

Abstract
Pestiviruses, a group of enveloped positive strand RNA viruses belonging to the family Flaviviridae, express their genes via a polyprotein that is subsequently processed by proteases. The structural protein region contains typical signal peptidase cleavage sites. Only the site at the C terminus of the glycoprotein Erns is different because it does not contain a hydrophobic transmembrane region but an amphipathic helix functioning as the Erns membrane anchor. Despite the absence of a hydrophobic region, the site between the C terminus of Erns and E1, the protein located downstream in the polyprotein, is cleaved by signal peptidase, as demonstrated by mutagenesis and inhibitor studies. Thus, ErnsE1 is processed at a novel type of signal peptidase cleavage site showing a different membrane topology. Prevention of glycosylation or introduction of mutations into the C-terminal region of Erns severely impairs processing, presumably by preventing proper membrane interaction or disturbing a conformation critical for the protein to be accepted as a substrate by signal peptidase.
Keywords: Membrane/Proteins, Protein/Viral, Protein/Processing, Viruses/Flavi, Enzyme Mechanisms, Protein Conformation, Protein Folding, Amphipathic Helix, Signal Peptidase
Introduction
Classical swine fever virus (CSFV)2 belongs to the genus Pestivirus, which also includes the animal viruses bovine viral diarrhea virus and border disease virus of sheep. The genus Pestivirus is part of the family Flaviviridae which also comprises the genera Flavivirus and Hepacivirus (1).
Pestiviruses are positive strand RNA viruses with a single-stranded genome of ∼12.3 kb in length that contains a single open reading frame coding for a polyprotein of about 4000 amino acids (2). The polyprotein is co- and posttranslationally processed by cellular and viral proteases into at least 12 mature proteins (3–12), arranged in the polyprotein in the following order: NH2-Npro, C, Erns, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B-COOH. C, Erns, E1, and E2 are part of the virion (13, 14), with C forming the capsid, and Erns, E1, and E2 representing glycoproteins associated with the virus envelope. Erns and E2 elicit neutralizing antibody responses that can lead to protective immunity (15–18).
The pestivirus Erns protein is a highly unusual protein. It forms a disulfide-linked dimer of ∼90 kDa, about half of which is due to glycosylation (9, 13, 19). The protein displays homology to the RNases of the T2/S superfamily (20–22). In different test systems, it was shown that Erns indeed has RNase activity, a feature that is unique among viral glycoproteins (22–25). The protein is essential for virus growth (21), but the RNase activity is dispensable. Viruses, in which the RNase activity has been knocked out, are clinically attenuated (26, 27). A role for Erns in immune evasion has been proposed (28–31).
Erns can be found in virus-free cell culture fluid of infected cells and in the blood of infected animals (9, 29, 32). It lacks a hydrophobic region that could serve as a transmembrane anchor, and there is also no consensus sequence for glycosylphosphatidylinositol anchor addition. It is nevertheless bound to the virion and associated with intracellular membranes (9, 13, 14, 32, 33). Erns membrane binding is achieved by the C-terminal region forming an amphipathic helix that is inserted in plane into the membrane (32–34).
The topology of the Erns membrane anchor raises interesting questions concerning the protease that cleaves the polyprotein at the Erns C terminus. Processing of the region of the polyprotein encompassing the viral structural proteins and the first two nonstructural proteins p7 and NS2 is attributed to cellular proteases. The processing sites at the N termini of Erns, E2, p7, and NS2 comply with the demands for signal peptidase cleavage sites, namely a positively charged N-terminal region and a central hydrophobic region (h-region), followed by a more hydrophilic part (c-region) containing the cleavage site with small and uncharged residues at positions −3 and −1, as suggested by von Heijne (35, 36). The results of sequence and mutagenesis analyses clearly support the idea of SPase cleavage at these sites in the viral polyprotein (5, 6, 9).3 In contrast, the site separating Erns and E1 does not comply with the above described features of a SPase cleavage site. The −3/−1 residues at the Erns/E1 cleavage site are in agreement with a sequence processed by SPase, but a hydrophobic region upstream of the cleavage site is missing. This fact raises the question of whether SPase cleaves this site as proposed in some reports or which other protease is responsible for processing the polyprotein here.
In the work presented here, we analyzed the cleavage at the Erns C terminus with different approaches, including mutagenesis and inhibitor studies. For the first time, we provide clear evidence that processing between Erns and E1 is executed by SPase, which reveals the existence of a new type of SPase cleavage site not described so far.
EXPERIMENTAL PROCEDURES
Cells and Viruses
BHK-21 cells (kindly provided by T. Rümenapf) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and nonessential amino acids. The modified vaccinia virus strain Ankara containing the phage T7 RNA polymerase (MVA-T7) (37) was kindly provided by B. Moss (National Institutes of Health, Bethesda, MD).
Construction of Recombinant Plasmids
Restriction and subcloning were done according to standard procedures (38). Unless stated otherwise, all restriction and modifying enzymes were purchased from New England Biolabs (Frankfurt, Germany) and Fermentas GmbH (Sankt Leon-Roth, Germany). Plasmid pCITE 2a(+) was obtained from AGS (Heidelberg, Germany). It contains a T7 RNA polymerase promotor followed by a picornavirus internal ribosomal entry site. The synthetic DNA oligonucleotides were purchased from Metabion (München, Germany).
From the infectious cDNA clone of the CSFV strain Alfort/Tübingen (39), coding sequences were amplified with the pairs of primers NcoI (neu)/E1–3SXbaR, NcoI (neu)/E0–3SXbaI, E05SIII/E1–3SXbaR, and E05SIII/E0–3SXbaI and inserted into the NcoI/XbaI sites of pCITE 2a(+), yielding the constructs Npro-E1, Npro-Erns, SSeqErns-E1, and SSeqErns, respectively.
To obtain construct SSeqE1-E2, the cDNA of E1E2 was amplified from plasmid p578 (39). In addition, the coding sequence of the Erns signal peptide (SSeq) (nucleotides 1120–1173 of the full-length cDNA clone pA/CSFV (39)) was also incorporated in the SSeqErns-E1 and SSeqErns constructs. This sequence was amplified by PCR with the primers IB72, IB73, and E05SIII as follows. First, a PCR fragment was obtained from plasmid p578 with the primers IB72 and HPS38.2. This PCR product was then amplified with IB73 and HPS 38.2 and used for the generation of a further PCR fragment by amplification with E05SIII and E2 d(−). This PCR fragment was restricted with NcoI and XbaI and inserted into the NcoI/XbaI sites of pCITE 2a(+).
The construct mellSSeqErns-E1 containing the signal peptide of mellitin (mellSSeq) was generated in a similar way as described above. The coding sequence of the mellitin signal peptide was obtained from Qiagen (Hilden, Germany) and amplified in three steps by PCR as described before. First, a PCR fragment was amplified from plasmid SSeqErns-E1 with the primers IB131 and pCITErev. This PCR product was amplified with IB132 and pCITErev to obtain the second PCR fragment that contains part of the mellSSeq-coding sequence. The rest of the coding sequence was introduced by the third PCR with the second PCR fragment as template and the primers IB133 and E1–3SXbaR. The final product was cut with NcoI and XbaI and inserted into pCITE 2a(+), cut with NcoI/XbaI.
The constructs ppLSSeqErns-E1 and ppLSSeqErns-E1* are also based on SSeqErns-E1, with V5 tag and preprolactin signal peptide preceding ErnsE1. The sequence coding for the preprolactin signal peptide coding sequence (ppLSSeq) was amplified from a preprolactin vector (kindly provided by Heiner Niemann) by PCR with the primers IB97 and IB98 for ppLSSeqErns-E1 or with IB97 and IB100 for ppLSSeqErns-E1*. In order to introduce the sequence coding for a V5 tag, the resulting PCR fragments were used as templates in a further PCR with primers IB94 and IB98 for ppLSSeqErns-E1 or IB94 and IB100 for ppLSSeqErns-E1*. The resulting PCR products were cut with NcoI and EcoRI. The primers IB99 and E1–3SXbaR were used to amplify the ErnsE1-coding fragment from the SSeqErns-E1 plasmid. After restriction with EcoRI and XbaI, the resulting fragment was ligated together with the previously obtained fragment coding for V5-ppLSSeq and with pCITE 2a(+), cut with NcoI/XbaI.
