physiological conditions and the topic requires further delineation. Although no interaction between VEGF and HSPG was considered, for the sake of simplification of the model, VEGF may bind (Gitay‐Goren et al. 1992; Houck et al. 1992) and interact with HSPG (Selleck 2006) in endothelial cells. If this is the case, the diffusion range of injury‐released growth factors would be limited within an even shorter range than that predicted by our model. Furthermore, as the biology of VEGFR1 is not as well delineated as that of VEGFR2, our model concentrated on the former; thus, our model can be refined as more data become available regarding VEGFR1.
CONCLUSION
Our mathematical model and experimental data demonstrate that intercellular signalling distance in non‐excitable cells can be extended beyond its normally limited range by altering the cell microenvironment. The model predicts that the extent of cell signal propagation can be changed by altering at least one of the following parameters; initial calcium concentration in the endoplasmic reticulum, IP3 degradation rate, half‐saturation constant for IP3 activation of IP3R and/or rate of IP3 generation by PLCδ. Experimental data confirmed a part of this prediction by altering the microenvironment of non‐excitable cells. Signal propagation distance was increased as the basal level of PLCδ was overexpressed. Such a control paradigm for intercellular signal control might well explain certain cellular responses in various disease states. Future work focusing on altering PLCδ levels in cells may lead to extension of pharmacological effects of locally administrated drugs beyond their previously known normal diffusion ranges.
Supporting information
Appendix: Model equations and Glossary
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
ACKNOWLEDGEMENT
This study was supported in part by grants from the National Institutes of Health (GM/HL 49039).
REFERENCES
- Albasanz JL, Dalfo E, Ferrer I, Martin M (2005) Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer's disease and dementia with Lewy bodies correlates with stage of Alzheimer's‐disease‐related changes. Neurobiol. Dis. 20, 685–693. [DOI] [PubMed] [Google Scholar]
- Allen V, Swigart P, Cheung R, Cockcroft S, Katan M (1997) Regulation of inositol lipid‐specific phospholipase cdelta by changes in Ca2+ ion concentrations. Biochem. J. 327, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A, D’Amore PA (1991) Density‐dependent expression of hyaluronic acid binding to vascular cells in vitro . Microvasc. Res. 41, 239–251. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Verselis VK (1992) Biophysics of gap junctions. Semin. Cell Biol. 3, 29–47. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361, 315–325. [DOI] [PubMed] [Google Scholar]
- Castilla MA, Arroyo MV, Aceituno E, Aragoncillo P, Gonzalez‐Pacheco FR, Texeiro E, Bragado R, Caramelo C (1999) Disruption of cadherin‐related junctions triggers autocrine expression of vascular endothelial growth factor in bovine aortic endothelial cells: effects on cell proliferation and death resistance. Circ. Res. 85, 1132–1138. [DOI] [PubMed] [Google Scholar]
- Cohen KD, Jackson WF (2005) Membrane hyperpolarization is not required for sustained muscarinic agonist‐induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation 12, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colden‐Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL (1987) Bradykinin‐induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circ. Res. 61, 632–640. [DOI] [PubMed] [Google Scholar]
- Crljen V, Visnjic D, Banfic H (2004) Presence of different phospholipase C isoforms in the nucleus and their activation during compensatory liver growth. FEBS Lett. 571, 35–42. [DOI] [PubMed] [Google Scholar]
- De Smedt H, Parys JB (1995) [Molecular and functional diversity of inositol triphosphate‐induced Ca(2+) release]. Verh. K. Acad. Geneeskd. Belg. 57, 423–458. [PubMed] [Google Scholar]
