

Abstract
The zebrafish system offers many unique opportunities for the study of molecular biology. To date, only random mutagenesis, and not directed gene knockouts, have been demonstrated in this system. To more fully develop the potential of the zebrafish system, an approach to effectively inhibit the expression of any targeted gene in the developing zebrafish embryo has been developed. This approach uses a transient, cytoplasmic, T7 expression system, injected into the fertilized zebrafish egg to rapidly produce high levels of a ribozyme directed against the mRNA encoded by the targeted gene to inhibit its expression. In a demonstration of this strategy, expression of the recessive dominant zebrafish no tail gene was effectively inhibited by using this strategy to yield a phenotype identical to that resulting from a known defective mutation in this same gene. This, ribozyme-mediated, message deletion strategy may have use in determining the function of genetic coding sequences of unknown function.
Keywords: gene function determination, T7 RNA polymerase, transient expression
The human genome project and allied interests will soon have elucidated the sequence of the entire human genome (1, 2). Although this anticipated advance is exciting, it is also misleading because knowledge of the sequences of ORFs and genetic coding regions without a knowledge of the function of the gene products of this vast array of putative genes provides only very limited insight into the human genome. Full knowledge of the genome requires knowledge of the function of each of the gene products of the putative genetic coding sequences. Although gene function determination is ongoing within the field of molecular genetics, the rate at which the function of a gene can be determined is many orders of magnitude slower than the rate at which a gene can be sequenced. Therefore, a massive backlog of genetic sequences in search of a function looms on the horizon.
One of the most useful means of determining gene function is by “knocking out” or disrupting the unknown coding sequence in an animal model and observing which structure(s) or function(s) is deleted in the resulting “knockout” model (3, 4). Although gene knockout experiments have led to the elegant elucidation of the function of many genes in the mouse, this method is particularly cumbersome and time-consuming (5, 6). Recent studies using ribozymes directed against the mRNA expression product of a targeted gene have shown the ability to down-regulate or “knockdown” the expression of these targeted genes (7). Specifically, Zhao and Pick have generated loss-of-function phenotypes of the fushi tarazu gene in transgenic Drosophila expressing a ribozyme directed against this developmentally important gene (8). Encouraged by these results, we undertook the development of a strategy for determination of gene function in a zebrafish system that we believe will be useful in determining the functions of many mammalian and human genes because there is evidence for strong conservation of nucleotide sequence between analogous zebrafish and mammalian genes (9–15). Rather than attempting to disrupt the gene as in gene knockout experiments, we have chosen to attempt to selectively destroy or significantly diminish the mRNA encoded by a targeted coding sequence. In our experiments, we have used ribozymes (16–19) targeted to sequences in the 5′ region of the encoded message to cleave it and render it nonfunctional. Because the targeted gene is not destroyed or disrupted as in a knockout experiment, but its expression is severely diminished, we describe this approach as a gene “knockdown” strategy.
We have chosen to use the zebrafish system to develop this knockdown strategy because this model system offers several significant advantages over the mouse model yet is also designed with a complex vertebrate species probably containing a majority of those genes found in higher vertebrates such as humans. Zebrafish are oviparous, use external fertilization, and produce large numbers of eggs (20). Their embryogenesis is relatively rapid and requires no attention from the parental fish (20). They develop from the fertilized egg to free swimming fry in 3–4 days (20). Their embryos are nearly transparent, allowing researchers to view the entire process of development from fertilization to hatching (20). These qualities separate zebrafish from the most popular research vertebrate, the mouse. In addition, our strategy attempts to take advantage of an aspect of gene transfer in fish eggs that is distinct from gene transfer in mice and other mammalian species. Whereas a very high percentage of mammalian fertilized eggs microinjected with DNA sequences integrate these sequences into the mammalian chromosome, in fish, such stable chromosomal integration is rare (21–23). But, in fish, a large number of the injected DNA sequences are maintained in a reasonably long term, transient fashion as episomal genetic elements (22, 23), a phenomenon not usually observed in mammalian embryonic gene transfer. Therefore, although the generation of stable transgenic fish or gene knockout fish, by simple integration or by homologous recombination, is extremely difficult, the fish is the ideal species in which to express high levels of a transient transgene product during early embryonic development and for much of the remainder of the first week of life, from the multiple episomal genetic elements still present after egg microinjection.