QuikChange mutagenesis (Stratagene, Heidelberg, Germany) was employed according to the supplier's instructions to introduce substitutions, insertions or deletions. The constructs SSeqErns-E1 and Npro-Erns served as templates for all mutagenesis approaches for characterization of the Erns/E1 cleavage site.
The cloned PCR products were all verified by nucleotide sequencing with the BigDye Terminator Cycle Sequencing Kit (PE Applied Biosystems, Weiterstadt, Germany). Sequence analysis and alignments were done with Genetics Computer Group software (40). Further details of the cloning procedures and the sequences of the primers used for cloning and mutagenesis are available on request.
Transient Expression of Proteins
BHK-21 cells were infected with vaccinia virus MVA-T7, subsequently transfected with the desired cDNA construct using SuperFect (Qiagen), and labeled with 35S-amino acids as described earlier (32, 33). Where mentioned, tunicamycin (Roche Applied Science) or BFA (Calbiochem) was added to the medium during starving and labeling periods to a concentration of 10 μg/ml (tunicamycin) or 1 μg/ml (BFA).
Preparation and Fractionation of Cell Extracts
Crude extracts of transiently transfected cells were prepared under denaturing conditions as described before (32). Alternatively, transfected BHK-21 cells were harvested via fractionation essentially as described (32, 33, 41). Briefly, the tissue culture fluid of infected cells containing the secreted proteins was removed as the first fraction. The cells were harvested by scraping them into 1.5 ml of PBS and then passaged 10 times through a 27-gauge needle. Nuclei and cell debris obtained by low speed centrifugation (700 × g, 3 min) were collected in the pellet, and the supernatant of this centrifugation step was used to recover the membrane fraction by high speed centrifugation (107,000 × g; rotor TLA100.3; Beckmann TL100 centrifuge, for 25 min). The water-soluble proteins were found in the supernatant of this centrifugation step. All samples were resuspended in 1× radioimmune precipitation buffer (20 mm Tris, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% deoxycholate, 0.1% SDS, 1 mg/liter bovine serum albumin, pH 7.6), denatured by the addition of 1% SDS, and heated to 95 °C before they were chilled on ice. After sonication, the samples were brought to a final concentration of 0.3% SDS by the addition of 1× radioimmune precipitation buffer and were used further in immunoprecipitation experiments.
Immunoprecipitation and Quantification of Erns
Immunoprecipitation of proteins was carried out as described (32) with 5 μl of rabbit Erns antiserum or 100 μl of mAb 24/16 (anti-Erns). After incubation of samples with Staphylococcus aureus (42), the bound immune complexes were eluted in sample buffer by heating to 95 °C. The S. aureus bacteria were removed by centrifugation, and the supernatants were analyzed by 10% SDS-PAGE. Where specified, the precipitated proteins were treated before electrophoresis with 1 μl of endoglycosidase H or PNGase F (New England Biolabs) for 1 h at 37 °C as suggested by the supplier.
The gels were exposed to PhosphorImaging plates and scanned with a Fujifilm Bas 1500 scanner (Raytest, Straubenhardt, Germany). Signal intensities were determined using TINA 2.0 software (Raytest). The amount of Erns cleaved or uncleaved contained in all four steps of fractionation was taken as 100%.
The overwhelming amount of the viral glycoproteins analyzed here was found in the membrane fractions obtained after the low speed or high speed centrifugation. The soluble fraction contained only minimal amounts of viral proteins (data not shown). Therefore, only the results obtained for the tissue culture fluid and the membrane-containing fractions are shown in the present report.
Cell-free Translation
A coupled transcription/translation system (TNT® T7 coupled reticulocyte lysate system, Promega) supplemented with canine microsomal membranes (Promega) was used to study ErnsE1 cleavage in vitro. The assays were performed as suggested by the supplier in a final volume of 12,5 μl. Proteins were labeled with [35S]methionine. In some cases, inhibitors were added at the final concentration indicated in the legend to Fig. 4. MeOSuc-Ala-Ala-Pro-Val chloromethyl ketone (signal peptidase inhibitor (SP-I)) was obtained from Sigma. The protease inhibitors N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (for γ-secretase), leupeptin (for serine/cysteine proteases), and 1,10-phenanthroline (for site-2 protease) were purchased from Sigma, and the signal peptide peptidase inhibitor 1,3-di-(N-carboxybenzoyl-l-leucyl-l-leucyl)amino acetone was from Calbiochem. The glycosylation acceptor peptide Ac-NYT-NH2 was kindly provided by Mark O. Lively.
FIGURE 4.

Influence of different protease inhibitors on ErnsE1 processing. A, SDS-PAGE with products of in vitro translation experiments with construct SSeqErns-E1 (shown on top) in the presence of the protease inhibitors specified by letters above the gel (A, SP-I, MeOSuc-Ala-Ala-Pro-Val chloromethyl ketone used at 1.5–2.5 mm; B, signal peptide peptidase inhibitor, 1,3-di-(N-carboxybenzoyl-l-leucyl-l-leucyl)amino acetone, used at 10 μm; C, γ-secretase inhibitor, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester, used at 100 μm; D, serine/cysteine protease inhibitor, leupeptin, used at 25 μm; E, site-2 protease-metalloprotease inhibitor, 1,10-phenanthroline, used at 5 mm). Note that the concentration at which 1,10-phenanthroline was used in this study was shown to inhibit also the site 1 intramembrane protease (71). Translation of the RNA in the absence of xinhibitor or in the absence of inhibitor and microsomal membranes, as specified by the code at the top of the gel, served as controls. Lane 5, showing the products obtained in the presence of inhibitor E, was exposed 5 times longer than the other lanes. See also the legend to Fig. 1. Effects of SP-I on processing of construct SSeqE1-E2 (B) or a preprolactin construct (C) were also determined. For the preprolactin construct, which includes the first 86 amino acids of the precursor protein, the signal peptide of 30 amino acids with the SPase and the signal peptide peptidase (SPPase) cleavage sites are specified. Protein size marker bands are shown with the molecular masses given in kDa.
For in vitro translation of preprolactin, the RNA was generated by in vitro transcription using SP6 polymerase (New England Biolabs), basically as described before (43). The SP-I for the inhibition of the preprolactin cleavage was used at a concentration of 2.5 mm. For all of the other assays, SP-I was added to a final concentration of 1.5 mm.
Translation was carried out at 30 °C for 1 h or for 1 h 30 min. Thereafter, the samples were transferred on ice and provided with antibodies for immunoprecipitation. Where indicated, Proteinase K was added after completion of translation to destroy proteins that had not been translocated. For control purposes, Triton X-100 was added in some cases. The protease protection assays were conducted as described (32).
RESULTS
ErnsE1 Is Cleaved in the ER
The protease processing the ErnsE1 site in the pestivirus polyprotein is not known, mainly because the amino acid sequence at the cleavage site does not fit into the schemes of the possible processing enzymes. As a first step toward identification of the responsible protease, we wanted to determine where in the cell the processing occurs. Erns is preceded by a signal peptide responsible for translocation of this protein and the following E1 into the ER. Processing of this fusion protein could occur either in the ER or in another compartment of the secretory pathway through which the protein is transported on its way out of the cell. Brefeldin A (BFA) can be used to prevent protein transport from the ER to the Golgi, resulting in accumulation of translocated proteins in the ER (44–48). Cells transiently expressing the region of the pestivirus polyprotein encompassing Npro/C/Erns/E1 (construct Npro-E1; Fig. 1A) were treated with BFA. Immunoprecipitation with an antiserum against Erns showed that, in addition to the ErnsE1 precursor, also the processed Erns protein was present (Fig. 1B, lane 1). Due to the altered glycosylation in consequence of the BFA treatment, the bands migrated slightly differently than those in the control without BFA (Fig. 1B, lanes 1 and 2). To support this hypothesis, the precipitated proteins were treated with endoglycosidase H to remove the non-complex carbohydrates from the proteins. After this treatment, the products obtained in the presence of BFA were not distinguishable from the control (Fig. 1B, lanes 3 and 4). Cleavage of ErnsE1 in the presence of BFA indicates that the processing occurs in the ER because BFA blocks ER to Golgi transport (44, 46–48). However, because BFA treatment also leads to redistribution of Golgi proteins into the ER (45), the involvement of Golgi components cannot be excluded by this assay. The results obtained with the endoglycosidase H assay localize ErnsE1 processing to the ER or cis-Golgi compartment.