- Demer LL, Wortham CM, Dirksen ER, Sanderson MJ (1993) Mechanical stimulation induces intercellular calcium signaling in bovine aortic endothelial cells. Am. J. Physiol. 264, H2094–H2102. [DOI] [PubMed] [Google Scholar]
- Dodge AB, Lu X, D’Amore PA (1993) Density‐dependent endothelial cell production of an inhibitor of smooth muscle cell growth. J. Cell. Biochem. 53, 21–31. [DOI] [PubMed] [Google Scholar]
- Domenighetti AA, Beny JL, Chabaud F, Frieden M (1998) An intercellular regenerative calcium wave in porcine coronary artery endothelial cells in primary culture. J. Physiol. 513, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd CJ, Cooney CL, Nugent MA (1999) Heparan sulfate mediates bFGF transport through basement membrane by diffusion with rapid reversible binding. J. Biol. Chem. 274, 5236–5244. [DOI] [PubMed] [Google Scholar]
- Drumheller PD, Hubbell JA (1991) Local modulation of intracellular calcium levels near a single‐cell wound in human endothelial monolayers. Arterioscler. Thromb. 11, 1258–1265. [DOI] [PubMed] [Google Scholar]
- Elsner J, Dichmann S, Dobos GJ, Kapp A (1996) Actin polymerization in human eosinophils, unlike human neutrophils, depends on intracellular calcium mobilization. J. Cell. Physiol. 167, 548–555. [DOI] [PubMed] [Google Scholar]
- Emerson GG, Segal SS (2000) Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ. Res. 87, 474–479. [DOI] [PubMed] [Google Scholar]
- Etienne‐Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO (2000) ICAM‐1‐coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 165, 3375–3383. [DOI] [PubMed] [Google Scholar]
- Ettenson DS, Gotlieb AI (1995) Basic fibroblast growth factor is a signal for the initiation of centrosome redistribution to the front of migrating endothelial cells at the edge of an in vitro wound. Arterioscler. Thromb. Vasc. Biol. 15, 515–521. [DOI] [PubMed] [Google Scholar]
- Ettenson DS, Koo EW, Januzzi JL, Edelman ER (2000) Endothelial heparan sulfate is necessary but not sufficient for control of vascular smooth muscle cell growth. J. Cell. Physiol. 184, 93–100. [DOI] [PubMed] [Google Scholar]
- Europe‐Finner GN, Newell PC (1986) Inositol 1,4,5‐trisphosphate and calcium stimulate actin polymerization in Dictyostelium discoideum. J. Cell Sci. 82, 41–51. [DOI] [PubMed] [Google Scholar]
- Filion RJ, Popel AS (2004) A reaction‐diffusion model of basic fibroblast growth factor interactions with cell surface receptors. Ann. Biomed. Eng. 32, 645–663. [DOI] [PubMed] [Google Scholar]
- Fu Y, Cheng JX, Hong SL (1994) Characterization of cytosolic phospholipases C from porcine aortic endothelial cells. Thromb. Res. 73, 405–417. [DOI] [PubMed] [Google Scholar]
- Gilula NB, Reeves OR, Steinbach A (1972) Metabolic coupling, ionic coupling and cell contacts. Nature 235, 262–265. [DOI] [PubMed] [Google Scholar]
- Gitay‐Goren H, Soker S, Vlodavsky I, Neufeld G (1992) The binding of vascular endothelial growth factor to its receptors is dependent on cell surface‐associated heparin‐like molecules. J. Biol. Chem. 267, 6093–6098. [PubMed] [Google Scholar]
- Gomes P, Srinivas SP, Vereecke J, Himpens B (2006) Gap junctional intercellular communication in bovine corneal endothelial cells. Exp. Eye Res. 83, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Hinman LE, Beilman GJ, Groehler KE, Sammak PJ (1997) Wound‐induced calcium waves in alveolar type II cells. Am. J. Physiol. 273, L1242–L1248. [DOI] [PubMed] [Google Scholar]
- Hofer T, Venance L, Giaume C (2002) Control and plasticity of intercellular calcium waves in astrocytes: a modeling approach. J. Neurosci. 22, 4850–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N (1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267, 26031–26037. [PubMed] [Google Scholar]
- Kamouchi M, Droogmans G, Nilius B (1999) Membrane potential as a modulator of the free intracellular Ca2+ concentration in agonist‐activated endothelial cells. Gen. Physiol. Biophys. 18, 199–208. [PubMed] [Google Scholar]