To initially test this knockdown strategy, we chose the zebrafish no tail (ntl) gene (9, 24) as a target to “knockdown” its expression because homozygous mutant fish lacking this functional gene do not form a tail (25) and this phenotype is easily observable and provides an excellent assay for the disruption of a specific gene function.
MATERIALS AND METHOD
Selection of Targeted Sites on the ntl mRNA.
The mRNA sequence of the ntl gene (9) was analyzed with the rnadraw program (26) to generate the most stable secondary structure of the ntl mRNA molecule. Two CUC and one GUC sites on three loops of the generated ntl mRNA’s secondary structure were selected as potential targeted sites.
Construction of Plasmids.
Two complementary oligonucleotides were synthesized for each of the three targeted sites on the ntl mRNA molecule. These two oligonucleotides were allowed to anneal to yield a double-stranded DNA fragment with SalI and PstI half-sites on its 5′ and 3′ termini, respectively. This SalI/PstI fragment was then inserted into the SalI/PstI sites of plasmid pGval (18) to produce plasmids pGvaRz365, pGvaRz435, and pGvaRz564. In the pGvaRz vectors, the ribozyme gene(s) is under the control of a human adenovirus type 2-associated vaRNA I (Va) gene promoter, which is an RNA polymerase III promoter. These three plasmids were subjected to XbaI/SacI digestion, and an ≈600-bp fragment containing the va promoter and ribozyme sequences was generated and inserted into the EcoRI/SacI sites of the pTM-1 vector (27) lacking the encephalomyocarditis virus internal ribosome entry site sequence (both of the XbaI and EcoRI sites were blunt-ended by Klenow filling-in before SacI cleavage). The resulted plasmids were named pT7vaRz(ntl)365, pT7vaRz(ntl)435, and pT7vaRz(ntl)564. In addition, two other plasmids encoding the ntl435 ribozyme with selected point mutations [Rz(ntl)435–11 and Rz(ntl)435–22] and one with a scrambled sequence [Rz(ntl)435s] were generated (see Table 1) for use in control experiments. Expression of the ribozyme sequences in these plasmids is under the control of the two arranged in tandem T7 and va I promoters as shown in Fig. 1.
Table 1.
Zebrafish egg injections and resulting phenotypic change
| Injected material | Egg injected, n | Egg survived 96 h postfertilization, n | Phenotypic change
|
||
|---|---|---|---|---|---|
| None | Partial | Full | |||
| Uninjected | — | 501 | 501 | 0 | 0 |
| pT7va* + pT7T7 + T7 RNAP | 508 | 211 | 211 | 0 | 0 |
| pT7vaRz(ntl)435†+ pT7T7 + T7 RNAP | 799 | 316 | 256 | 38 (12%)‡ | 22 (7%)‡ |
| pT7vaRz(ntl)435-11† + pT7T7 + T7 RNAP | 257 | 85 | 85 | 0 | 0 |
| pT7vaRz(ntl)435-22† + pT7T7 + T7 RNAP | 426 | 135 | 135 | 0 | 0 |
| pT7vaRz(ntl)435-S† + pT7T7 + T7 RNAP | 631 | 220 | 220 | 0 | 0 |
The data shown were the results of several individual injection experiments. Each injection resulted in quantitatively similar percentages of the phenotypic changes in the injected zebrafish embryos. See text for discussion. The phenotypic changes are as defined and shown in Fig. 2.
pT7va is a control plasmid produced by deletion of ribozyme coding sequences from pT7vaRz.
The complementary flanking sequences (binding arms) are as follows:
Rz(ntl)435: 5′-CCTCCGTT.......AGTTTATT-3′
Rz(ntl)435-11: 5′-CCTCCCTT.......AGATTATT-3′
Rz(ntl)435-22:5′-CCACCCTT.......AGATTTTT-3′
Rz(ntl)435-S: 5′-CTCTACTT.......TTGCTGTA-3′
The percentage is calculated by dividing the number of the phenotypic change by the number of the survived.