FIGURE 1.
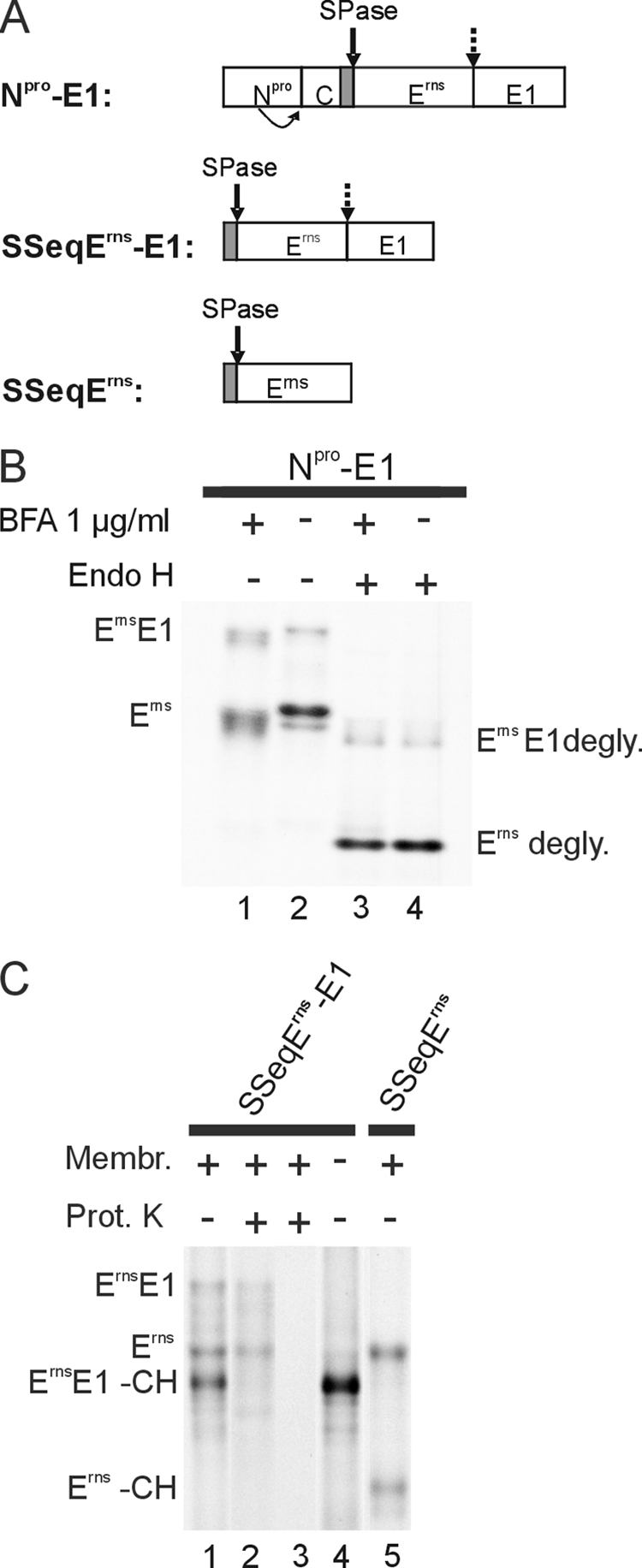
ErnsE1 is cleaved in the ER. A, a schematic drawing of the cDNA constructs used in the expression studies presented in B and C. The names of the constructs are given on the left. The bars show the expressed regions of the viral polyprotein with the designations of the mature viral proteins. Signal peptides are shown as gray bars. A bent arrow indicates autocatalytic cleavage of Npro at its C terminus. A solid arrow points to known SPase cleavage sites, whereas the site investigated here is marked with a broken arrow. B, SDS-PAGE with products precipitated with a rabbit serum against Erns from cells transiently expressing the Npro-E1 construct in the absence or presence of BFA as indicated at the top of the gel. The precipitated proteins were loaded directly onto the gel or pretreated with endoglycosidase H (Endo H) as indicated above the gel. C, SDS-PAGE with products precipitated with the Erns-specific mAb 24/16 after in vitro translation of the constructs specified at the top. Translation was done in the presence or absence of canine microsomal membranes as indicated. To prove translocation of proteins into membrane vesicles, aliquots of the translation products obtained after translation in the presence of membranes were treated with Proteinase K (lane 2) or Proteinase K and Triton X-100 (lane 3) as described (32). The names of the different products are indicated on the left and right of gels in B and C. degly., the removal of carbohydrate side chains by deglycosylation; -CH, glycosylation did not occur because of absence of membranes or inefficient translocation. Prot. K, Proteinase K.
Processing of the ErnsE1 precursor can also be observed after in vitro translation (Fig. 1C). Cleavage of the fusion protein is dependent on the presence of microsomal membranes (Fig. 1C, lanes 1 and 4, respectively), indicating that translocation of the protein into the membranous compartment is essential, which in consequence indicates that a protease within a membranous compartment cleaves the protein. In fact, ErnsE1 is translocated in this assay into ER-derived vesicles before processing. This point is proven by a protease protection approach showing that the unglycosylated ErnsE1 precursor (ErnsE1-CH in Fig. 1C) was destroyed by Proteinase K, whereas the bands representing glycosylated ErnsE1 and fully processed Erns were protected unless the membranes were destroyed with Triton X-100 (compare lanes 1, 2, and 3 in Fig. 1C). Because transport of proteins to vesicles originating from other membrane compartments is not known for microsomes, it can be concluded that processing of the ErnsE1 precursor takes place in the ER.
ErnsE1 Cleavage Can Be Blocked by Mutations at the Cleavage Site
Experimental support for the hypothesis that a protein is cleaved by SPase can be obtained by mutagenesis analysis for the −1- and −3-positions of the cleavage site. As a general rule, these positions have to be occupied by small and uncharged amino acids in an SPase cleavage site. The presence of large and/or charged residues at −1 or −3 blocks SPase processing (35).
The −1- and −3-positions of the Erns/E1 cleavage site are occupied by alanine (C-terminal sequence of Erns, “GAYA”). A variety of mutants were established in which the alanine residues were replaced by other amino acids (Fig. 2A). The effects of these mutations were analyzed in transient expression studies by quantification of the uncleaved versus cleaved ErnsE1. As would be expected for an SPase cleavage site, the exchange of Arg, Phe, or Leu for Ala resulted in almost complete prevention of ErnsE1 processing (Fig. 2, B and C). Interestingly, also constructs with Gly, Cys, or Ser at position −1 (Fig. 2B) and Val, Thr, Ser, or Gly at position −3 (Fig. 2C) showed a severe reduction of cleavage efficiency. This finding was not in agreement with the described requirements for a SPase cleavage site.
FIGURE 2.

ErnsE1 processing efficiency of constructs with mutations at positions −1 and −3 of the cleavage site. A, mutations introduced into construct SSeqErns-E1. The sequence around the Erns/E1 cleavage site (broken arrow) is given, and positions −1 and −3 are indicated. Below, the names of the different constructs with the indicated mutations at −3 or −1 are underlined. B and C, results obtained after expression of the mutations at position −1 (B) or −3 (C). Top, SDS-PAGE with the proteins precipitated with the rabbit Erns serum from cell culture fluid (lanes 1) or the membrane fractions of cells (lanes 2) expressing the constructs given above or no plasmid (Mock). Lane M contains a protein size marker, and the size of the visible bands in kDa is given on the left. On the right, the names of the precipitated proteins are given. Below, a diagram shows the quantification of the results with the intracellular amounts of cleaved Erns (white bars) and uncleaved ErnsE1 precursor (gray bars) given here as a percentage of the total recovered Erns protein. Mean values of at least three independent experiments are given, and the S.D. is indicated. Note that the construct designated “GAYA” represents the wild type.