- Kato H, Fukami K, Shibasaki F, Homma Y, Takenawa T (1992) Enhancement of phospholipase C delta 1 activity in the aortas of spontaneously hypertensive rats. J. Biol. Chem. 267, 6483–6487. [PubMed] [Google Scholar]
- Kim YH, Park TJ, Lee YH, Baek KJ, Suh PG, Ryu SH, Kim KT (1999) Phospholipase C‐delta1 is activated by capacitative calcium entry that follows phospholipase C‐beta activation upon bradykinin stimulation. J. Biol. Chem. 274, 26127–26134. [DOI] [PubMed] [Google Scholar]
- Klepeis VE, Cornell‐Bell A, Trinkaus‐Randall V (2001) Growth factors but not gap junctions play a role in injury‐induced Ca2+ waves in epithelial cells. J. Cell Sci. 114, 4185–4195. [DOI] [PubMed] [Google Scholar]
- Kosugi T, Osanai T, Kamada T, Nakano T, Okumura K (2003) Phospholipase C activity is enhanced in skin fibroblasts obtained from patients with essential hypertension. J. Hypertens. 21, 583–590. [DOI] [PubMed] [Google Scholar]
- Laskey RE, Adams DJ, Cannell M, Van Breemen C (1992) Calcium entry‐dependent oscillations of cytoplasmic calcium concentration in cultured endothelial cell monolayers. Proc. Natl. Acad. Sci. USA 89, 1690–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ridefelt P, Hakansson L, Venge P (1999) Regulation of human eosinophil migration across lung epithelial monolayers by distinct calcium signaling mechanisms in the two cell types. J. Immunol. 163, 5649–5655. [PubMed] [Google Scholar]
- Luckhoff A, Busse R (1990) Calcium influx into endothelial cells and formation of endothelium‐derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 416, 305–311. [DOI] [PubMed] [Google Scholar]
- MacGabhann F, Popel AS (2004) Model of competitive binding of vascular endothelial growth factor and placental growth factor to VEGF receptors on endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 286, H153–H164. [DOI] [PubMed] [Google Scholar]
- Margolis B, Rhee SG, Felder S, Mervic M, Lyall R, Levitzki A, Ullrich A, Zilberstein A, Schlessinger J (1989) EGF induces tyrosine phosphorylation of phospholipase C‐II: a potential mechanism for EGF receptor signaling. Cell 57, 1101–1107. [DOI] [PubMed] [Google Scholar]
- Matsushima H, Shimohama S, Fujimoto S, Takenawa T, Kimura J (1995) Changes in platelet phospholipase C protein level and activity in Alzheimer's disease. Neurobiol. Aging 16, 895–900. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Muthukrishnan L, Warder E, D’Amore PA (1989) Growth factors are released by mechanically wounded endothelial cells. J. Cell Biol. 109, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSherry IN, Spitaler MM, Takano H, Dora KA (2005) Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium 38, 23–33. [DOI] [PubMed] [Google Scholar]
- Mizuguchi M, Yamada M, Kim SU, Rhee SG (1991) Phospholipase C isozymes in neurons and glial cells in culture: an immunocytochemical and immunochemical study. Brain Res. 548, 35–40. [DOI] [PubMed] [Google Scholar]
- Moerenhout M, Himpens B, Vereecke J (2001) Intercellular communication upon mechanical stimulation of CPAE‐endothelial cells is mediated by nucleotides. Cell Calcium 29, 125–136. [DOI] [PubMed] [Google Scholar]
- Naito M, Hayashi T, Kuzuya M, Funaki C, Asai K, Kuzuya F (1989) Vascular endothelial cell migration in vitro roles of cyclic nucleotides, calcium ion and cytoskeletal system. Artery 17, 21–31. [PubMed] [Google Scholar]
- Nakano T, Osanai T, Tomita H, Sekimata M, Homma Y, Okumura K (2002) Enhanced activity of variant phospholipase C‐delta1 protein (R257H) detected in patients with coronary artery spasm. Circulation 105, 2024–2029. [DOI] [PubMed] [Google Scholar]
- Newmark P (1977) Membranes and receptors. Nature 267, 107–109. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y (1995) Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 9, 484–496. [PubMed] [Google Scholar]
- Nomoto K, Tomita N, Miyake M, Xhu DB, Logerfo PR, Weinstein IB (1995) Expression of phospholipases gamma 1, beta 1, and delta 1 in primary human colon carcinomas and colon carcinoma cell lines. Mol. Carcinog. 12, 146–152. [DOI] [PubMed] [Google Scholar]