Figure 1.
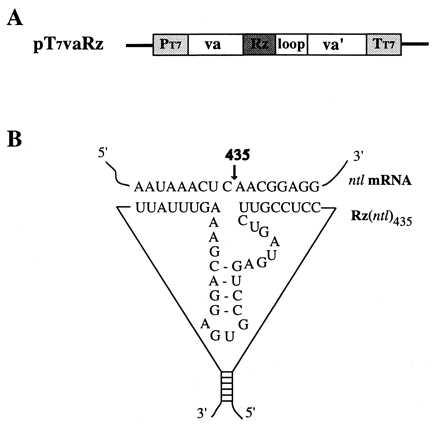
The ntl ribozyme expression vector and schematic secondary structure of ntl ribozyme RNA. (A) Ntl ribozyme expression vector pT7vaRz. The vector pT7vaRz(ntl) was constructed first by an insertion of a ribozyme sequence (Rz) against the ntl mRNA into a va I RNA expression cassette; the cassette containing the ribozyme sequence (va-Rz-loop-va’) subsequently was cloned into a pTM-1 expression vector (27) lacking the encephalomyocarditis virus internal ribosome entry site sequence in such a way that the expression of Rz is under the control of both a T7 promoter (PT7) and an adenovirus va I internal promoter, a pol III promoter (upstream va). The PT7-driven transcription stops at the T7 terminator (TT7) whereas the va I-driven transcription terminates at the 3′ end of the loop. (B) The sequences flanking Rz(ntl) were designed in such a way that the 5′ and 3′ ends of the ntl ribozyme form a stable stem structure (18). Both the ribozyme and its targeted, complementary ntl mRNA sequences, as well as the cleavage site (435), also are shown.
Microinjection.
Whole plasmid DNA was used for microinjection because it is believed that circular plasmid DNA can persist and express in the fish embryos longer than in the linear form (22, 23). The microinjection solution consisted of a sodium–Tris buffer (44 mM NaCl/5 mM Tris⋅HCl, pH 7.0), 90 ng/μl pT7vaRz DNA, with or without 10 ng/μl pT7T7 plasmid DNA and 2.5 unit/μl of T7 RNA polymerase. The microinjection was performed by using methods developed earlier and previously described (21, 23). Each fertilized egg at the single cell stage, 0.5 h postfertilization, received ≈2 nl of microinjection solution after chorion removal by treatment with a 0.25% trypsin solution. After microinjection, the injected embryos were maintained in Holtfreter’s solution (0.35% NaCl/0.01% KCl/0.01% CaCl2) at 28.5°C.
RNA Preparation and Northern Blotting Analyses.
Total RNA from 50 injected or uninjected zebrafish embryos at 5.2, 12, and 24 h postfertilization was isolated by using the RNAzol method (28). After ethanol precipitation and drying, total RNA was dissolved in diethyl-pyrocarbonate water, and its concentration was quantitated spectrophotometrically. Northern blottings were performed by using a NorthernMax Kit from Ambion (Austin, TX). RNA (8 μg) from each sample was loaded to a 1% agarose–formaldehyde gel. After gel transfer and washing, the nylon membrane was hybridized with ntl probe and exposed to x-ray film for 72 h. Subsequently, the membrane was rehybridized with the β-actin and ribozyme probes and was exposed to x-ray films for 12 h.
DNA Fragments and Probe Preparation.
A 900-bp ntl cDNA fragment (114–1014) was amplified from zebrafish embryo total RNA by reverse transcriptase-PCR by using a pair of primers and then cloned into the EcoRV sites of the pCR-3 vector (Promega). This plasmid was named “pCRntl.” A 900-bp fragment was recovered after EcoRI/XhoI cleavage of pCRntl and was used as the template for production of the ntl gene probe. A 600-bp fragment containing the va I promoter and the ribozyme gene was recovered after EcoRI/MluI digestion of pT7vaRz plasmid and was used as a probe for ribozyme RNA detection. A 2-kb fragment of the mouse β-actin gene was used as a β-actin probe to detect zebrafish β-actin mRNA, which served as an internal control for quantitation of the total RNA of each sample in Northern blots. 32P-labeled probes were synthesized by using a random primer kit (GIBCO/BRL).