For control purposes, we wanted to test the same changes in the context of a typical SPase cleavage site and therefore introduced the mutations at the C-terminal end of the signal peptide preceding Erns in the viral polyprotein (Fig. 3). We first changed the C terminus of the wild type signal peptide, which ends with the residues PVAA, into GAYA (construct Npro-Erns/GAYA) to display the sequence found at the Erns/E1 site (Fig. 3A). Afterward, several exemplary exchanges of the above described mutagenesis scheme were introduced into the latter sequence. In transient expression studies, results were obtained that would be expected for a typical SPase cleavage site with Leu at −1 or −3 and Val at −1 blocking processing but Val, Gly, or Ser at −3 and Cys at −1 having basically no influence on processing efficiency (Fig. 3, B and C). A direct comparison of the effects of different mutations introduced at the N-terminal or C-terminal cleavage sites of Erns makes these differences obvious (Fig. 3C and Table 1).
FIGURE 3.

Processing efficiency of constructs with mutations at positions −1 and −3 in a standard signal peptide cleavage site. A, the original sequence context in the internal signal peptide located between capsid protein C and Erns in the CSFV polyprotein (construct Npro-Erns, upper part) and the sequence of the mutant Npro-Erns/GAYA with the C-terminal part of the signal peptide replaced by the GAYA motif found at the Erns C terminus. Below, the names of the different constructs with the indicated mutations at −3 or −1 of Npro-Erns/GAYA are given. B, the results obtained after transient expression, immunprecipitation, and SDS-PAGE of the mutated proteins. C, diagrams showing the processing efficiency determined for the different mutants presented in B in comparison with the results obtained for the equivalent mutations in the context of the Erns C terminus demonstrated in Fig. 2. On top, an explanation of the different types of bars is given. Below, the results for mutations affecting positions −1 (left diagram) or −3 (right diagram) are shown. Each section of the diagrams presents one type of mutation with its effect on processing of the standard SPase cleavage site at the N terminus of Erns (N) or on processing at the Erns C terminus (C). See also the legend to Fig. 2.
TABLE 1.
Amino acids at positions −1 and −3 in signal peptides cleaved by SPase and in the Erns/E1 cleavage site
The upper part of the table presents the residues found in standard SPase cleavage sites with the incidence decreasing from left to right (35, 60, 70). Below, the residues tested in the context of the Erns/E1 cleavage site are listed with the cleavage efficiency decreasing from left to right. The intensity of the decrease is indicated as follows: >, decrease; ≫, strong decrease; ⋙, very strong decrease of processing efficiency.
| Positions | Residues |
|---|---|
| Signal peptides | |
| Position −1 | Ala, Gly, Cys, Ser, Thr |
| Position −3 | Ala, Val, Ser, Cys, Thr |
| Site-directed mutagenesis atm the Erns/E1 site | |
| Position −1 | Ala ≫ Gly ⋙ Cys > Ser > Leu > Val > Arg |
| Position −3 | Ala > Val ≫ Thr > Ser ⋙ Gly > Leu > Phe > Arg |
Taken together, the results of the mutagenesis studies as summarized in Table 1 cannot answer the question of whether the Erns/E1 site is cleaved by SPase, but it has to be stressed that all changes known to impair SP cleavage also block the ErnsE1 processing. The finding that also exchanges expected to be neutral in a typical SPase cleavage site reduce the processing efficiency severely might indicate that additional requirements have to be fulfilled to allow SPase activity in this special case.
The Influence of Protease Inhibitors on ErnsE1 Cleavage
Experiments with specific inhibitors can help to identify the protease active on a given substrate. Therefore, construct SSeqErns-E1 was translated in a cell-free system in the presence of different known protease inhibitors. Among the huge number of different substances blocking proteases, we selected inhibitors specific for intramembrane-cleaving proteases or signal peptidase to cover the likely candidates that could be responsible for ErnsE1 cleavage. Cell-free translation of construct SSeqErns-E1 and immunoprecipitation with anti-Erns monoclonal antibody 24/16 (mAb 24/16) resulted in detection of an unglycosylated precursor product of ∼43 kDa (Fig. 4A, lane 7). In the presence of microsomal membranes and absence of inhibitor, two more bands were detected that represent the glycosylated precursor ErnsE1 of about 66 kDa and the cleaved glycosylated Erns protein of ∼48 kDa (lane 6). The addition of inhibitors against signal peptide peptidase, γ-secretase, site-2 protease-metalloprotease or of leupeptin as a serine/cysteine protease inhibitor did slightly influence the ratio between the different products but did not principally change the outcome (Fig. 4A, lanes 2–5). In contrast, the SP-I MeOSuc-Ala-Ala-Pro-Val chloromethyl ketone (49–52) was able to prevent processing so that the band representing processed Erns was missing (Fig. 4A, lane 1).
As controls for the SPase inhibition by SP-I, its influence on processing of two proteins containing typical SPase cleavage sites was tested. SSeqE1-E2 encodes a typical signal peptide, followed by CSFV E1 and E2. For this construct, the detection of the unprocessed glycosylated E1E2 precursor of about 70 kDa (E1E2 in Fig. 4B) after immunoprecipitation with an anti-E2 monoclonal antibody proves blocking of SPase processing. This result was obtained when SSeqE1-E2 was translated in the presence of SP-I (Fig. 4B, lane 1). However, the precursor was absent, when translation was done in the presence of membranes but in the absence of inhibitor (Fig. 4B, lane 2). Because free glycosylated E2 has about the same size as unglycosylated E1E2, the product of the processing is not readily detected but becomes visible after removal of N-linked carbohydrates with PNGase F (Fig. 4B, lane 3).
As a second control, construct ppL86AA was used, which encodes the N-terminal 86 amino acids of preprolactin. SPase processing leads to release of the corresponding prolactin fragment. Again, this fragment was almost not detected in the presence of SP-I (Fig. 4C). Taken together, the inhibitor studies strongly support the conclusion that processing of ErnsE1 is executed by SPase.
The Type of Signal Peptide and Its Removal Are Not Crucial for ErnsE1 Processing
The internal signal peptide responsible for translocation of Erns into the ER might have special features important for ErnsE1 processing. More importantly, processing of a protein at more than one site can occur in a hierarchic order, where one cleavage has to precede another processing step. This principle is usually seen with SPase and signal peptide peptidase cleavages of signal peptides with signal peptide peptidase activity being dependent on the completion of the SPase cleavage (53–55). Similarly, hierarchical cleavage patterns are quite common for processing of viral polyproteins. Therefore, the possibility could not be excluded that the observed repression of ErnsE1 processing by SP-I represents an indirect effect resulting from prevention of SP removal from the Erns N terminus. We therefore analyzed the influence of different signal peptides and of abrogation of signal peptide processing on ErnsE1 cleavage.
When the original signal peptide of Erns in construct SSeqErns-E1 was replaced by a preprolactin translocation signal fused with an N-terminal V5 tag (construct ppLSSeqErns-E1) or by an insect signal peptide from the mellitin gene (construct mellSSeqErns-E1) translocation, glycosylation and processing of the pestivirus glycoprotein precursor occurred in the same way as in the wild type construct (Fig. 5A, lanes 2, 5, and 8). Also, the inhibition of the cleavage by SP-I was obvious by the absence of the processed Erns band (Fig. 5A, lanes 1, 4, and 7).
FIGURE 5.

Importance of the type of signal peptide and of signal peptide cleavage on ErnsE1 processing. A, the upper part shows schemes of the analyzed constructs in which ErnsE1 was combined with different signal peptides. Below, SDS-PAGE with proteins precipitated with mAb 24/16 after in vitro translation of the constructs indicated above the gels. Translation was done in the presence or absence of microsomal membranes or SP-I, as indicated. B, top, constructs pplSSeqErns-E1 and variant pplSSeqErns-E1* with the cleavage site SP/Erns blocked by mutation, are shown. Below, SDS-PAGE with products of in vitro translation precipitated with mAb 24/16 against Erns or an anti-V5 mAb, as specified above the gel. See also the legend to Fig. 1.