- Nugent MA, Edelman ER (1992) Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: a mechanism for cooperactivity. Biochemistry 31, 8876–8883. [DOI] [PubMed] [Google Scholar]
- Nugent MA, Karnovsky MJ, Edelman ER (1993) Vascular cell‐derived heparan sulfate shows coupled inhibition of basic fibroblast growth factor binding and mitogenesis in vascular smooth muscle cells. Circ. Res. 73, 1051–1060. [DOI] [PubMed] [Google Scholar]
- Peng Z, Dang A, Arendshorst WJ (2007) Increased expression and activity of phospholipase C in renal arterioles of young spontaneously hypertensive rats. Am. J. Hypertens. 20, 38–43. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Spray DC, Chanson M, Montesano R, Orci L, Meda P (1989) Junctional communication is induced in migrating capillary endothelial cells. J. Cell Biol. 109, 3027–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG (2001) Regulation of phosphoinositide‐specific phospholipase C. Annu. Rev. Biochem. 70, 281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammak PJ, Hinman LE, Tran PO, Sjaastad MD, Machen TE (1997) How do injured cells communicate with the surviving cell monolayer? J. Cell Sci. 110, 465–475. [DOI] [PubMed] [Google Scholar]
- Selleck SB (2006) Signaling from across the way: transactivation of VEGF receptors by HSPGs. Mol. Cell 22, 431–432. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Homma Y, Suenaga T, Fujimoto S, Taniguchi T, Araki W, Yamaoka Y, Takenawa T, Kimura J (1991) Aberrant accumulation of phospholipase C‐delta in Alzheimer brains. Am. J. Pathol. 139, 737–742. [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Matsushima H, Fujimoto S, Takenawa T, Taniguchi T, Kameyama M, Kimura J (1995) Differential involvement of phospholipase C isozymes in Alzheimer's disease. Gerontology 41 (Suppl. 1), 13–19. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Perry G, Richey P, Takenawa T, Whitehouse PJ, Miyoshi K, Suenaga T, Matsumoto S, Nishimura M, Kimura J (1993) Abnormal accumulation of phospholipase C‐delta in filamentous inclusions of human neurodegenerative diseases. Neurosci. Lett. 162, 183–186. [DOI] [PubMed] [Google Scholar]
- Sibley DR, Benovic JL, Caron MG, Lefkowitz RJ (1988) Phosphorylation of cell surface receptors: a mechanism for regulating signal transduction pathways. Endocr. Rev. 9, 38–56. [DOI] [PubMed] [Google Scholar]
- Sneyd J, Charles AC, Sanderson MJ (1994) A model for the propagation of intercellular calcium waves. Am. J. Physiol. 266, C293–C302. [DOI] [PubMed] [Google Scholar]
- Sneyd J, Wetton BT, Charles AC, Sanderson MJ (1995) Intercellular calcium waves mediated by diffusion of inositol trisphosphate: a two‐dimensional model. Am. J. Physiol. 268, C1537–C1545. [DOI] [PubMed] [Google Scholar]
- Thore S, Dyachok O, Gylfe E, Tengholm A (2005) Feedback activation of phospholipase C via intracellular mobilization and store‐operated influx of Ca2+ in insulin‐secreting beta‐cells. J. Cell Sci. 118, 4463–4471. [DOI] [PubMed] [Google Scholar]
- Tran PO, Hinman LE, Unger GM, Sammak PJ (1999) A wound‐induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Exp. Cell Res. 246, 319–326. [DOI] [PubMed] [Google Scholar]
- Waltenberger J, Claesson‐Welsh L, Siegbahn A, Shibuya M, Heldin CH (1994) Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269, 26988–26995. [PubMed] [Google Scholar]
- Whitaker M, Patel R (1990) Calcium and cell cycle control. Development 108, 525–542. [DOI] [PubMed] [Google Scholar]
- Wiesner TF, Berk BC, Nerem RM (1996) A mathematical model of cytosolic calcium dynamics in human umbilical vein endothelial cells. Am. J. Physiol. 270, C1556–C1569. [DOI] [PubMed] [Google Scholar]
- Yamada M, Mizuguchi M, Rhee SG, Kim SU (1991) Developmental changes of three phosphoinositide‐specific phospholipase C isozymes in the rat nervous system. Brain Res. Dev. Brain Res. 59, 7–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix: Model equations and Glossary
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item