RESULTS
To provide high levels of a ribozyme targeted against the ntl mRNA in both the nucleus and cytoplasm of zebrafish cells, because it is not presently clear in which of these compartments of the cell ribozymes have their principal activity, an expression cassette was constructed that incorporates both a strong T7 promoter system (27) driving high level cytoplasmic ribozyme expression and the strong adenovirus va I, pol III promoter (18) driving high level nuclear ribozyme expression. The structure and mode of construction of pT7vaRz(ntl) plasmids is described in Fig. 1. Initially, three different ribozyme constructs were generated targeting the 5′ region of the ntl mRNA molecule. By using the rnadraw program (26) to predict the secondary structure of the ntl mRNA, open loops and open regions of the RNA structure containing the GUC or CUC sequence required for ribozyme cleavage were identified and targeted. Three such regions surrounding a CUC at nucleotide 365, a CUC at nucleotide 435, and a GUC at nucleotide 564 were chosen to design ribozymes targeted against. The three plasmids expressing these ribozymes were designated pT7vaRz(ntl)365, pT7vaRz(ntl)435, and pT7vaRz(ntl)564.
Zebrafish eggs were generated, collected, and injected with plasmids at the 1-cell stage by using methods established by one of us (Y.F.X.) (22, 23). Each egg received ≈200 pg of ribozyme-encoding plasmid. Because cytoplasmic expression from the T7 promoter of each of the pT7vaRz(ntl) plasmids requires co-injection with a pT7T7 plasmid prebound to T7 RNA polymerase (27), the presence or absence of this source of T7 RNA polymerase would determine whether or not cytoplasmic ribozyme expression was present within the developing zebrafish egg. Therefore, duplicate experiments with and without 20 pg of pT7T7 plasmid with prebound T7 RNA polymerase were carried out for each of the ribozyme-encoding plasmids pT7vaRz(ntl)365, pT7vaRz(ntl)435, and pT7vaRz(ntl)564 to determine whether cytoplasmic or nuclear ribozyme expression was most effective at disrupting gene expression. Of the six separate experiments, only the three that included the 20 pg of pT7T7 plasmid with prebound T7 RNA polymerase resulted in fry with any evidence of the no tail phenotype (data not shown). Whereas plasmids pT7vaRz(ntl)365 and pT7vaRz(ntl)564 co-injected with the pT7T7 plasmid system showed the no tail phenotype to a limited extent, only plasmid pT7vaRz(ntl)435 showed the full no tail phenotype. Indeed, 7% of fry developing from zebrafish fertilized eggs co-injected with pT7vaRz(ntl)435 plasmid and pT7T7 plasmid pre-bound to T7 RNA polymerase showed a phenotype indistinguishable from the ntl mutant line of zebrafish (Fig. 2). In addition, a higher percentage (12%) of the fry developing from these injected eggs showed a degree of the no tail phenotype (partial) sufficient to indicate the function of the ntl gene (Fig. 2 and Table 1). In an attempt to determine whether antisense oligonucleotides also might yield a similar effect, we injected 300 fertilized zebrafish eggs with a 21-base antisense deoxyoligonucleotide complementary to the (ntl)435 target sequence at molar concentrations 200-fold higher than the injected pT7vaRz(ntl)435 plasmid and were unable to see any abnormal phenotype in the resulting 150 fry. To confirm the specificity of the targeting Rz(ntl)435 ribozyme-flanking sequences for the ntl message, fertilized zebrafish eggs also were injected with plasmids encoding Rz(ntl)435 ribozymes with point mutations in these flanking sequences or with scrambled flanking sequences (Table 1). A single point mutation in the flanking sequences, a double point mutation, and complete scrambling of these sequences did not yield any observation of altered tail phenotype at all (Table 1).
Figure 2.