To test whether blocking of signal peptide cleavage prevents processing of the Erns/E1 site, the von Heijne motif of the preprolactin signal peptide in construct ppLSSeqErns-E1 was mutated (construct ppLSSeqErns-E1*, serine at position −1 of the SPase cleavage site replaced by proline). After in vitro translation, the products were immunoprecipitated with mAb 24/16 against Erns or with an anti-V5 mAb. Fully processed Erns could be precipitated with mAb 24/16 after translation of this construct in the absence of the SP-I as for the wild type construct (Fig. 5B, lanes 9 and 3, respectively). Importantly, the processed Erns as well as glycosylated ErnsE1 precursor were also detected with the anti-V5 mAb (Fig. 5B, lane 10). This finding proved that removal of the signal peptide was inhibited as desired. Once again, the addition of SP-I prevented release of Erns, regardless whether the signal peptide mutation was present or not (Fig. 5B, lanes 7 or 1, respectively). In summary, these data show that neither the nature of the signal peptide nor its processing have a significant influence on processing at the Erns/E1 site.
ErnsE1 Cleavage Is Dependent on Glycosylation
A typical signal peptide representing a substrate for SPase is composed of a basic sequence, followed by a hydrophobic transmembrane region and a C-terminal polar region containing the von Heijne motif at positions −3 and −1. The data described above show that the Erns/E1 site is processed by SPase although only the C-terminal polar region is present, whereas the two preceding parts of a typical cleavage site are replaced by an amphipathic helix. The mutation analysis showed that for ErnsE1 cleavage, the requirements with regard to the −1/−3-positions are more stringent than for regular signal peptide processing. These findings could indicate that protein conformation might have a major influence on the processing.
Because of its high content of carbohydrates, folding of Erns is most likely heavily influenced by glycosylation. We therefore analyzed whether prevention of N-glycosylation has an impact on ErnsE1 processing. Constructs Npro-Erns and SSeqErns-E1 were translated in vitro in the presence or absence of Ac-NYT-NH2, a tripeptide substrate of glycosyltransferase that can competitively block glycosylation of other substrates (Fig. 6A) (48, 56). To provide a size marker for unglycosylated and glycosylated Erns, construct SSeqErns was translated without the addition of the inhibitor (Fig. 6A, lane 1). After translation, Erns or fusion proteins containing Erns were precipitated with mAb 24/16. For Npro-Erns, processed Erns was clearly detected as an unglycosylated protein band of ∼26 kDa in the presence of the competitive inhibitor (marked with a white arrowhead in Fig. 6A, lane 2). This finding proves that, despite the inhibition of glycosylation, SPase processing of the internal signal peptide connecting capsid protein C and Erns occurs. In contrast, cleaved unglycosylated Erns was not detected after translation of SSeqErns-E1 in the presence of the inhibitor (Fig. 6, lane 5). To prove that the inhibitor did not prevent translocation of the proteins, proteinase protection assays were conducted. Unglycosylated ErnsE1 was precipitated with the Erns-specific antibody when SSeqErns-E1 was translated in the presence of membranes and treated with Proteinase K (Fig. 6A, lane 8). Because this band was somewhat hidden in a smear, a further control was conducted without immunoprecipitation. In this experiment, the unprocessed ErnsE1 precursor was detected very clearly, regardless of whether the sample was treated with Proteinase K or not (Fig. 6A, lanes 9 and 10, respectively) but was not present when proteinase treatment was done with a sample translated in the absence of membranes (Fig. 6A, lane 11).
FIGURE 6.

ErnsE1 processing under prevention of glycosylation. A, results obtained after in vitro translation. Top, the constructs used in the studies are presented schematically. Below, SDS-PAGE gels are shown with products precipitated with mAb 24/16 after in vitro translation of the indicated products in the presence or absence of the N-glycosylation acceptor (competitive inhibitor) AC-NYT-NH2 (lanes 1–8). Lanes 8–11 show the results of Proteinase K protection assays, with lanes 9–11 containing products of in vitro translation without immunoprecipitation. The presence or absence of Proteinase K is indicated at the top of the gel. Lane 11 shows a control established by translation in the absence of membranes and inhibitor followed by Proteinase K treatment. Please note that in lanes 9–11 a strong unspecific band migrating a bit faster than Erns. The white arrow marks unglycosylated Erns cleaved by SPase from the Npro/C/Erns precursor. B, results of transient expression assays conducted with the constructs shown on top. The gels show the proteins precipitated with the given antibodies (Erns, mAb 24/16 against Erns; C, rabbit serum against C protein; E2, mAb A18 against E2) from the extracts of cells transfected with the indicated constructs. The transfected cells were treated with tunicamycin as indicated above the gels. Some of the precipitated proteins were deglycosylated with PNGase F (lanes 3, 6, and 10). See also the legends to Figs. 1 and 2. Please note that E1 is visible in lane 11 due to coprecipitation with E2 because of formation of a disulfide-linked heterodimer in infected cells (13). Prot. K, Proteinase K.
As a second approach, different CSFV expression constructs were transiently expressed in BHK-21 cells in the presence of the N-glycosylation inhibitor tunicamycin. In addition to SSeqErns-E1, constructs Npro-E1 and SSeqE1-E2 were used, which contain the polyprotein region covering Npro/C/SP/Erns/E1 or SP/E1/E2, respectively (Fig. 6B). Proteins expressed from the transiently transfected cDNA constructs were labeled with radioactive amino acids in situ and subsequently precipitated with mAb 24/16 for Erns, A18 for E2, or rabbit antiserum D1* for C. Before SDS-PAGE, part of the precipitates were treated with PNGase F to remove the N-linked carbohydrates.
Fully processed Erns without carbohydrates has a size of about 26 kDa. A corresponding band was detected for SSeqErns-E1 or Npro-E1 when the constructs were expressed in the absence of tunicamycin, and the precipitated proteins were treated with PNGase F (Fig. 6B, lanes 3 and 6). In contrast, only the 43 kDa band of nonglycosylated ErnsE1 was detected when expression was done in the presence of tunicamycin (Fig. 6B, lanes 1 and 4). Thus, processing of the Erns/E1 site was inhibited by tunicamycin treatment. However, this effect was specific for this individual processing site because the cleavage at the typical SPase sites at the N terminus of Erns or at the E1/E2 border occurred also when glycosylation was prevented. This fact could be concluded from the detection of the C protein (Fig. 6B, lane 7) or processed deglycosylated E2 (Fig. 6B, lane 9) when expression was done under tunicamycin treatment. Tunicamycin is a widely used substance that inhibits N-glycosylation but not translocation or SPase activity. Accordingly, processing of the C/Erns and the E1/E2 sites was observed, whereas processing of the ErnsE1 precursor was blocked in the presence of the drug. Taken together, the results show that processing of ErnsE1 can only occur when the protein is glycosylated, a finding that strongly supports the conclusion that the overall conformation of the protein is especially important for processing of ErnsE1.
The Integrity of the Erns Membrane Anchor Is Crucial for ErnsE1 Processing
The prevention of N-glycosylation certainly has a major effect on the structure of a protein containing nine potential sites for N-glycosylation. To investigate whether also less invasive alterations of the Erns structure can influence ErnsE1 processing, further mutants were tested. The C-terminal region of Erns is of special interest because it represents the membrane anchor of the protein that interacts with the lipid bilayer via an amphipathic helix, thereby possibly presenting the cleavage site to SPase. Three different deletion mutants were established on the basis of SSeqErns-E1. Each mutant protein contained a deletion of 14 amino acids of the Erns C-terminal region located upstream (Erns-Del 158–171), at the N-terminal border (Erns-Del 172–185), or in the center (Erns-Del 200–213) of the region mapped as membrane anchor (Fig. 7A). After transient expression of the latter two constructs, a severe reduction of ErnsE1 processing was observed in comparison with the wild type construct. For Erns-Del 172–185, processing seemed to be completely blocked because no Erns could be detected. In contrast, construct Erns-Del 158–171 showed about wild type processing efficiency (Fig. 7B). Similar results were obtained for two further mutants with 14 amino acid deletions located further upstream of position 158 (data not shown).
FIGURE 7.

Effect of short deletions or alanine insertions in the Erns C-terminal region on ErnsE1 processing. A, construct SSeqErns-E1 is shown with the C-terminal region of Erns presented as an enlargement with the sequence given in one-letter code (residue number in the mature Erns given at the top). The deletions and the positions of the inserted alanine residues (underlined) are demonstrated with the names of the corresponding expression constructs specified on the left. In analogy to the bovine viral diarrhea virus Erns, the amphipathic helix should start around position 180, as defined by an alanine insertion scanning approach (32). B, SDS-PAGE with the proteins precipitated with mAb 24/16 from tissue culture fluid (lanes 1) and cell extracts (lanes 2) of cells transfected with the indicated constructs. C, controls that were either done with Proteinase K protection assays using products of in vitro translation (lanes 1, 2, 4, and 5) or proteins precipitated from transfected cells (similar to B) that were deglycosylated with PNGase F. *, the membranes were not added as in the in vitro translation assays but were present because of expression in cells. See also the legends to Figs. 1 and 2.
A more subtle effect on the arrangement of the Erns amphipathic helix can be achieved by insertion of single alanine residues into the helix, resulting in a ∼100° twist around the helix axis. This alteration provokes a significant disturbance of the amphipathic character of the helix without affecting the overall helical structure. Insertion of alanine at position 191 of the Erns sequence was shown before to reduce the efficiency of membrane binding from about 87% (wild type) to ∼30% (32). When this mutation was introduced into SSeqErns-E1, the transient expression analysis revealed a severe reduction of ErnsE1 processing (Fig. 7B, construct Erns-Ala191). The amphipathic character of the helix can in part be restored when 3 or 4 alanine residues are inserted. Indeed, a higher processing efficiency was observed for construct Erns-3×Ala191 compared with Erns-Ala191. However, the restoring effect was rather low, which fits with the results obtained in the analysis of membrane association of equivalent mutants at position 191 (32).
Secretion of the cleaved Erns of mutants Erns-Del 200–213, Erns-3×Ala191, and Erns-Ala191 was more efficient than for the wild type or the Erns-Del 158–171 protein but less impressive than after expression of the mutated Erns alone (32), due to the overall much lower amount of cleaved Erns compared with the yield after expression of Erns alone. As expected, the ErnsE1 fusion protein is not secreted at all because the transmembrane region at the C terminus of E1 ensures efficient membrane binding of the fusion protein. To prove that the introduced mutations do not impair protein translocation, proteinase protection assays were conducted for constructs Erns-Ala191 and Erns-Del 200–213. These assays showed that both the glycosylated ErnsE1 precursor and the fully processed Erns protein were protected from the Proteinase K, whereas the unglycosylated precursor (ErnsE1-CH) was destroyed (Fig. 7C, lanes 1 and 2 or lanes 4 and 5). The fact that the formerly mentioned bands represent glycosylated proteins and thus must have been translocated was also proven by deglycosylation with PNGase F of transiently expressed proteins precipitated from transfected BHK-21 cells (Fig. 7C, lanes 3 and 6). Moreover, the PNGase F-treated samples demonstrate again the poor processing of the two mutant constructs because the Erns-CH band is much less prominent than the precursor band ErnsE1-CH.
Importantly, all of the different mutations with alterations affecting membrane interaction of the Erns C-terminal helix showed significantly reduced processing of the ErnsE1 precursor, whereas changes outside of the helix had no significant effect.
DISCUSSION
Posttranslational modification of viral envelope proteins is usually done by the host cellular system that is normally responsible for trimming and maturation of cellular (surface) proteins. The possible modifications include removal of signal peptides by SPase and signal peptide peptidase cleavage, protein glycosylation, acylation, phosphorylation, and post-ER maturation cleavage by, for example, Golgi proteases. These processes are essential to obtain functional viral envelope proteins.
Positive strand RNA viruses express their proteins via polyproteins that are subsequently processed into the mature products. The enveloped members of this virus group employ host systems also for cleavage of the polyprotein region giving rise to the viral structural proteins. To enable such a processing scheme, the structural polyprotein adopts a multimembrane-spanning topology, in which individual proteins are separated by transmembrane regions, often organized in a head to tail arrangement of stop transfer sequence and signal peptide. These structures are then cleaved at typical SPase cleavage sites in the signal peptide moiety. Upon completion of the processing process, the transmembrane regions form the membrane anchor of the N-terminal cleavage product. The same concept is also found in pestiviruses with SPase cleavage occurring at typical SPase cleavage sites at the N termini of glycoproteins Erns and E2 and the non-structural proteins p7 and NS2. The only site that does not conform to SPase cleavage site requirements is the one between Erns and E1. This site has long been known to be special because processing of ErnsE1 is delayed, so that ErnsE1 can always be detected as a precursor in infected cells (9). It could be hypothesized that the ErnsE1 fusion protein has a biological function in the virus life cycle, but the fact that viruses with an artificial deletion of the Erns-coding sequence can be efficiently complemented with Erns in trans argues against this hypothesis (21, 57, 58). On the other hand, it has recently been shown that processing of ErnsE1 is crucial for pestivirus viability (59).
Because the Erns/E1 processing site lacks a hydrophobic h-region, which is regarded as a crucial part of a SPase cleavage site, it was speculated whether the delayed processing at this site was due to processing in a downstream compartment of the secretory pathway (9, 59) or resulted from inefficient SPase cleavage in consequence of the unusual structure. As reported here, the ErnsE1 precursor is cleaved in the ER. Moreover, the data show that this site is cleaved by SPase despite its unusual features. This conclusion is based on the facts that processing can be blocked by a SPase inhibitor that was used before in a variety of analyses (49–52) and by typical exchanges of the amino acids at the −1- or −3-positions of the cleavage site. However, the mutagenesis analyses indicated again that the Erns/E1 cleavage site is unusual. Introduction of cysteine at position −1 considerably impaired ErnsE1 processing, whereas the same exchange in a regular SPase cleavage site had basically no effect. Similarly, a glycine at position −3 in the context of the Erns/E1 site had almost the same inhibitory effect as a leucine in −3, whereas glycine at −3 was neutral when tested in the context of a typical SPase cleavage site. These results make it obvious that sequence requirements exceeding those known for a typical SPase cleavage site have to be fulfilled in order to allow processing of ErnsE1. This hypothesis is supported by the results obtained with mutants with alterations introduced rather far upstream of the Erns/E1 cleavage site. Two deletion mutants and a variant with a single alanine insertion 36 residues upstream of the cleavage site showed considerably reduced or even no processing of the precursor. In part, these results can simply be explained by the fact that mutations disturbing the C-terminal amphipathic helix lead to reduced membrane binding of the Erns C terminus, thereby impairing also the contact of the cleavage site with SPase. This is true for the alanine insertion at position 191 (32) and certainly has to be taken into account for the two deletions introduced into the C-terminal region. However, the argument of reduced membrane binding can hardly explain the results obtained with the point mutations at positions −3 and −1. Moreover, it also has to be questioned why the inhibition of protein glycosylation was able to prevent ErnsE1 processing because this manipulation does not directly affect the C terminus of the protein. Membrane binding of a unglycosylated artificial fusion protein containing the amphipathic helix of Erns has been demonstrated (33). It therefore seems justified to speculate that, at least in the latter cases, structural constraints prevent proper interaction between enzyme and substrate, so that it cannot be cleaved.
When signal peptides, the regular substrates of SPase, were compared, a high degree of variation was detected that concerned both the length and sequences of the N-terminal region, h-region, and c-regions (35, 60). Thus, one could conclude that SPase is highly flexible with regard to its substrate as long as the general scheme and the −1, −3 rule are obeyed. It even can be questioned whether most of the typical features of a signal peptide are actually important for its acceptance as a substrate by SPase or whether these features are only necessary for the initiation of translocation. In an oversimplified view, one could regard only the c-region as the sequence interacting with the protease, with the −1, −3 residues being responsible for substrate recognition. However, such relaxed conditions would result in a huge number of SPase cleavage sites and thus contradict the fact that in reality SPase shows a high degree of substrate specificity (60, 61). It is therefore obvious that, in addition to the c-region, further parameters are necessary to define whether a certain sequence represents an SPase substrate. One of these parameters could be the context of substrate and protease with the translocon. However, it has been shown that purified SPase can execute a specific posttranslational cleavage of a substrate in vitro (62–66). In such assay mixtures, regular interaction of substrate and SPase with the translocon is highly unlikely. Similarly, the substrate investigated here is most likely not associated like a standard signal peptide with the Sec61 complex. The Erns/E1 cleavage site has to be inserted into the membrane after complete translocation of Erns and E1 because a fully glycosylated ErnsE1 precursor can be detected within infected cells. Both glycosylation and generation of the E1 C terminus by SPase cleavage of the E1/E2 site can only be executed after completion of E1 translocation. The ErnsE1 precursor is subsequently processed, as shown in pulse-chase experiments (9). The membrane interaction of the Erns amphipathic helix results in an in plane configuration of the Erns/E1 cleavage site region instead of the regular transmembrane situation with the signal peptide still interacting with the translocon. The different topology of the ErnsE1 site is demonstrated in Fig. 8, where the processing of the pestivirus polyprotein region containing Erns, E1, and the N-terminal part of E2 is illustrated. The C terminus of E1 contains two transmembrane regions, a stop transfer sequence and a signal sequence responsible for translocation of E2. The signal sequence is processed by SPase in a typical transmembrane topology, whereas the Erns/E1 site is preceded by the in plane amphipathic helix.