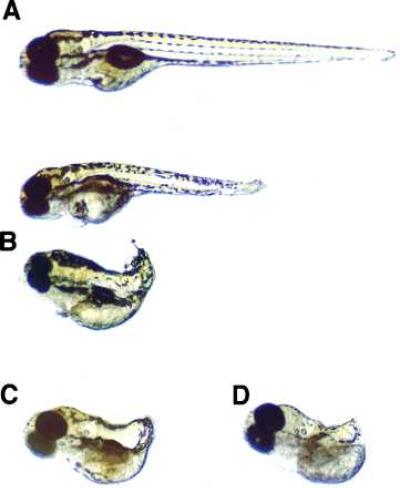
Phenotypic changes after development of zebrafish eggs that had been injected with pT7vaRz(ntl)435, pT7T7, and T7 RNA polymerase. (A) Normal zebrafish at 96 h of embryogenesis. (B) Two examples of partial no tail phenotypic changes at 96 h of embryogenesis in zebrafish developing from eggs injected with pT7vaRz(ntl)435, pT7T7, and T7 RNA polymerase. (C) An example of full no tail phenotypic changes at 96 h of embryogenesis in zebrafish developing from eggs injected with pT7vaRz(ntl)435, pT7T7, and T7 RNA polymerase. (D) A homozygous no tail mutant zebrafish (ntlb195) (23) at 96 h of embryogenesis.
To confirm both the presence of the ntl targeted ribozyme and the destruction of ntl mRNA in zebrafish embryos injected with the ribozyme-expressing pT7vaRz(ntl)435/pT7T7 plasmid combination we undertook a Northern analysis of the RNA complement of these embryos. RNA was isolated from injected zebrafish embryos at 5.2 h after fertilization, the time period when ntl message is reported to be at its highest level (9), as well as at 12 and 24 h postfertilization, and used for a Northern hybridization experiment by using a labeled probe specific for the ntl message (Fig. 3A). Fig. 3A clearly shows the marked decrease in ntl mRNA in the injected embryos compared with the noninjected control embryos. At 5.2 h, the ribozyme-injected group expressed 63% less ntl mRNA than did the uninjected control group as determined by the relative intensities of autoradiographic signals of ntl mRNA on Northern blots by densitometric analysis. Because the ntl message appears and directs its function principally at 5.2 h into development (9), well before the phenotypic manifestation of its action, tail formation, occurs, it is not possible to perform Northern hybridization analysis on only those 7% of the embryos that show the complete no tail phenotype. Therefore, because all injected embryos, including those that eventually showed the normal phenotype (81%) and those that only showed partial no tail phenotype (12%), were included in the embryos from which the mRNA was isolated, we could not expect a complete loss of the ntl mRNA. The ≈63% decrease in ntl mRNA shown in Fig. 3A for injected embryos was consistent with our phenotypic observations of later stage injected zebrafish fry. In addition to Northern analysis of the ntl mRNA, we also used a probe specific for the ntl435 ribozyme to carry out Northern hybridization analysis of the appearance of ntl435 ribozyme in zebrafish embryos at various times during their development. Fig. 3B shows large amounts of ntl435 ribozyme in injected zebrafish embryos from 5.2 to 24 h after fertilization, with the highest levels occurring at 12 h postinjection, clearly indicating the presence of substantial amounts of ribozyme during the period of embryogenesis of the zebrafish.
Figure 3.
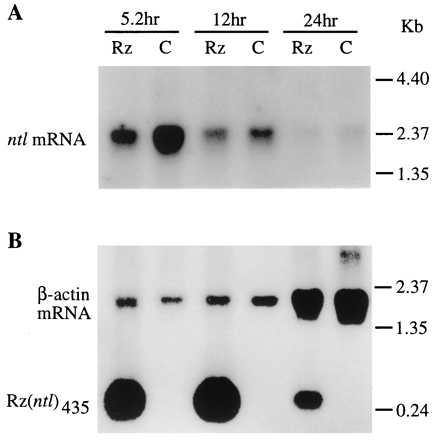
Northern blot analyses of the reduction of ntl mRNA and the presence of ribozyme in the pT7vaRz(ntl)435-injected zebrafish embryos. Fertilized zebrafish eggs were injected with pT7vaRz(ntl)435, pT7T7, and T7 RNA polymerase as described in Fig. 2. Total RNA was isolated from 50 injected and uninjected embryos at 5.2, 12, and 24 h postfertilization. Each RNA sample (8 μg) was subjected to electrophoresis in a 1% agarose–formaldehyde gel. After gel transfer, the membrane containing the transferred RNAs was hybridized with appropriate probes as described in Materials and Methods. (A) Northern blot with the ntl probe. Rz, RNA isolated from the injected embryos; C, RNA isolated from uninjected embryos. (B) The same membrane rehybridized with ribozyme and actin probes. The actin mRNAs serve as the internal RNA quantitation references for each time point.
DISCUSSION
We have shown that the expression of a ribozyme within the developing zebrafish embryo, targeted against a mRNA encoding a specific gene product, can effectively knockdown the level of that message sufficient to block the action of the targeted gene product. We demonstrated this by targeting the message encoded by a zebrafish recessive dominant gene (ntl) (25) with a known function contributing to the formation of the fish’s tail and have shown the complete blockage of tail formation in a significant percentage of fry hatched from embryos injected with plasmids encoding a ntl targeted ribozyme. This result suggests that a ribozyme gene knockdown strategy could be as useful as the established gene knockout strategy to identify and elucidate gene function while being much more efficient.
Furthermore, it seems that this ribozyme-mediated gene knockdown strategy depends on the T7 cytoplasmic expression system used in these experiments. The expression plasmid used in the experiments described herein contained a dual promoter system such that ribozyme expression could be driven by a nuclear promoter, the adenovirus va I, a pol III promoter, or the T7 promoter in a cytoplasmic mode. The importance of the T7 cytoplasmic expression system is suggested because zebrafish developing from eggs that were not co-injected with the pT7T7 plasmid with prebound T7 RNA polymerase, a prerequisite for T7 cytoplasmic expression, showed no alteration of normal tail phenotype despite the potent adenovirus va I, pol III promoter, driving ribozyme expression in plasmids in these fish. This observation also suggests that ribozymes can effectively act against RNA molecules within the cytoplasm because T7 promoter-mediated ribozyme expression is restricted to the cytoplasm (27). Whether the T7 expression system facilitates effective ribozyme cleavage of target RNA as a result of its cytoplasmic site of synthesis or because of its immediate and high level of expression (27) or both remains unclear.
An additional significant finding is that no other developmental or structural abnormalities were observed in any zebrafish developing from eggs injected with the anti-ntl ribozyme expression vector. Furthermore, point mutations in the anti-ntl ribozyme flanking sequences that target the ntl message completely eliminated the appearance of any abnormal tail phenotypes in fish developing from eggs injected with these mutant ribozymes (Table 1). These findings support the remarkable specificity of the ribozyme-mediated gene knockdown strategy used herein.
The results presented here suggest that the determination of zebrafish analogues to human coding sequences, followed by their knockdown by using the strategy outlined herein, could result in a rapid means of identifying their function. Because only knowledge of short sequences of the target mRNA molecule is necessary for the design of the knockdown ribozyme, only minimal sequence information need be determined for zebrafish analogues. By identifying highly conserved motifs within a gene or a family of genes, it may be possible to predict short sequence targets within a zebrafish analogue for sequence determination and targeting. This strategy may hold the potential of greatly increasing the efficiency of determination of gene function.
Acknowledgments
We thank A. Lieber and M. A. Kay, who provided us with the pGvaL plasmid, Dr. Thomas Quertermous for critically reading this manuscript, and G. Westlake and T. Creamer for their valuable technical assistance.