FIGURE 8.

Schematic presentation of the membrane topology and processing scheme of the pestivirus polyprotein region encompassing Erns, E1, and the N-terminal part of E2 (not drawn to scale). Transmembrane regions and the Erns C-terminal amphipathic helix are shown as cylinders, whereas the rest of the proteins are indicated by black lines. The two gray horizontal lines represent the ER- or cytosol-facing surfaces of the ER membrane. The translocon is indicated as a light gray structure. SPase cleavage sites are marked by arrows and SPase. The signal sequence upstream of Erns responsible for Erns translocation is not shown. Note that the transmembrane region upstream of the E2 protein is a regular signal sequence with a typical transmembrane topology that stands in marked contrast to the in plane configuration of the Erns amphipathic helix preceding the Erns/E1 SPase cleavage site.
It is known for signal peptides that the position of the cleavage site relative to the membrane surface is important for substrate recognition by SPase because artificial lengthening of the h-region of a signal peptide prevents SP cleavage (67). Moreover, it has been reported that the typical α-helical conformation of the h-region must not extend into the c-terminal part of the c-region to ensure that the −1, −3 residues are located in a region of extended conformation (66, 68, 69). Thus, a parameter for substrate recognition should be the correct presentation of a suitable cleavage site in a given “cleavage space” close to the membrane surface. For ErnsE1, this hypothesis means that the amphipathic helix has to be inserted into the membrane in such a way that the cleavage site is presented and orientated correctly in this cleavage space. This requirement can explain why alteration of the ectodomain structure by prevention of glycosylation, changes affecting the amphipathic helix itself, or even subtle influences like point mutations can affect cleavage efficiency of this substrate because all of these alterations can disturb the conformation of the complex composed of the C-terminal amphipathic helix and the membrane. Even a subtle change of the conformation of this complex can affect the positioning of the cleavage site with respect to the enzyme.
These ideas indicate that a variety of demands have to be fulfilled to achieve the cleavage of a substrate like the ErnsE1 precursor. To our present knowledge, the Erns/E1 site represents the first SPase cleavage site in which the membrane-traversing h-region is missing and is replaced by an amphipathic helix embedded in plane into the membrane. However, in the past, putative virus-specific biological features often turned out to be also used by the cells themselves, representing alternatives in the standard repertoire of cell biology that are designed to serve special demands.
Pestivirus Erns represents a very fascinating viral protein. It is an essential component of the virus particle, engaged in the infection process, but it is also involved in repression of the host's immune response to a pestivirus infection (26–31). The immune repressive function of Erns is connected with its enzymatic activity, namely its ability to hydrolyze RNA. So far, the target of the RNase is not known. Different theories have been put forward, and in all of them the fact that Erns is secreted from the infected cell plays a central role. Massive loss of the protein from the cell harboring replicating virus would be deleterious because it would interfere with the formation of infectious virus progeny. Thus, an equilibrium between retention and secretion has to be established that retains sufficient amounts of the protein within the infected cell. It is tempting to speculate that the unusual way by which this protein is bound to the membrane is one of the features playing a role in the establishment of this equilibrium. This unusual membrane anchor has imposed another problem on the virus, namely to assure processing of the unusual structure at the Erns/E1 border. As shown in this report, evolution has led to the point that this site is cleaved by SPase despite the absence of the hydrophobic h-region and, more importantly, a totally different membrane topology of the substrate. It can therefore be hypothesized that the interaction of this substrate with the enzyme is more sensible to mutations and structural changes than the standard substrate. More information on the structure of the Erns C terminus in the context of a membrane environment is urgently needed to fully understand this interesting substrate/enzyme interaction. Most likely, the results of these future analyses will also shed some light onto the still quite unclear basis of SPase cleavage specificity in general and will probably provide yet further interesting open questions about the fascinating Erns protein.
Acknowledgments
We thank Maren Ziegler, Petra Wulle, and Janett Wieseler for excellent technical assistance. We are very grateful to Mark O. Lively for helpful discussions in the early phase of the project and for providing the glycosylation acceptor Ac-NYT-NH2. We also thank Robert Stark and Heinz-Jürgen Thiel for providing the rabbit anti Erns serum.
This work was supported by Deutsche Forschungsgemeinschaft Grant Me1367/4.
I. Bintintan and G. Meyers, unpublished results.
- CSFV
- classical swine fever virus (formerly known as hog cholera virus)
- BFA
- brefeldin A
- BHK-21 cells
- baby hamster kidney cells
- mAb
- monoclonal antibody
- PNGase F
- peptide:N-glycosidase F
- SP
- signal peptide
- SPase
- signal peptidase
- SP-I
- signal peptidase inhibitor
- ER
- endoplasmic reticulum
- h-region
- hydrophobic region
- c-region
- hydrophilic region containing the cleavage site.
REFERENCES
- 1.Heinz F. X., Collett M. S., Purcell R. H., Gould E. A., Howard C. R., Houghton M., Moormann J. M., Rice C. M., Thiel H. J. (2000) in Virus Taxonomy: Seventh Report of the International Committee on Taxonomy of Viruses (van Regenmortel M. H. V., Fauquet C. M., Bishop D. H. L., Carstens E. B., Estes M. K., Lemon S. M., Maniloff J., Mayo M. A., McGeoch D. J., Pringle C. R., Wickner R. B. eds) pp. 859–878, Academic Press, Inc., San Diego, CA [Google Scholar]
- 2.Lindenbach B. D., Thiel H.-J., Rice C. M. (2007) in Fields Virology (Knipe D. M., Howley P. M. eds) pp. 1101–1152, Lippincott-Raven Publishers, Philadelphia [Google Scholar]
- 3.Collett M. S., Larson R., Belzer S. K., Retzel E. (1988) Virology 165, 200–208 [DOI] [PubMed] [Google Scholar]
- 4.Collett M. S., Moennig V., Horzinek M. C. (1989) J. Gen. Virol. 70, 253–266 [DOI] [PubMed] [Google Scholar]
- 5.Elbers K., Tautz N., Becher P., Stoll D., Rümenapf T., Thiel H. J. (1996) J. Virol. 70, 4131–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harada T., Tautz N., Thiel H. J. (2000) J. Virol. 74, 9498–9506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heimann M., Roman-Sosa G., Martoglio B., Thiel H. J., Rümenapf T. (2006) J. Virol. 80, 1915–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lackner T., Müller A., König M., Thiel H. J., Tautz N. (2005) J. Virol. 79, 9746–9755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rümenapf T., Unger G., Strauss J. H., Thiel H. J. (1993) J. Virol. 67, 3288–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tautz N., Elbers K., Stoll D., Meyers G., Thiel H. J. (1997) J. Virol. 71, 5415–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiskerchen M., Belzer S. K., Collett M. S. (1991) J. Virol. 65, 4508–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J., Mendez E., Caron P. R., Lin C., Murcko M. A., Collett M. S., Rice C. M. (1997) J. Virol. 71, 5312–5322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel H. J., Stark R., Weiland E., Rümenapf T., Meyers G. (1991) J. Virol. 65, 4705–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiland F., Weiland E., Unger G., Saalmüller A., Thiel H. J. (1999) J. Gen. Virol. 80, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 15.Bolin S., Moennig V., Kelso Gourley N. E., Ridpath J. (1988) Arch. Virol. 99, 117–123 [DOI] [PubMed] [Google Scholar]
- 16.Donis R. O., Corapi W., Dubovi E. J. (1988) J. Gen. Virol. 69, 77–86 [DOI] [PubMed] [Google Scholar]
- 17.Weiland E., Stark R., Haas B., Rümenapf T., Meyers G., Thiel H. J. (1990) J. Virol. 64, 3563–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiland E., Ahl R., Stark R., Weiland F., Thiel H. J. (1992) J. Virol. 66, 3677–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.König M., Lengsfeld T., Pauly T., Stark R., Thiel H. J. (1995) J. Virol. 69, 6479–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horiuchi H., Yanai K., Takagi M., Yano K., Wakabayashi E., Sanda A., Mine S., Ohgi K., Irie M. (1988) J. Biochem. 103, 408–418 [DOI] [PubMed] [Google Scholar]
- 21.Hulst M. M., Moormann R. J. (2001) Methods Enzymol. 342, 431–440 [DOI] [PubMed] [Google Scholar]
- 22.Schneider R., Unger G., Stark R., Schneider-Scherzer E., Thiel H. J. (1993) Science 261, 1169–1171 [DOI] [PubMed] [Google Scholar]
- 23.Hausmann Y., Roman-Sosa G., Thiel H. J., Rümenapf T. (2004) J. Virol. 78, 5507–5512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hulst M. M., Himes G., Newbigin E., Moormann R. J. (1994) Virology 200, 558–565 [DOI] [PubMed] [Google Scholar]
- 25.Windisch J. M., Schneider R., Stark R., Weiland E., Meyers G., Thiel H. J. (1996) J. Virol. 70, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer C., Von Freyburg M., Elbers K., Meyers G. (2002) J. Virol. 76, 8494–8503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyers G., Saalmüller A., Büttner M. (1999) J. Virol. 73, 10224–10235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iqbal M., Poole E., Goodbourn S., McCauley J. W. (2004) J. Virol. 78, 136–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magkouras I., Mätzener P., Rümenapf T., Peterhans E., Schweizer M. (2008) J. Gen. Virol. 89, 2501–2506 [DOI] [PubMed] [Google Scholar]
- 30.Mätzener P., Magkouras I., Rümenapf T., Peterhans E., Schweizer M. (2009) Virus Res. 140, 15–23 [DOI] [PubMed] [Google Scholar]
- 31.Meyers G., Ege A., Fetzer C., von Freyburg M., Elbers K., Carr V., Prentice H., Charleston B., Schürmann E. M. (2007) J. Virol. 81, 3327–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tews B. A., Meyers G. (2007) J. Biol. Chem. 282, 32730–32741 [DOI] [PubMed] [Google Scholar]
- 33.Fetzer C., Tews B. A., Meyers G. (2005) J. Virol. 79, 11901–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langedijk J. P. (2002) J. Biol. Chem. 277, 5308–5314 [DOI] [PubMed] [Google Scholar]
- 35.von Heijne G. (1990) J. Membr. Biol. 115, 195–201 [DOI] [PubMed] [Google Scholar]
- 36.von Heijne G. (1986) Nucleic Acids Res. 14, 4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyatt L. S., Moss B., Rozenblatt S. (1995) Virology 210, 202–205 [DOI] [PubMed] [Google Scholar]
- 38.Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 39.Meyers G., Thiel H. J., Rümenapf T. (1996) J. Virol. 70, 1588–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devereux J., Haeberli P., Smithies O. A. (1984) Nucleic Acids Res. 12, 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shmulevitz M., Duncan R. (2000) EMBO J. 19, 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kessler S. W. (1981) Methods Enzymol. 73, 442–459 [DOI] [PubMed] [Google Scholar]
- 43.Meyers G., Tautz N., Becher P., Thiel H. J., Kümmerer B. M. (1996) J. Virol. 70, 8606–8613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujiwara T., Oda K., Yokota S., Takatsuki A., Ikehara Y. (1988) J. Biol. Chem. 263, 18545–18552 [PubMed] [Google Scholar]
- 45.Lippincott-Schwartz J., Yuan L. C., Bonifacino J. S., Klausner R. D. (1989) Cell 56, 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misumi Y., Misumi Y., Miki K., Takatsuki A., Tamura G., Ikehara Y. (1986) J. Biol. Chem. 261, 11398–11403 [PubMed] [Google Scholar]
- 47.Oda K., Hirose S., Takami N., Misumi Y., Takatsuki A., Ikehara Y. (1987) FEBS Lett. 214, 135–138 [DOI] [PubMed] [Google Scholar]
- 48.Wieland F. T., Gleason M. L., Serafini T. A., Rothman J. E. (1987) Cell 50, 289–300 [DOI] [PubMed] [Google Scholar]
- 49.Casanova C. L., Xue G., Taracha E. L., Dobbelaere D. A. (2006) Mol. Biochem. Parasitol. 149, 144–154 [DOI] [PubMed] [Google Scholar]
- 50.Nilsson I., Johnson A. E., von Heijne G. (2002) FEBS Lett. 516, 106–108 [DOI] [PubMed] [Google Scholar]
- 51.Nilsson I., Johnson A. E., von Heijne G. (2003) J. Biol. Chem. 278, 29389–29393 [DOI] [PubMed] [Google Scholar]
- 52.van Geest M., Nilsson I., von Heijne G., Lolkema J. S. (1999) J. Biol. Chem. 274, 2816–2823 [DOI] [PubMed] [Google Scholar]
- 53.Martoglio B. (2003) Biochem. Soc. Trans. 31, 1243–1247 [DOI] [PubMed] [Google Scholar]
- 54.Weihofen A., Lemberg M. K., Ploegh H. L., Bogyo M., Martoglio B. (2000) J. Biol. Chem. 275, 30951–30956 [DOI] [PubMed] [Google Scholar]
- 55.Weihofen A., Binns K., Lemberg M. K., Ashman K., Martoglio B. (2002) Science 296, 2215–2218 [DOI] [PubMed] [Google Scholar]
- 56.Lau J. T., Welply J. K., Shenbagamurthi P., Naider F., Lennarz W. J. (1983) J. Biol. Chem. 258, 15255–15260 [PubMed] [Google Scholar]
- 57.Reimann I., Semmler I., Beer M. (2007) Virology 366, 377–386 [DOI] [PubMed] [Google Scholar]
- 58.Widjojoatmodjo M. N., van Gennip H. G., Bouma A., van Rijn P. A., Moormann R. J. (2000) J. Virol. 74, 2973–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wegelt A., Reimann I., Zemke J., Beer M. (2009) J. Gen. Virol. 90, 2462–2467 [DOI] [PubMed] [Google Scholar]
- 60.von Heijne G. (1985) J. Mol. Biol. 184, 99–105 [DOI] [PubMed] [Google Scholar]
- 61.von Heijne G. (1984) J. Mol. Biol. 173, 243–251 [DOI] [PubMed] [Google Scholar]
- 62.Jackson R. C., Blobel G. (1977) Proc. Natl. Acad. Sci. U.S.A. 74, 5598–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jackson R. C., Blobel G. (1980) Ann. N.Y. Acad. Sci. 343, 391–404 [DOI] [PubMed] [Google Scholar]
- 64.Lively M. O. (1989) Curr. Opin. Cell Biol. 1, 1188–1193 [DOI] [PubMed] [Google Scholar]
- 65.Lively M. O., Newsome A. L., Nusier M. (1994) Methods Enzymol. 244, 301–314 [DOI] [PubMed] [Google Scholar]
- 66.Paetzel M., Karla A., Strynadka N. C., Dalbey R. E. (2002) Chem. Rev. 102, 4549–4580 [DOI] [PubMed] [Google Scholar]
- 67.Nilsson I., Whitley P., von Heijne G. (1994) J. Cell Biol. 126, 1127–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karla A., Lively M. O., Paetzel M., Dalbey R. (2005) J. Biol. Chem. 280, 6731–6741 [DOI] [PubMed] [Google Scholar]
- 69.von Heijne G. (1998) Nature 396, 111, 113. [DOI] [PubMed] [Google Scholar]
- 70.Choo K. H., Ranganathan S. (2008) BMC Bioinformatics 9, Suppl. 12, S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seidah N. G., Mowla S. J., Hamelin J., Mamarbachi A. M., Benjannet S., Touré B. B., Basak A., Munzer J. S., Marcinkiewicz J., Zhong M., Barale J. C., Lazure C., Murphy R. A., Chrétien M., Marcinkiewicz M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1321–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]