References
- 1.Cox D R, Green E D, Lander E S, Cohen D, Myers R M. Science. 1994;265:2031–2032. doi: 10.1126/science.8091223. [DOI] [PubMed] [Google Scholar]
- 2.Guyer M S, Collins F S. Proc Natl Acad Sci USA. 1995;92:10841–10848. doi: 10.1073/pnas.92.24.10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capecchi M R. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 4.Hasty P, Ramiro R-S, Krumlauf R, Bradley A. Nature (London) 1991;350:243–246. doi: 10.1038/350243a0. [DOI] [PubMed] [Google Scholar]
- 5.Shastry B S. Mol Cell Biochem. 1994;136:171–182. doi: 10.1007/BF00926078. [DOI] [PubMed] [Google Scholar]
- 6.Galli-Taliadoros L A, Sedgwick J D, Wood S A, Korner H. J Immunol Methods. 1995;181:1–15. doi: 10.1016/0022-1759(95)00017-5. [DOI] [PubMed] [Google Scholar]
- 7.Sokol D L, Murray J D. Trans Res. 1996;5:363–371. doi: 10.1007/BF01980201. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J J, Pick L. Nature (London) 1993;365:448–451. doi: 10.1038/365448a0. [DOI] [PubMed] [Google Scholar]
- 9.Schulte-Merker S, Ho R K, Herrmann B G, Nusslein-Volhard C. Development. 1992;116:1021–1032. doi: 10.1242/dev.116.4.1021. [DOI] [PubMed] [Google Scholar]
- 10.Herrmann B G, Labeit S, Pouska A, King T R, Lehrach H. Nature (London) 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- 11.Smith J C, Price B M J, Green J B A, Weigel D, Herrmann B G. Cell. 1990;67:79–87. doi: 10.1016/0092-8674(91)90573-h. [DOI] [PubMed] [Google Scholar]
- 12.Blum M, Gaunt S J, Cho K W Y, Steinbeisser H, Blumberg B, Bittner D, De Robertis E M. Cell. 1992;69:1097–1106. doi: 10.1016/0092-8674(92)90632-m. [DOI] [PubMed] [Google Scholar]
- 13.Izpisua-Belmonte J C, De Robertis E M, Storey K G, Stern C D. Cell. 1993;76:645–659. doi: 10.1016/0092-8674(93)90512-o. [DOI] [PubMed] [Google Scholar]
- 14.Blumberg B, Wright C V E, De Robertis E M, Cho K W Y. Science. 1991;253:194–196. doi: 10.1126/science.1677215. [DOI] [PubMed] [Google Scholar]
- 15.Stachel S E, Grunwald D J, Myers P. Development. 1993;117:1261–1274. doi: 10.1242/dev.117.4.1261. [DOI] [PubMed] [Google Scholar]
- 16.Kruger K, Grabowski P J, Zaug A J, Sands J, Gottschling D E, Cech T R. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 17.Sarver N, Cantin E M, Chang P S, Zaia J A, Ladne P A, Stephens D A, Rossi J. Science. 1990;247:1222–1225. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- 18.Lieber A, Strauss M. Mol Cell Biol. 1995;15:540–551. doi: 10.1128/mcb.15.1.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieber A, Kay M A. J Virol. 1996;70:3153–3158. doi: 10.1128/jvi.70.5.3153-3158.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachidanio rerio) Eugene, OR: Univ. of Oregon Press; 1993. [Google Scholar]
- 21.Zhu Z, Xu K, Li G, Xie Y, He L. Kexue Tongbao Acad Sin. 1986;31:988–990. [Google Scholar]
- 22.Zhu Z, Xu K, Xie Y, Li G, He L. Sci Sin. 1989;2:147–155. [Google Scholar]
- 23.Xie Y, Liu D, Zou J, Li G, Zhu Z. Aquaculture. 1993;111:207–213. [Google Scholar]
- 24.Schulte-Merker S, van Eeden F J M, Halpern M E, Kimmel C B, Nusslein-Volhard C. Development. 1994;120:1009–1015. doi: 10.1242/dev.120.4.1009. [DOI] [PubMed] [Google Scholar]
- 25.Halpern M E, Ho R K, Walker C, Kimmel C B. Cell. 1993;75:99–111. [PubMed] [Google Scholar]
- 26.Matzra O, Wennborg A. Comp Appl Bios. 1996;12:247–249. doi: 10.1093/bioinformatics/12.3.247. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Li Y, Xiong K, Wagner T E. Nucleic Acids Res. 1994;22:2114–2120. doi: 10.1093/nar/22.11.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]